Tracing Baculovirus AcMNPV Infection Using a Real-Time Method Based on ANCHORTM DNA Labeling Technology



Abstract
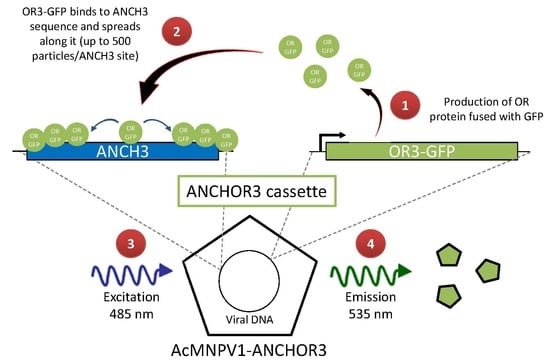
1. Introduction
2. Materials and Methods
2.1. Virus and Cells
2.2. Construction of Baculovirus Transfer Vectors
2.3. Construction of Recombinant AcMNPV–ANCHOR3 Baculovirus
2.4. Spodoptera Exigua Infection
2.5. Fluorescence Imaging and Quantification
3. Results
3.1. Construction of AcMNPV–ANCHOR3 Viruses
3.2. Structural and Biological Characterisation of AcMNPV–ANCHOR Virus
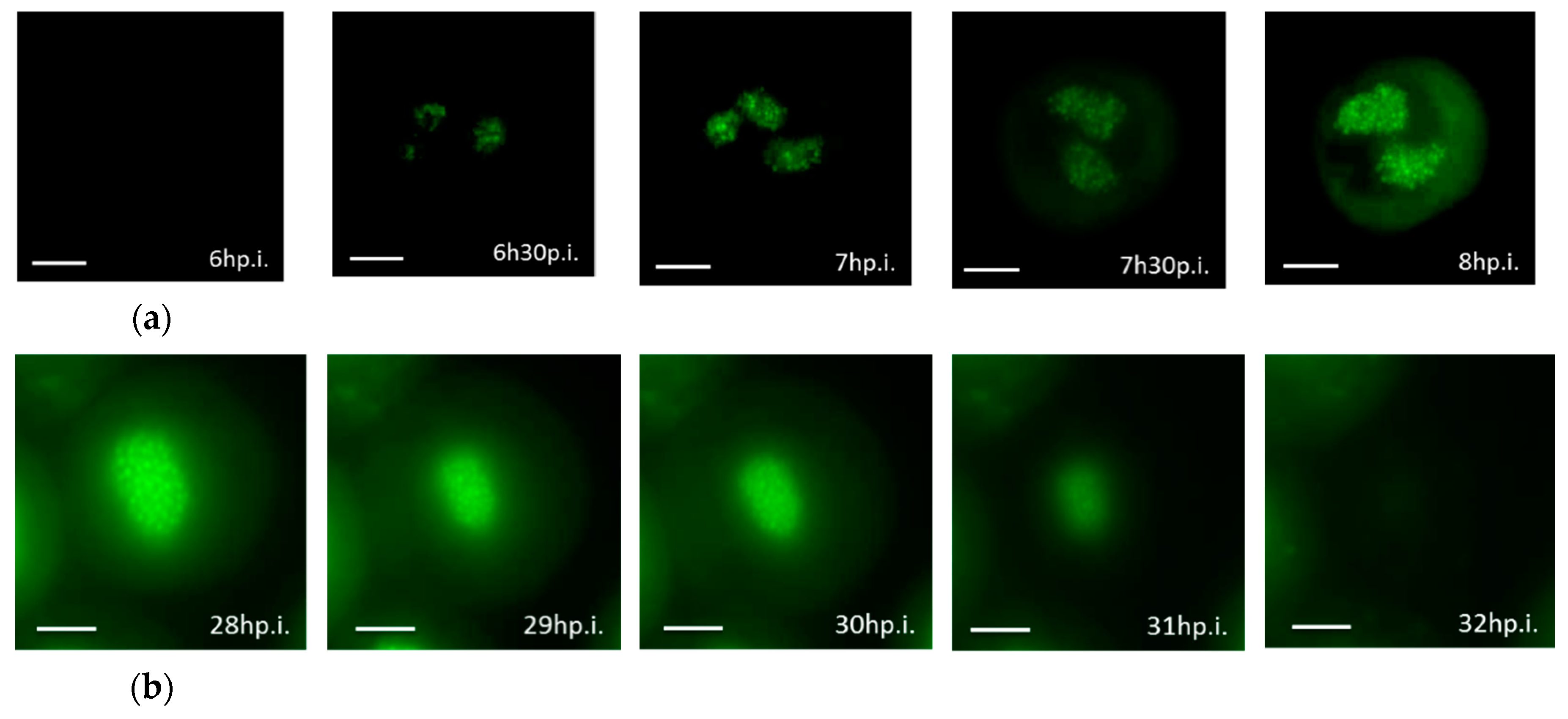
3.3. Real-time Visualization of AcMNPV–ANCHOR
3.4. Budded Virus Quantification by Fluorimetry
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Slack, J.; Arif, B.M. The baculoviruses occlusion-derived virus: Virion structure and function. Adv. Virus Res. 2006, 69, 99–165. [Google Scholar]
- Jehle, J.A.; Lange, M.; Wang, H.; Hu, Z.; Wang, Y.; Hauschild, R. Molecular identification and phylogenetic analysis of baculoviruses from Lepidoptera. Virology 2006, 346, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Organisation for Economic Co-operation and Development. Consensus Document on Information Used in the Assessment of Environmental Applications Involving Baculoviruses; No.20; Organisation for Economic Co-operation and Development: Paris, France, 2002. [Google Scholar]
- Moscardi, F. Assessment of the application of baculoviruses for control of lepidoptera. Ann. Rev. Entomol. 1999, 44, 257–289. [Google Scholar] [CrossRef]
- Lacey, L.A.; Thomson, D.; Vincent, C.; Arthurs, S.P. Codling moth granulovirus: A comprehensive review. Biocontrol Sci. Technol. 2008, 18, 639–663. [Google Scholar] [CrossRef]
- Miller, L.K. The Baculoviruses; Springer: New York, NY, USA, 1997. [Google Scholar]
- Summers, M.D. Milestones leading to the genetic engineering of baculoviruses as expression vector systems and viral pesticides. Adv. Virus Res. 2006, 68, 3–73. [Google Scholar] [PubMed]
- Chambers, A.C.; Aksular, M.; Graves, L.P.; Irons, S.L.; Possee, R.D.; King, L.A. Overview of the baculovirus expression system. Curr. Protoc. Protein Sci. 2018, 91, 5.4.1–5.4.6. [Google Scholar] [CrossRef] [PubMed]
- Rohrmann, G.F. Baculovirus Molecular Biology, 4th ed.; National Center for Biotechnology Information: Bethesda, MD, USA, 2019. Available online: https://www.ncbi.nlm.nih.gov/books/NBK543458/ (accessed on 16 October 2019).
- Smith, G.E.; Summers, M.D.; Fraser, M.J. Production of human beta interferon in insect cells infected with a baculovirus expression vector. Mol. Cell. Biol. 1983, 3, 2156–2165. [Google Scholar] [CrossRef] [PubMed]
- George, S. Use and Control of Co-Expression in the Baculovirus-Insect Cell System for the Production of Multiple Proteins and Complex Biologics. Ph.D. Thesis, University of Waterloo, Waterloo, ON, Canada, 2016. [Google Scholar]
- Graham, T.G.W.; Wang, X.; Song, D.; Etson, C.M.; van Oijen, A.M.; Rudner, D.Z.; Loparo, J.J. ParB spreading requires DNA bridging. Genes Dev. 2014, 28, 1228–1238. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.; Cattoni, D.I.; Walter, J.-C.; Rech, J.; Parmeggiani, A.; Nollmann, M.; Bouet, J.-Y. Stochastic self-assembly of ParB proteins builds the bacterial DNA segregation apparatus. Cell Syst. 2015, 1, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Germier, T.; Kocanova, S.; Walther, N.; Bancaud, A.; Shaban, H.A.; Sellou, H.; Politi, A.Z.; Ellenberg, J.; Gallardo, F.; Bystricky, K. Real-time chromatin dynamics at the single gene level during transcription activation. bioRxiv 2017, 113, 111–179. [Google Scholar]
- Saad, H.; Gallardo, F.; Dalvai, M.; Tanguy-le-Gac, N.; Lane, D.; Bystricky, K. DNA dynamics during early double-strand break processing revealed by non-intrusive imaging of living cells. PLoS Genet. 2014, 10, e1004187. [Google Scholar] [CrossRef] [PubMed]
- Mariamé, B.; Kappler-Gratias, S.; Kappler, M.; Balor, S.; Gallardo, F.; Bystricky, K.; Jung, J.U. Real-time visualization and quantification of human cytomegalovirus replication in living cells using the ANCHOR DNA labeling technology. J. Virol. 2018, 92, 571–589. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, T.; Quentin-Froignant, C.; Carlon-Andres, I.; Lagadec, F.; Rayne, F.; Ragues, J.; Kehlenbach, R.H.; Zhang, W.; Ehrhardt, A.; Bystricky, K.; et al. In vivo labelling of adenovirus DNA identifies chromatin anchoring and biphasic genome replication. J. Virol. 2018, 92, e00795-18. [Google Scholar] [CrossRef] [PubMed]
- Ayres, M.D.; Howard, S.C.; Kuzio, J.; López-Ferber, M.; Possee, R.D. The complete DNA sequence of Autographa californica nuclear polyhedrosis virus. Virology 1994, 202, 586–605. [Google Scholar] [CrossRef] [PubMed]
- Vaughn, J.L.; Goodwin, R.H.; Tompkins, G.J.; Mccawley, A. The establishment of two cell lines from the insect Spodoptera frugiperda (Lepidoptera; Noctuidae). In Vitro 1977, 13, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Cerutti, M.; Chaabihi, H.; Devauchelle, G.; Gauthier, L.; Kaczorek, M.; Lefranc, M.-P.; Poul, M.-A. Recombinant Baculovirus and Use Thereof in the Production of Monoclonal Antibodies. France Patent WO95/20672, 3 August 1995. [Google Scholar]
- Cerutti, M.; Juliant, S.; Mutuel, D.; Devauchelle, G. Baculovirus Recombinants Exprimant Plusieurs Gènes Hétérologues. France Patent FR2891552, 5 October 2005. [Google Scholar]
- Cerutti, M.; Juliant, S.; Thery, S. Système d’expression Baculovirus. France Patent FR3054841, 5 August 2016. [Google Scholar]
- Weyer, U.; Knight, S.; Possee, R.D. Analysis of the very late gene expression by Autographa californica nuclear polyhedrosis virus and the further development of multiple expression vectors. J. Gen. Virol. 1990, 71, 1525–1534. [Google Scholar] [CrossRef] [PubMed]
- King, L.; Hitchman, R.; Possee, R.D. Recombinant baculovirus isolation. In Baculovirus and Insect Cell Expression Protocols; Murhammer, D.W., Ed.; Humana Press: New York, NY, USA, 2016; pp. 77–93. [Google Scholar]
- Kitts, P.A.; Possee, R.D. A method for producing recombinant baculovirus expression vectors at high frequency. BioTechniques 1993, 14, 810–817. [Google Scholar] [PubMed]
- Vida, M.G. De Bombyce; apud Ludovicum Vicentinum: Roma, Italy, 1527. [Google Scholar]
- Erlandson, M.A.; Toprak, U.; Hegedus, D.D. Role of the peritrophic matrix in insect-pathogen interactions. J. Insect Physiol. 2019, 117, 103894. [Google Scholar] [CrossRef] [PubMed]
- Okano, K.; Mikhailov, V.S.; Maeda, S. Colocalization of baculovirus IE-1 and two DNA-binding proteins, DBP and LEF-3, to viral replication factories. J. Virol. 1999, 73, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Vanarsdall, A.L.; Okano, K.; Rohrmann, G.F. Characterization of the Role of Very Late Expression Factor 1 in Baculovirus Capsid Structure and DNA Processing. J. Virol. 2006, 80, 1724–1733. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | A3 | A4 | A5 | Mean | SD |
|---|---|---|---|---|---|
| AcMNPV–Wt | 5.80 × 107 | 9.10 × 106 | 1.93 × 107 | 2.88 × 107 | 2.58 × 107 |
| AcMNPV1–ANCHOR3 | 2.42 × 107 | 3.12 × 106 | 1.27 × 107 | 1.33 × 107 | 1.06 × 107 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hinsberger, A.; Graillot, B.; Blachère Lopez, C.; Juliant, S.; Cerutti, M.; King, L.A.; Possee, R.D.; Gallardo, F.; Lopez Ferber, M. Tracing Baculovirus AcMNPV Infection Using a Real-Time Method Based on ANCHORTM DNA Labeling Technology. Viruses 2020, 12, 50. https://doi.org/10.3390/v12010050
Hinsberger A, Graillot B, Blachère Lopez C, Juliant S, Cerutti M, King LA, Possee RD, Gallardo F, Lopez Ferber M. Tracing Baculovirus AcMNPV Infection Using a Real-Time Method Based on ANCHORTM DNA Labeling Technology. Viruses. 2020; 12(1):50. https://doi.org/10.3390/v12010050
Chicago/Turabian StyleHinsberger, Aurélie, Benoît Graillot, Christine Blachère Lopez, Sylvie Juliant, Martine Cerutti, Linda A. King, Robert D. Possee, Franck Gallardo, and Miguel Lopez Ferber. 2020. "Tracing Baculovirus AcMNPV Infection Using a Real-Time Method Based on ANCHORTM DNA Labeling Technology" Viruses 12, no. 1: 50. https://doi.org/10.3390/v12010050
APA StyleHinsberger, A., Graillot, B., Blachère Lopez, C., Juliant, S., Cerutti, M., King, L. A., Possee, R. D., Gallardo, F., & Lopez Ferber, M. (2020). Tracing Baculovirus AcMNPV Infection Using a Real-Time Method Based on ANCHORTM DNA Labeling Technology. Viruses, 12(1), 50. https://doi.org/10.3390/v12010050