Discovery and Prevalence of Divergent RNA Viruses in European Field Voles and Rabbits

 , , , , , and
, , , , , and
Abstract
1. Introduction
2. Materials and Methods
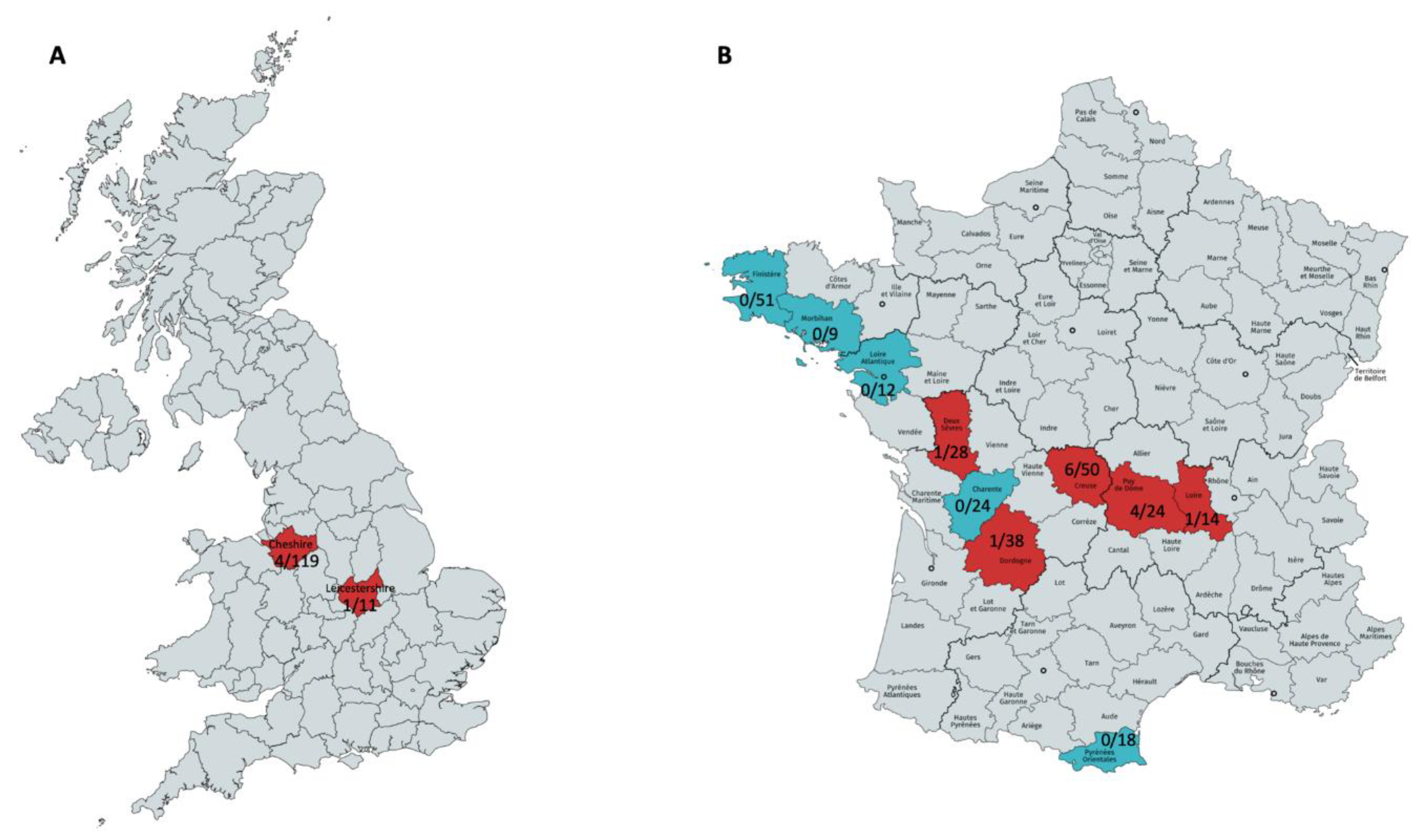
2.1. Samples
2.2. Nucleic Acid Preparation
2.3. High-Throughput Sequencing
2.4. Analysis of Virus Abundance
2.5. Contig Confirmation PCR
2.6. Phylogenetic Analysis
3. Results
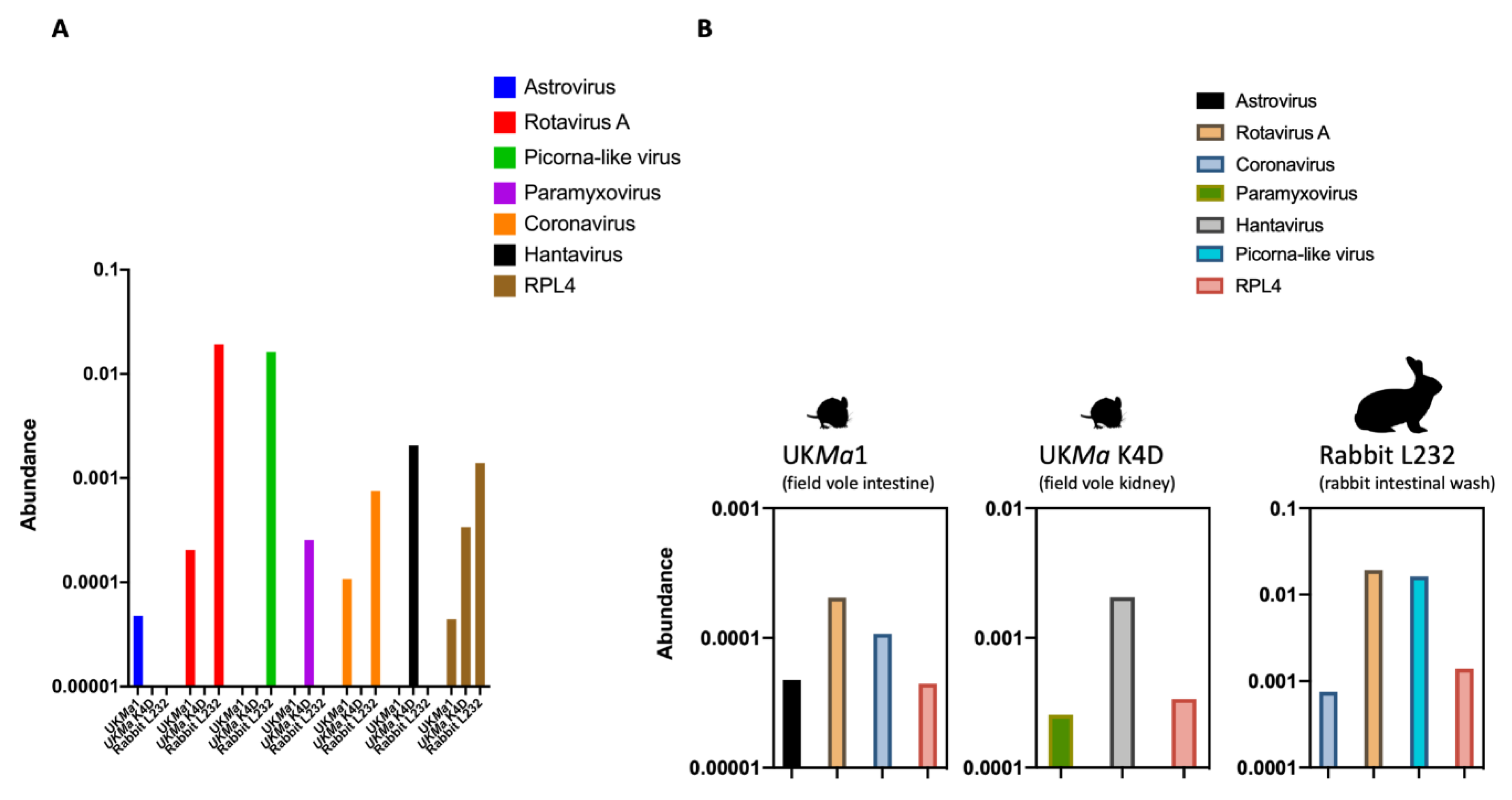
3.1. Abundance of Viral Reads
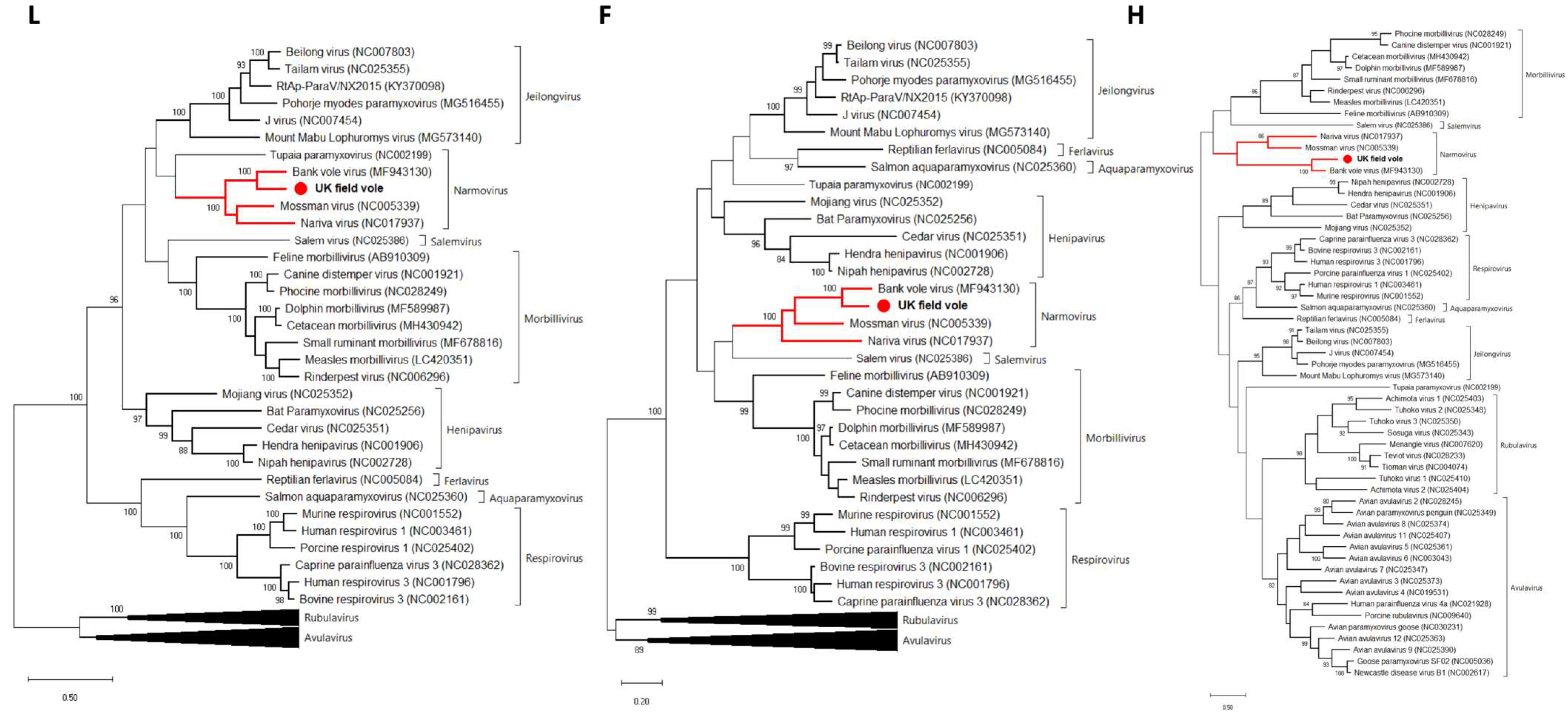
3.2. Paramyxovirus
3.3. Rotavirus A
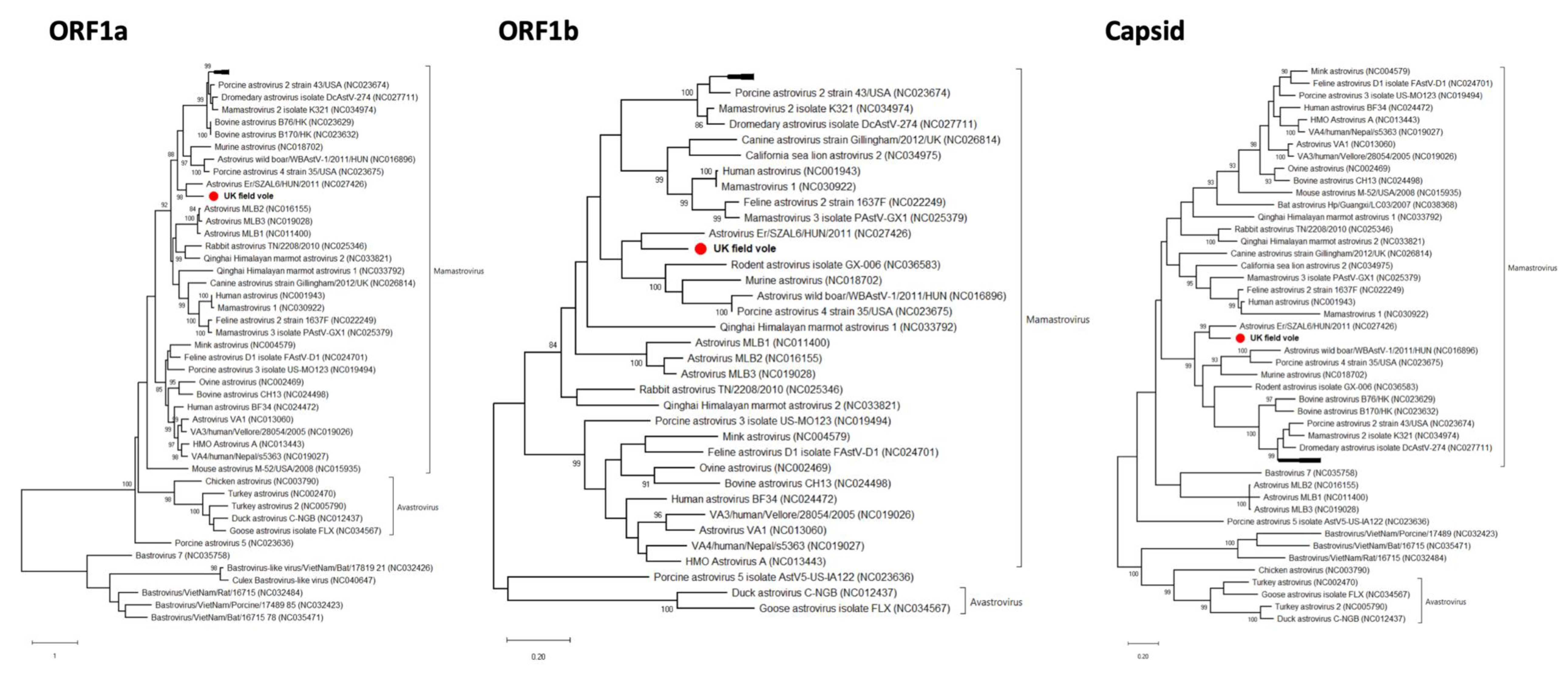
3.4. Astrovirus
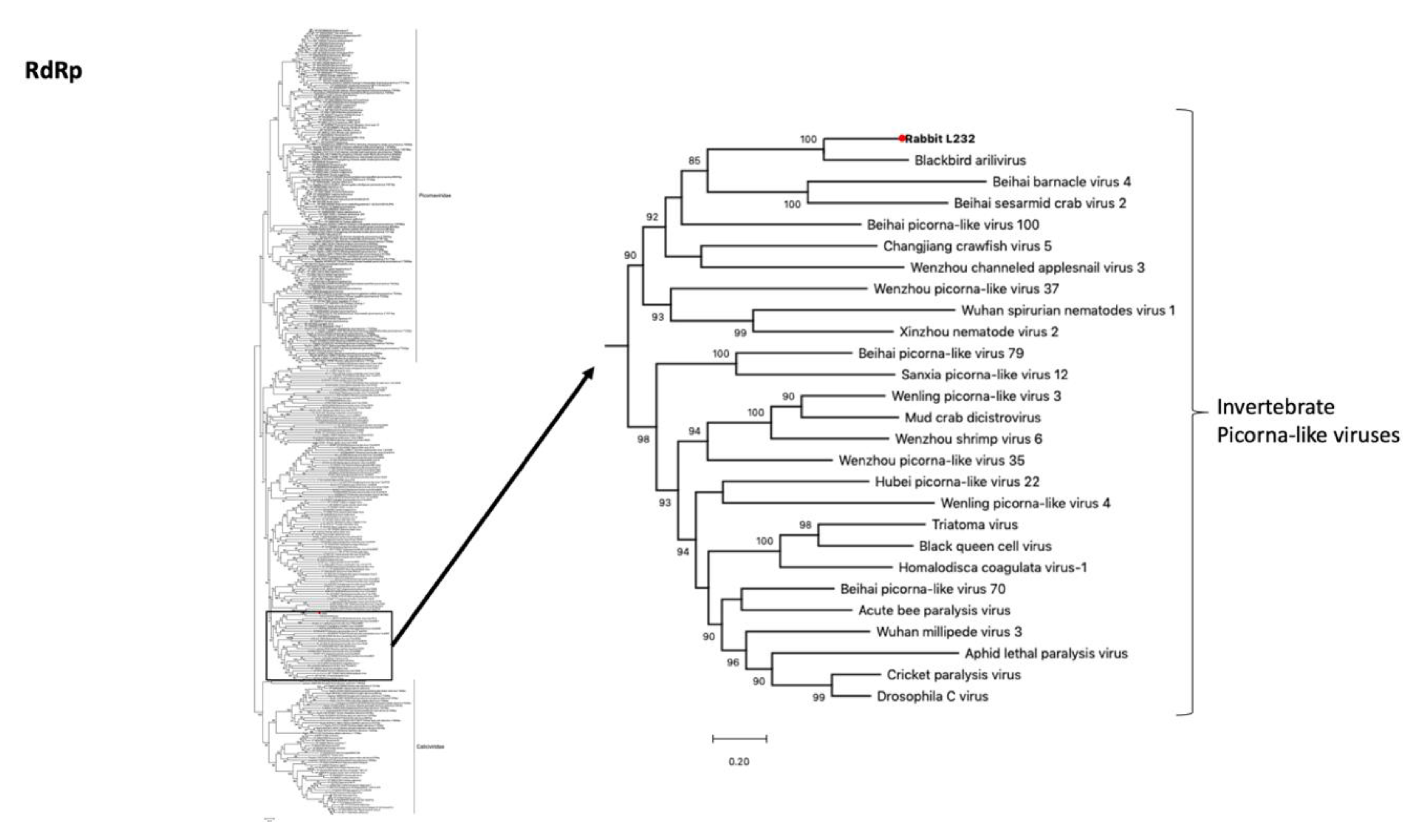
3.5. Picorna-Like Virus
3.6. Plant Viruses
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Koonin, E.V.; Senkevich, T.G.; Dolja, V.V. The ancient Virus World and evolution of cells. Biol. Direct 2006, 1, 29. [Google Scholar] [CrossRef]
- Shi, M.; Lin, X.D.; Tian, J.H.; Chen, L.J.; Chen, X.; Li, C.X.; Qin, X.D.; Jun, L.; Cao, J.P.; Eden, J.S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539. [Google Scholar] [CrossRef]
- Shi, M.; Lin, X.D.; Chen, X.; Tian, J.H.; Chen, L.J.; Li, K.; Wang, W.; Eden, J.S.; Shen, J.J.; Liu, L.; et al. The evolutionary history of vertebrate RNA viruses. Nature 2018, 556, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Lauber, C.; Seitz, S.; Mattei, S.; Suh, A.; Beck, J.; Herstein, J.; Börold, J.; Salzburger, W.; Kaderali1, L.; Briggs, J.A.G.; et al. Deciphering the Origin and Evolution of Hepatitis B Viruses by Means of a Family of Non-enveloped Fish. Viruses. Cell Host Microbe 2017, 22, 387–399. [Google Scholar] [CrossRef] [PubMed]
- Li, C.X.; Shi, M.; Tian, J.H.; Lin, X.D.; Kang, Y.J.; Chen, L.J.; Qin, X.C.; Xu, J.; Holmes, E.C.; Zhang, Y.Z. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. eLife 2015, 4, e05378. [Google Scholar] [CrossRef] [PubMed]
- Webster, C.L.; Waldron, F.M.; Robertson, S.; Crowson, D.; Ferrari, G.; Quintana, J.F.; Brouqui, J.M.; Bayne, E.H.; Longdon, B.; Buck, A.H.; et al. The Discovery, Distribution, and Evolution of Viruses Associated with Drosophila melanogaster. PLoS Biol. 2015, 13, e1002210. [Google Scholar] [CrossRef]
- Wille, M.; Netter, H.; Littlejohn, M.; Yuen, L.; Shi, M.; Eden, J.S.; Klaassen, M.; Holmes, E.C.; Hurt, A.C. A Divergent Hepatitis D-Like Agent in Birds. Viruses 2018, 10, 720. [Google Scholar] [CrossRef]
- Shi, M.; Lin, X.D.; Vasilakis, N.; Tian, J.H.; Li, C.X.; Chen, L.J.; Eastwood, G.; Diao, X.N.; Chen, M.H.; Chen, X.; et al. Divergent Viruses Discovered in Arthropods and Vertebrates Revise the Evolutionary History of the Flaviviridae and Related Viruses. J. Virol. 2016, 90, 659–669. [Google Scholar] [CrossRef]
- Luis, A.D.; Hayman, D.T.; O’Shea, T.J.; Cryan, P.M.; Gilbert, A.T.; Pulliam, J.R.; Mills, J.N.; Timonin, M.E.; Willis, C.K.; Cunningham, A.A.; et al. A comparison of bats and rodents as reservoirs of zoonotic viruses: Are bats special? Proc. Biol. Sci. 2013, 280, 20122753. [Google Scholar] [CrossRef]
- Wu, Z.; Lu, L.; Du, J.; Yang, L.; Ren, X.; Liu, B.; Jiang, J.; Yang, J.; Dong, J.; Sun, L.; et al. Comparative analysis of rodent and small mammal viromes to better understand the wildlife origin of emerging infectious diseases. Microbiome 2018, 6, 178. [Google Scholar] [CrossRef]
- Tsoleridis, T.; Onianwa, O.; Horncastle, E.; Dayman, E.; Zhu, M.; Danjittrong, T.; WachtL, M.; Behnke, J.M.; Chapman, S.; Strong, V.; et al. Discovery of Novel Alphacoronaviruses in European Rodents and Shrews. Viruses 2016, 84, 24. [Google Scholar] [CrossRef] [PubMed]
- Firth, C.; Bhat, M.; Firth, M.A.; Williams, S.H.; Frye, M.J.; Simmonds, P.; Conte, J.M.; Ng, J.; Garcia, J.; Bhuva, N.P.; et al. Detection of zoonotic pathogens and characterization of novel viruses carried by commensal Rattus norvegicus in New York City. MBio 2014, 5, e01933-14. [Google Scholar] [CrossRef] [PubMed]
- Sachsenröder, J.; Braun, A.; Machnowska, P.; Ng, T.F.F.; Deng, X.; Guenther, S.; Bernstein, S.; Ulrich, R.G.; Delwart, E.; Johne, R. Metagenomic identification of novel enteric viruses in urban wild rats and genome characterization of a group A rotavirus. J. Gen. Virol. 2014, 95, 2734–2747. [Google Scholar] [CrossRef] [PubMed]
- Huchon, D.; Madsen, O.; Sibbald, M.J.; Ament, K.; Stanhope, M.J.; Catzeflis, F.; de Jong, W.W.; Douzery, E.J. Rodent phylogeny and a timescale for the evolution of Glires: Evidence from an extensive taxon sampling using three nuclear genes. Mol. Biol. Evol. 2002, 19, 1053–1065. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.G.; Kapusinszky, B.; Wang, C.; Rose, R.K.; Lipton, H.L.; Delwart, E.L. The fecal viral flora of wild rodents. PLoS Pathog 2011, 7, e1002218. [Google Scholar] [CrossRef]
- Ge, D.; Wen, Z.; Xia, L.; Zhang, Z.; Erbajeva, M.; Huang, C.; Yang, Q. Evolutionary history of lagomorphs in response to global environmental change. PLoS ONE 2013, 8, e59668. [Google Scholar] [CrossRef]
- Mahar, J.E.; Hall, R.N.; Shi, M.; Mourant, R.; Huang, N.; Strive, T.; Holmes, E.C. The discovery of three new hare lagoviruses reveals unexplored viral diversity in this genus. Virus. Evol. 2019, 5, vez005. [Google Scholar] [CrossRef]
- Nicholson, L.J.; Mahar, J.E.; Strive, T.; Zheng, T.; Holmes, E.C.; Ward, V.K.; Duckworth, J.A. Benign Rabbit Calicivirus in New Zealand. Appl. Environ. Microbiol. 2017, 83, e00090-17. [Google Scholar] [CrossRef]
- Lanave, G.; Martella, V.; Farkas, S.L.; Fehér, S.M.; Bodnar, L.; Lavazza, A.; Decaro, N.; Buonavoglia, C.; Bányai, K. Novel bocaparvoviruses in rabbits. Vet. J. 2015, 206, 131–135. [Google Scholar] [CrossRef]
- Monchatre-Leroy, E.; Boué, F.; Boucher, J.M.; Renault, C.; Moutou, F.; Ar Gouilh, M.; Umhang, G. Identification of Alpha and Beta Coronavirus in Wildlife Species in France: Bats, Rodents, Rabbits, and Hedgehogs. Viruses 2017, 9, 364. [Google Scholar] [CrossRef]
- Corman, V.M.; Hilgensloh, L.; Voigt, U.; Marklewitz, M.; Siebert, U.; Drosten, C.; Drexler, J.F. Hepatitis E Virus Infection in European Brown Hares, Germany, 2007–2014. Emerg. Infect. Dis. 2019, 25, 1233–1235. [Google Scholar] [CrossRef]
- Kerr, P.J.; Eden, Jo.; di Giallonardo, F.; Peacock, D.; Liu, J.; Strive, T.; Read, A.F.; Holmes, E.C. Punctuated Evolution of Myxoma Virus: Rapid and Disjunct Evolution of a Recent Viral Lineage in Australia. J. Virol. 2019, 93, e01994-18. [Google Scholar] [CrossRef]
- Lau, S.K.P.; Woo, P.C.Y.; Li, K.S.M.; Tsang, A.K.L.; Fan, R.Y.Y.; Luk, H.K.H.; Cai, Ji.; Chan, Kw.; Zheng, Bo.; Wang, M.; et al. Discovery of a novel coronavirus, China Rattus coronavirus HKU24, from Norway rats supports the murine origin of Betacoronavirus 1 and has implications for the ancestor of Betacoronavirus lineage A. J. Virol. 2015, 89, 3076–3092. [Google Scholar] [CrossRef]
- Wang, W.; Lin, X.D.; Guo, W.P.; Zhou, R.H.; Wang, M.R.; Wang, C.Q.; Ge, S.; Mei, S.H.; Li, M.H.; Shi, M.; et al. Discovery, diversity and evolution of novel coronaviruses sampled from rodents in China. Virology 2015, 474, 19–27. [Google Scholar] [CrossRef]
- Tsoleridis, T.; Chappell, J.G.; Onianwa, O.; Marston, D.A.; Fooks, A.R.; Monchatre-Leroy, E.; Umhang, G.; Müller, M.A.; Drexler, J.F.; Drosten, C.; et al. Shared Common Ancestry of Rodent Alphacoronaviruses Sampled Globally. Viruses 2019, 11, 125. [Google Scholar] [CrossRef]
- Drexler, J.F.; Corman, V.M.; Lukashev, A.N.; van den Brand, J.M.A.; Gmyl, A.P.; Brünink, S.; Rasche, A.; Seggewiβ, N.; Feng, H.; Leijten, L.M.; et al. Evolutionary origins of hepatitis A virus in small mammals. Proc. Natl. Acad. Sci. USA 2015, 112, 15190–15195. [Google Scholar] [CrossRef]
- Drexler, J.F.; Corman, V.M.; Müller, M.A.; Lukashev, A.N.; Gmyl, A.; Coutard, B.; Adam, A.; Ritz, D.; Leijten, L.M.; van Riel, D.; et al. Evidence for novel hepaciviruses in rodents. PLoS Pathog. 2013, 9, e1003438. [Google Scholar] [CrossRef]
- Chappell, J.G.; Tsoleridis, T.; Onianwa, O.; Drake, G.; Ashpole, I.; Dobbs, P.; Edema, W.; Kumi-Ansah, F.; Bennett, P.; Tarlinton, R.E.; et al. Retrieval of the Complete Coding Sequence of the UK-Endemic Tatenale Orthohantavirus Reveals Extensive Strain Variation and Supports its Classification as a Novel Species. bioRxiv 2019. [Google Scholar] [CrossRef]
- Thompson, D.J.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic. Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Wille, M.; Shi, M.; Klaassen, M.; Hurt, A.C.; Holmes, E.C. Virome heterogeneity and connectivity in waterfowl and shorebird communities. ISME J. 2019, 13, 2603–2616. [Google Scholar] [CrossRef]
- Zhang, Y.Z.; Chen, Y.M.; Wang, W.; Qin, X.C.; Holmes, E.C. Expanding the RNA Virosphere by Unbiased Metagenomics. Annu. Rev. Virol. 2019, 6, 119–139. [Google Scholar] [CrossRef]
- Alkhovsky, S.; Butenko, A.; Eremyan, A.; Shchetinin, A. Genetic characterization of bank vole virus (BaVV), a new paramyxovirus isolated from kidneys of bank voles in Russia. Arch. Virol. 2018, 163, 755–759. [Google Scholar] [CrossRef]
- Miller, P.J.; Boyle, D.B.; Eaton, B.T.; Wang, L. F. Full-length genome sequence of Mossman virus, a novel paramyxovirus isolated from rodents in Australia. Virology 2003, 317, 330–344. [Google Scholar] [CrossRef]
- Lambeth, L.S.; Yu, M.; Anderson, D.E.; Crameri, G.; Eaton, B.T.; Wang, L.F. Complete genome sequence of Nariva virus, a rodent paramyxovirus. Arch. Virol. 2009, 154, 199–207. [Google Scholar] [CrossRef]
- McDonald, S.M.; Nelson, M.I.; Turner, P.E.; Patton, J.T. Reassortment in segmented RNA viruses: Mechanisms and outcomes. Nat. Rev. Microbiol. 2016, 14, 448–460. [Google Scholar] [CrossRef]
- Matthijnssens, J.; Ciarlet, M.; Rahman, M.; Attoui, H.; Bányai, K.; Estes, M.K.; Gentsch, J.R.; Iturriza-Gómara, M.; Kirkwood, C.D.; Martella, V.; et al. Recommendations for the classification of group A rotaviruses using all 11 genomic RNA segments. Arch Virol 2008, 153, 1621–1629. [Google Scholar] [CrossRef]
- Simmonds, P.; Adams, M.J.; Benkő, M.; Breitbart, M.; Brister, J.R.; Carstens, E.B.; Davison, A.J.; Delwart, E.; Gorbalenya, A.E.; Harrach, B.; et al. Virus taxonomy in the age of metagenomics. Nat. Rev. Microbiol. 2017, 15, 161. [Google Scholar] [CrossRef]
- Holmes, E.C. Reagent contamination in viromics: All that glitters is not gold. Clin. Microbiol. Infect. 2019, 25, 1167–1168. [Google Scholar] [CrossRef]
- Chong, R.; Shi, M.; Grueber, C.E.; Holmes, E.C.; Hogg, C.J.; Belov, K.; Barrs, V.R. Fecal Viral Diversity of Captive and Wild Tasmanian Devils Characterized Using Virion-Enriched Metagenomics and Metatranscriptomics. J. Virol. 2019, 93, e00205–e00219. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, S.; Liu, D.; Zhou, C.; Li, W.; Lin, Y.; Wang, X.; Shen, Q.; Wang, H.; Li, C.; et al. The fecal virome of red-crowned cranes. Arch. Virol. 2019, 164, 3–16. [Google Scholar] [CrossRef]
- Siqueira, J.D.; Dominguez-Bello, M.G.; Contreras, M.; Lander, O.; Caballero-Arias, H.; Xutao, D.; Noya-Alarcon, O.; Delwart, E. Complex. virome in feces from Amerindian children in isolated Amazonian villages. Nat. Commun. 2018, 9, 4270. [Google Scholar] [CrossRef]
- Pankovics, P.; Boros, Á.; Kiss, T.; Delwart, E.; Reuter, G. Detection of a mammalian-like astrovirus in bird, European roller (Coracias garrulus). Infect. Genet. Evol. 2015, 34, 114–121. [Google Scholar] [CrossRef]
- Kiss, O.; Elek, Z.; Moskát, C. High. breeding performance of European Rollers Coracias garrulus in heterogeneous farmland habitat in southern Hungary. Bird Study 2014, 61, 496–505. [Google Scholar] [CrossRef]
- Van Borm, S.; Steensels, M.; Mathijs, E.; Yinda, C.K.; Matthijnssens, J.; Lambrecht, B. Complete coding sequence of a novel picorna-like virus in a blackbird infected with Usutu virus. Arch. Virol. 2018, 163, 1701–1703. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Animal Species | Organ | Origin | Virus | Gene | Fragment Size (nt) | Accession Number |
|---|---|---|---|---|---|---|---|
| UKMa1 | Microtus agrestis | Intestine | UK | Rotavirus A | VP1 (partial) | 2145 | MN626437 |
| UKMa1 | Microtus agrestis | Intestine | UK | Rotavirus A | VP2 (partial) | 987 | MN626438 |
| UKMa1 | Microtus agrestis | Intestine | UK | Rotavirus A | VP3 (partial) | 2306 | MN626439 |
| UKMa1 | Microtus agrestis | Intestine | UK | Rotavirus A | VP6 (partial) | 878 | MN626440 |
| UKMa1 | Microtus agrestis | Intestine | UK | Rotavirus A | NSP2 (partial) | 900 | MN626435 |
| UKMa1 | Microtus agrestis | Intestine | UK | Rotavirus A | NSP3 (partial) | 349 | MN626436 |
| UKMa1 | Microtus agrestis | Intestine | UK | Astrovirus | ORF1a (partial) | 1728 | MN626433 |
| UKMa1 | Microtus agrestis | Intestine | UK | Astrovirus | ORF1b (partial) | 1554 | MN626434 |
| UKMa1 | Microtus agrestis | Intestine | UK | Astrovirus | Capsid (partial) | 1052 | MN626432 |
| UKMa K4D | Microtus agrestis | Kidney | UK | Paramyxovirus | L (partial) | 2731 | MN626428 |
| UKMa K4D | Microtus agrestis | Kidney | UK | Paramyxovirus | H (partial) | 615 | MN626427 |
| UKMa K4D | Microtus agrestis | Kidney | UK | Paramyxovirus | F (partial) | 726 | MN626426 |
| UKMa K4D | Microtus agrestis | Kidney | UK | Paramyxovirus | M (partial) | 760 | MN626429 |
| UKMa K4D | Microtus agrestis | Kidney | UK | Paramyxovirus | P (partial) | 856 | MN626431 |
| UKMa K4D | Microtus agrestis | Kidney | UK | Paramyxovirus | N (partial) | 1046 | MN626430 |
| L232 | Oryctolagus cuniculus | Intestinal wash | FR | Rotavirus A | VP1 (partial) | 3268 | MN626420 |
| L232 | Oryctolagus cuniculus | Intestinal wash | FR | Rotavirus A | VP2 (partial) | 2645 | MN626424 |
| L232 | Oryctolagus cuniculus | Intestinal wash | FR | Rotavirus A | VP3 (partial) | 2536 | MN626421 |
| L232 | Oryctolagus cuniculus | Intestinal wash | FR | Rotavirus A | VP6 (partial) | 1336 | MN626422 |
| L232 | Oryctolagus cuniculus | Intestinal wash | FR | Rotavirus A | VP7 (partial) | 978 | MN626423 |
| L232 | Oryctolagus cuniculus | Intestinal wash | FR | Rotavirus A | NSP2 (partial) | 980 | MN626417 |
| L232 | Oryctolagus cuniculus | Intestinal wash | FR | Rotavirus A | NSP3 (partial) | 951 | MN626418 |
| L232 | Oryctolagus cuniculus | Intestinal wash | FR | Rotavirus A | NSP4 (partial) | 584 | MN626419 |
| L232 | Oryctolagus cuniculus | Intestinal wash | FR | Picorna-like virus | Non-structural polyprotein (partial) | 890 | MN626416 |
| L232 | Oryctolagus cuniculus | Intestinal wash | FR | Picorna-like virus | Non-structural polyprotein contig 2 (partial) | 2187 | MN626416 |
| L232 | Oryctolagus cuniculus | Intestinal wash | FR | Picorna-like virus | Structural polyprotein (partial) | 2349 | MN626416 |
| L232 | Oryctolagus cuniculus | Intestinal wash | FR | Sobemovirus | Polyprotein P2ab (partial) | 1653 | MN626425 |
| L232 | Oryctolagus cuniculus | Intestinal wash | FR | Luteovirus | Peptidase (partial) | 2067 | MN626414 |
| L232 | Oryctolagus cuniculus | Intestinal wash | FR | Luteovirus | RdRp (partial) | 1269 | MN626413 |
| L232 | Oryctolagus cuniculus | Intestinal wash | FR | Luteovirus | Coat protein (partial) | 573 | MN626415 |
| Virus | Gene/Segment | Animal | GenBank acc. No | Most Closely Related Virus | Nucleotide Sequence Identity (%) | Cut Off for Genotypes (%) * | Assigned Genotype |
|---|---|---|---|---|---|---|---|
| Rotavirus A | VP1 | Rabbit | MN626420 | Bovine rotavirus core protein | 96.24 | 83 | R2 |
| Field vole | MN626437 | Human rotavirus A strain B10 | 76.96 | R * | |||
| VP2 | Rabbit | MN626424 | Rotavirus A giraffe/UCD/IRL/2007 | 98.38 | 84 | C2 | |
| Field vole | MN626438 | Rotavirus A strain RVA/Cow-wt/ZAF/1605/2007/G6P [5] | 78.21 | C * | |||
| VP3 | Rabbit | MN626421 | Rotavirus A isolate RVA/Human-tc/MAR/ma31/2011/G8P [14] | 98.34 | 81 | M2 | |
| Field vole | MN626439 | BatRVA/KEN/BATp39/Rousettus aegyptiacus/2015 | 69.74 (in aa) | M * | |||
| VP6 | Rabbit | MN626422 | Rotavirus A strain RVA/roe deer-wt/SLO/D38-14/2014/G6P [15] | 98.8 | 85 | I2 | |
| Field vole | MN626440 | Human rotavirus A isolate Omsk07-79 | 78.46 | I * | |||
| VP7 | Rabbit | MN626423 | Rotavirus A strain RVA/Sheep-tc/ESP/OVR762/2002/G8P [14] | 97.75 | 80 | G8 | |
| NSP2 | Rabbit | MN626417 | Rotavirus A strain RVA/Human-wt/ITA/111-05-27/2005/G6P [14] | 96.99 | 85 | N2 | |
| Field vole | MN626435 | Rotavirus A RVA/Dog-tc/JPN/RS15/1982/G3P [3] | 76.01 | N * | |||
| NSP3 | Rabbit | MN626418 | Rotavirus A strain RVA/Human-wt/BEL/B10925/1997/G6P [14] | 98.86 | 85 | T6 | |
| Field vole | MN626436 | Rotavirus A RVA/Human-wt/JPN/12597/2014/G8P [14] | 76.72 (in aa) | T * | |||
| NSP4 | Rabbit | MN626419 | Rotavirus A strain dog-wt/GER/88977/2013/G8P1 | 98.43 | 85 | E2 | |
| Astrovirus | ORF1a | Field vole | MN626433 | Astrovirus Er/SZAL6/HUN/2011 | 46.3 (in aa) | - | - |
| ORF1b | MN626434 | Astrovirus Er/SZAL6/HUN/2011 | 70 (in aa) | - | - | ||
| Capsid | MN626432 | Astrovirus Er/SZAL6/HUN/2011 | 73.31 (in aa) | - | - | ||
| Picorna-like virus | nonstructural polyprotein | Rabbit | MN626416 | Blackbird arilivirus | 35.57 (in aa) | - | - |
| nonstructural polyprotein | MN626416 | Blackbird arilivirus | 48.75 (in aa) | - | - | ||
| structural polyprotein | MN626416 | Blackbird arilivirus | 34.24 (in aa) | - | - | ||
| Paramyxovirus | L | Field vole | MN626428 | Bank vole virus | 70.66 (in aa) | - | - |
| H | MN626427 | Bank vole virus | 61.19 (in aa) | - | - | ||
| F | MN626426 | Bank vole virus | 76.35 (in aa) | - | - | ||
| M | MN626429 | Bank vole virus | 86.96 (in aa) | - | - | ||
| P | MN626431 | Bank vole virus | 62.41 (in aa) | - | - | ||
| N | MN626430 | Bank vole virus | 89.66 (in aa) | - | - | ||
| Luteovirus | coat protein | Rabbit | MN626415 | Pea enation mosaic virus 1 | 53.90 (in aa) | - | - |
| Peptidase | MN626414 | Pea enation mosaic virus 1 | 38.88 (in aa) | - | - | ||
| RdRp | MN626413 | Alfalfa enamovirus 2 | 77.64 (in aa) | - | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsoleridis, T.; Chappell, J.G.; Monchatre-Leroy, E.; Umhang, G.; Shi, M.; Bennett, M.; Tarlinton, R.E.; McClure, C.P.; Holmes, E.C.; Ball, J.K. Discovery and Prevalence of Divergent RNA Viruses in European Field Voles and Rabbits. Viruses 2020, 12, 47. https://doi.org/10.3390/v12010047
Tsoleridis T, Chappell JG, Monchatre-Leroy E, Umhang G, Shi M, Bennett M, Tarlinton RE, McClure CP, Holmes EC, Ball JK. Discovery and Prevalence of Divergent RNA Viruses in European Field Voles and Rabbits. Viruses. 2020; 12(1):47. https://doi.org/10.3390/v12010047
Chicago/Turabian StyleTsoleridis, Theocharis, Joseph G. Chappell, Elodie Monchatre-Leroy, Gérald Umhang, Mang Shi, Malcolm Bennett, Rachael E. Tarlinton, C. Patrick McClure, Edward C. Holmes, and Jonathan K. Ball. 2020. "Discovery and Prevalence of Divergent RNA Viruses in European Field Voles and Rabbits" Viruses 12, no. 1: 47. https://doi.org/10.3390/v12010047
APA StyleTsoleridis, T., Chappell, J. G., Monchatre-Leroy, E., Umhang, G., Shi, M., Bennett, M., Tarlinton, R. E., McClure, C. P., Holmes, E. C., & Ball, J. K. (2020). Discovery and Prevalence of Divergent RNA Viruses in European Field Voles and Rabbits. Viruses, 12(1), 47. https://doi.org/10.3390/v12010047