Infection Load and Prevalence of Novel Viruses Identified from the Bank Vole Do Not Associate with Exposure to Environmental Radioactivity

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Virus Identification
2.3. qPCR
2.4. Statistical Analysis
3. Results
3.1. Virus Identification from the Next-Generation Sequencing Data
3.2. Adeno-Associated Viruses
3.3. Arterivirus
3.4. Hepaciviruses
3.5. Mosavirus
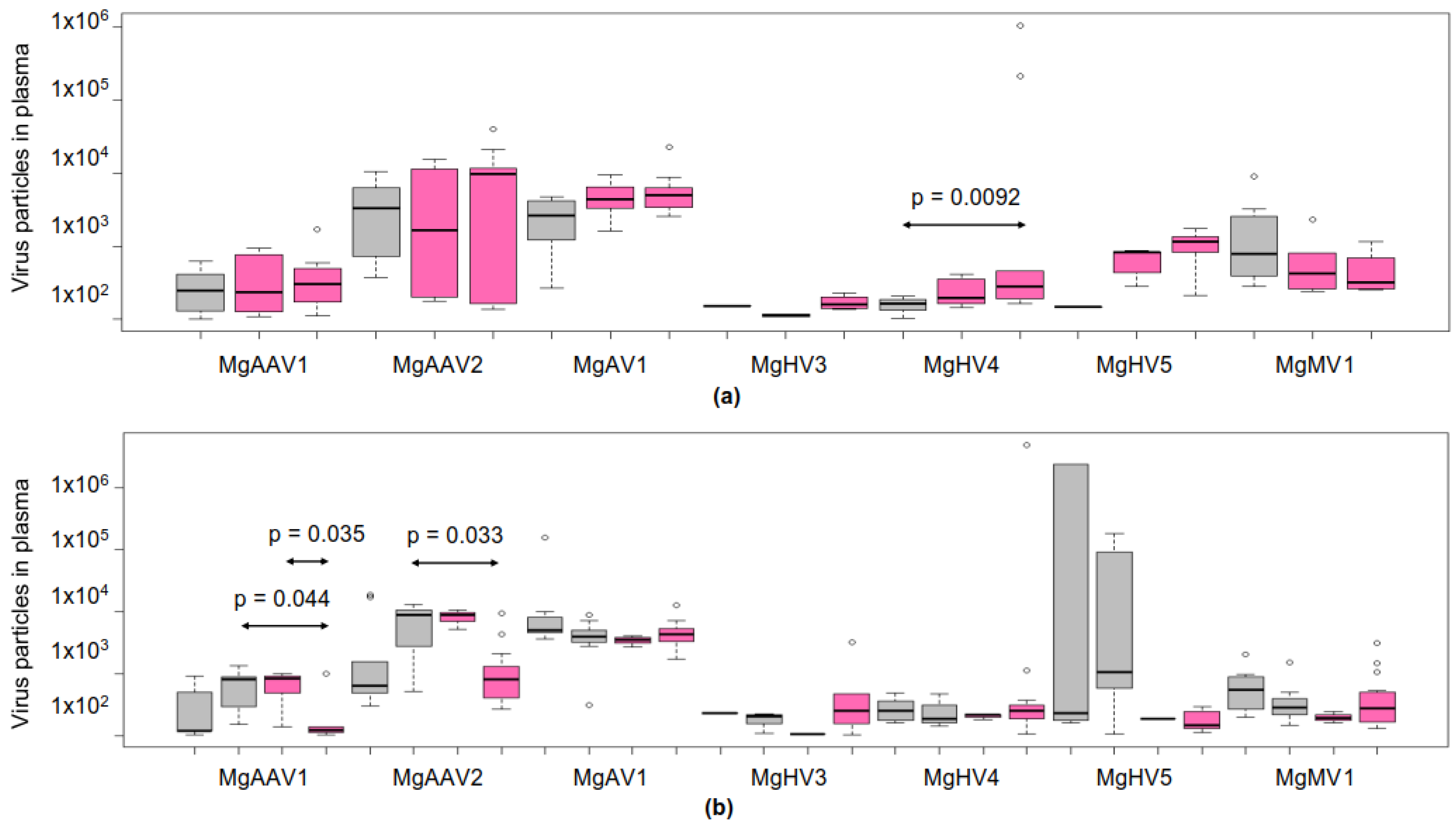
3.6. Viral Loads in Plasma
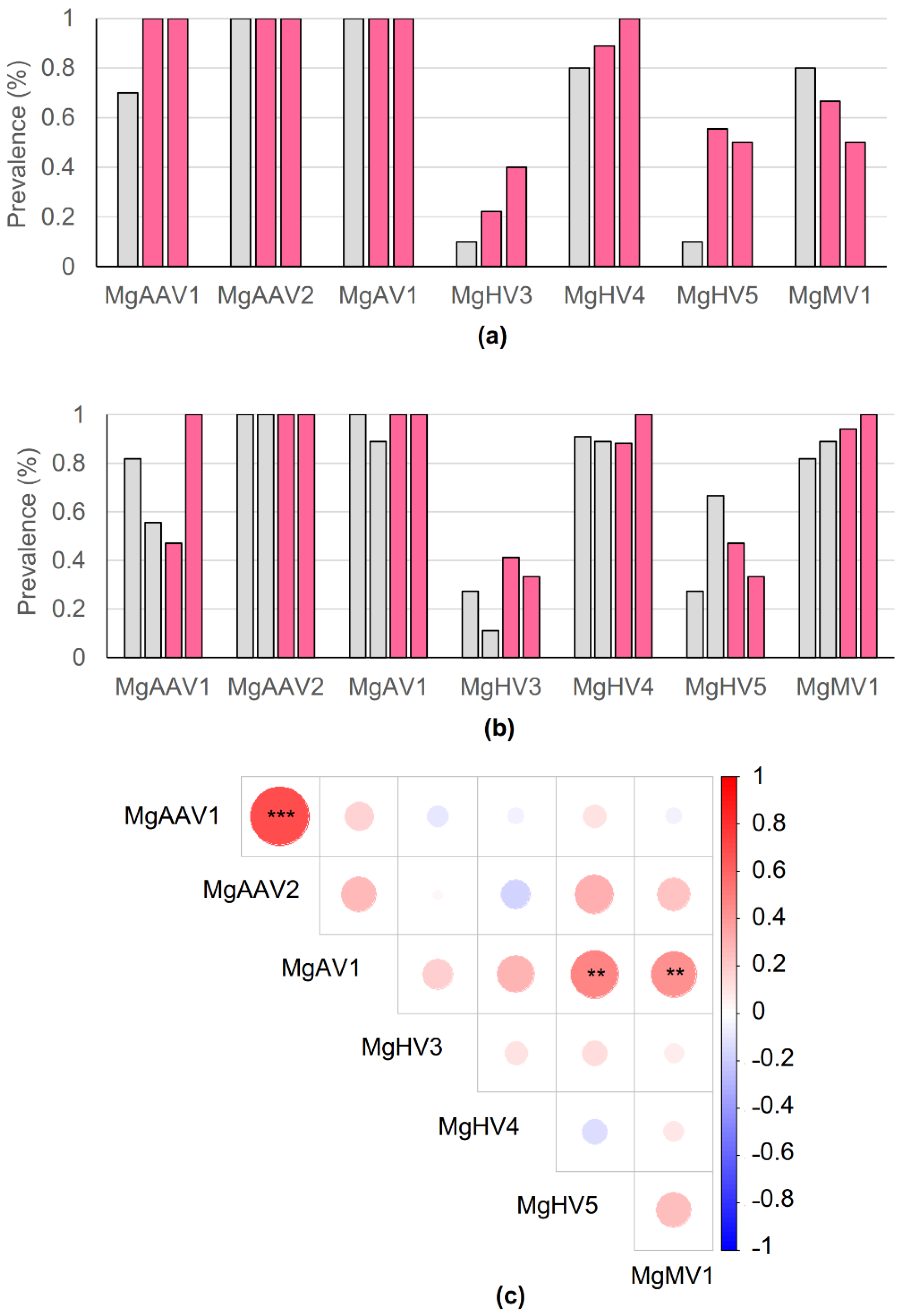
3.7. Viral Prevalence in the Populations
3.8. Plasma Loads of the Adeno-Associated Viruses Correlated Positively with Each Other
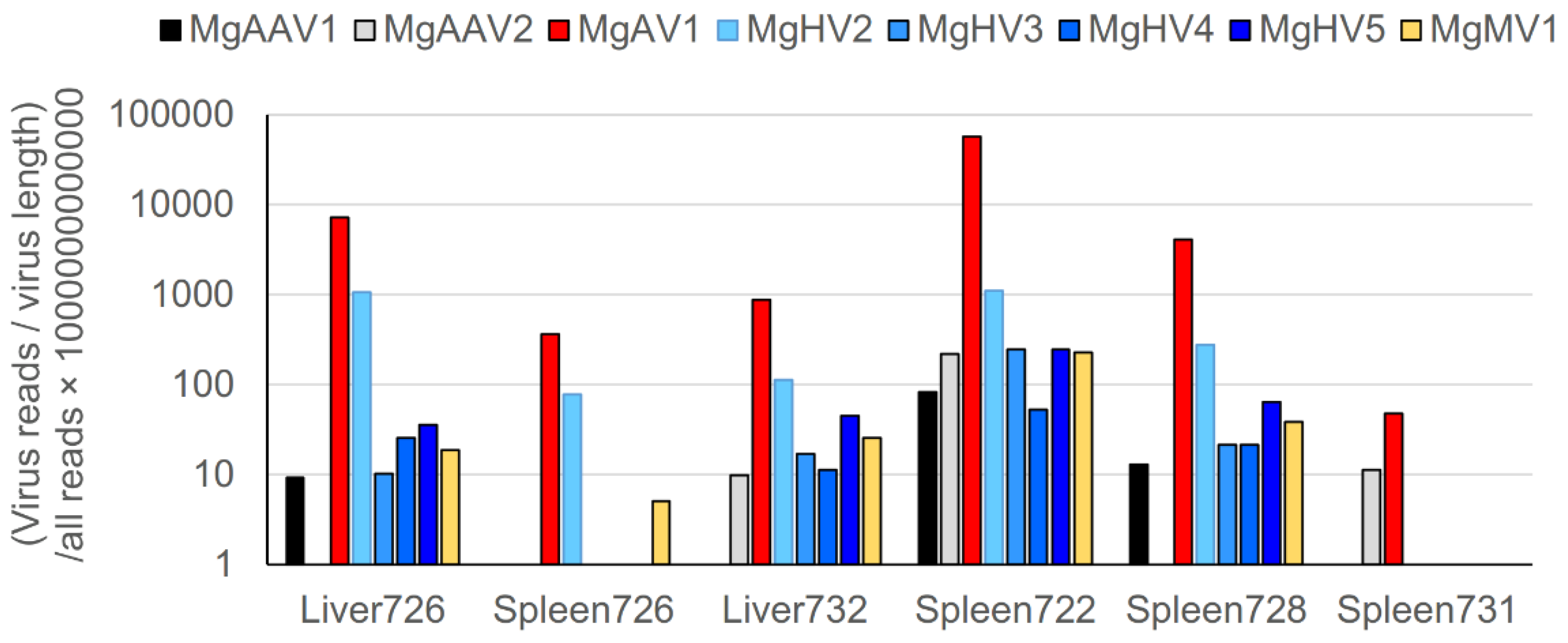
3.9. Virus Sequences in Bank Vole Liver and Spleen Transcriptomes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
Appendix A.1. DOSIMETRY
Appendix A.1.1. External Absorbed Dose Rate Estimations
Appendix A.1.2. Internal Absorbed Dose Rate Estimations
Appendix A.1.3. Total Absorbed Dose Rate Estimations
References
- Wu, Z.; Lu, L.; Du, J.; Yang, L.; Ren, X.; Liu, B.; Jiang, J.; Yang, J.; Dong, J.; Sun, L.; et al. Comparative analysis of rodent and small mammal viromes to better understand the wildlife origin of emerging infectious diseases. Microbiome 2018, 6, 178. [Google Scholar] [CrossRef]
- Phan, T.G.; Kapusinszky, B.; Wang, C.; Rose, R.K.; Lipton, H.L.; Delwart, E.L. The fecal viral flora of wild rodents. PLoS Pathog. 2011, 7, e1002218. [Google Scholar] [CrossRef]
- Han, B.A.; Schmidt, J.P.; Bowden, S.E.; Drake, J.M. Rodent reservoirs of future zoonotic diseases. Proc. Natl. Acad. Sci. USA 2015, 112, 7039–7044. [Google Scholar] [CrossRef] [PubMed]
- Avšič Županc, T.; Korva, M. Hantavirus Infections. Emerg. Infect. Dis. Clin. Case Stud. 2014, 21, e6–e16. [Google Scholar]
- Achazi, K.; Růžek, D.; Donoso-Mantke, O.; Schlegel, M.; Ali, H.S.; Wenk, M.; Schmidt-Chanasit, J.; Ohlmeyer, L.; Rühe, F.; Vor, T.; et al. Rodents as Sentinels for the Prevalence of Tick-Borne Encephalitis Virus. Vector-Borne Zoonotic Dis. 2011, 11, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Földes, F.; Madai, M.; Németh, V.; Zana, B.; Papp, H.; Kemenesi, G.; Bock-Marquette, I.; Horváth, G.; Herczeg, R.; Jakab, F. Serologic survey of the Crimean-Congo haemorrhagic fever virus infection among wild rodents in Hungary. Ticks Tick. Borne. Dis. 2019, 10, 101258. [Google Scholar] [CrossRef]
- Hazel, S.M.; Bennett, M.; Chantrey, J.; Bown, K.; Cavanagh, R.; Jones, T.R.; Baxby, D.; Begon, M. A longitudinal study of an endemic disease in its wildlife reservoir: Cowpox and wild rodents. Epidemiol. Infect. 2000, 124, 551–562. [Google Scholar] [CrossRef]
- Niklasson, B.; Kinnunen, L.; Hörnfeldt, B.; Hörling, J.; Benemar, C.; Olof Hedlund, K.; Matskova, L.; Hyypiä, T.; Winberg, G. A new picornavirus isolated from bank voles (Clethrionomys glareolus). Virology 1999, 255, 86–93. [Google Scholar] [CrossRef]
- Alkhovsky, S.; Butenko, A.; Eremyan, A.; Shchetinin, A. Genetic characterization of bank vole virus (BaVV), a new paramyxovirus isolated from kidneys of bank voles in Russia. Arch. Virol. 2018, 163, 755–759. [Google Scholar] [CrossRef]
- Vanmechelen, B.; Bletsa, M.; Laenen, L.; Lopes, A.R.; Vergote, V.; Beller, L.; Deboutte, W.; Korva, M.; Avšič Županc, T.; Goüy de Bellocq, J.; et al. Discovery and genome characterization of three new Jeilongviruses, a lineage of paramyxoviruses characterized by their unique membrane proteins. BMC Genom. 2018, 19, 617. [Google Scholar] [CrossRef]
- Tsoleridis, T.; Chappell, J.G.; Onianwa, O.; Marston, D.A.; Fooks, A.R.; Monchatre-Leroy, E.; Umhang, G.; Müller, M.A.; Drexler, J.F.; Drosten, C.; et al. Shared common ancestry of rodent alphacoronaviruses sampled globally. Viruses 2019, 11, 125. [Google Scholar] [CrossRef] [PubMed]
- Nainys, J.; Timinskas, A.; Schneider, J.; Ulrich, R.G.; Gedvilaite, A. Identification of two novel members of the tentative GenusWukipolyomavirus in Wild Rodents. PLoS ONE 2015, 10, e0140916. [Google Scholar] [CrossRef] [PubMed]
- Wágnerová, M.; Chalupková, A.; Hrabovská, Z.; Ancicová, L.; Mistríková, J. Possible role of different animal species in maintenance and spread of murine gammaherpesvirus 68 in the nature. Acta Virol. 2015, 59, 14–19. [Google Scholar] [CrossRef]
- Knowles, S.C.L.; Fenton, A.; Pedersen, A.B. Epidemiology and fitness effects of wood mouse herpesvirus in a natural host population. J. Gen. Virol. 2012, 93, 2447–2456. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, S.; Dutia, B.M.; Stewart, J.P.; Meredith, A.L.; Shaw, D.J.; Simmonds, P.; Sharp, C.P. Identification of novel anelloviruses with broad diversity in UK rodents. J. Gen. Virol. 2014, 95, 1544–1553. [Google Scholar] [CrossRef] [PubMed]
- Drexler, J.F.; Corman, V.M.; Müller, M.A.; Lukashev, A.N.; Gmyl, A.; Coutard, B.; Adam, A.; Ritz, D.; Leijten, L.M.; van Riel, D.; et al. Evidence for Novel Hepaciviruses in Rodents. PLoS Pathog. 2013, 9, e1003438. [Google Scholar] [CrossRef] [PubMed]
- Voutilainen, L.; Savola, S.; Kallio, E.R.; Laakkonen, J.; Vaheri, A.; Vapalahti, O.; Henttonen, H. Environmental change and disease dynamics: Effects of intensive forest management on Puumala hantavirus infection in boreal bank vole populations. PLoS ONE 2012, 7, e39452. [Google Scholar] [CrossRef]
- Tian, H.; Yu, P.; Bjørnstad, O.N.; Cazelles, B.; Yang, J.; Tan, H.; Huang, S.; Cui, Y.; Dong, L.; Ma, C.; et al. Anthropogenically driven environmental changes shift the ecological dynamics of hemorrhagic fever with renal syndrome. PLoS Pathog. 2017, 13, e1006198. [Google Scholar] [CrossRef]
- Lee, K.I.; Lee, J.S.; Jung, H.H.; Lee, H.Y.; Moon, S.H.; Kang, K.T.; Shim, Y.B.; Jang, J.W. Inactivation of enveloped and non-enveloped viruses in the process of chemical treatment and gamma irradiation of bovine-derived grafting materials. Xenotransplantation 2012, 19, 365–369. [Google Scholar] [CrossRef]
- Hume, A.J.; Ames, J.; Rennick, L.J.; Duprex, W.P.; Marzi, A.; Tonkiss, J.; Mühlberger, E. Inactivation of RNA viruses by gamma irradiation: A study on mitigating factors. Viruses 2016, 8, 204. [Google Scholar] [CrossRef]
- Ohagen, A.; Gibaja, V.; Horrigan, J.; Lunderville, D.; Jayarama, V.; Marcello, J.; Chapman, J.; Lazo, A. Induction of latent human cytomegalovirus by conventional gamma irradiation and prevention by treatment with INACTINE PEN110. Vox Sang. 2004, 87, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Agoni, L.; Lenz, J.; Guha, C. Variant Splicing and Influence of Ionizing Radiation on Human Endogenous Retrovirus K (HERV-K) Transcripts in Cancer Cell Lines. PLoS ONE 2013, 8, e76472. [Google Scholar] [CrossRef] [PubMed]
- Manome, Y.; Yao, X.J.; Kufe, D.W.; Cohen, E.A.; Fine, H.A. Selective effects of DNA damaging agents on HIV long terminal repeat activation and virus replication in vitro. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1996, 11, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Chia, H.C.; Chen, P.J.; Lee, P.H.; Cheng, A.L.; Hsu, H.C.; Cheng, J.C.H. Radiation-induced hepatitis B virus reactivation in liver mediated by the bystander effect from irradiated endothelial cells. Clin. Cancer Res. 2007, 13, 851–857. [Google Scholar] [CrossRef]
- Huang, W.; Zhang, W.; Fan, M.; Lu, Y.; Zhang, J.; Li, H.; Li, B. Risk factors for hepatitis B virus reactivation after conformal radiotherapy in patients with hepatocellular carcinoma. Cancer Sci. 2014, 105, 697–703. [Google Scholar] [CrossRef]
- Westphal, E.M.; Blackstock, W.; Feng, W.; Israel, B.; Kenney, S.C. Activation of lytic Epstein-Barr virus (EBV) infection by radiation and sodium butyrate in vitro and in vivo: A potential method for treating EBV-positive malignancies. Cancer Res. 2000, 60, 5781–5788. [Google Scholar]
- Nandakumar, A.; Uwatoko, F.; Yamamoto, M.; Tomita, K.; Majima, H.J.; Akiba, S.; Koriyama, C. Radiation-induced Epstein-Barr virus reactivation in gastric cancer cells with latent EBV infection. Tumor Biol. 2017, 39, 1–9. [Google Scholar] [CrossRef]
- McEntee, G.; Kyula, J.N.; Mansfield, D.; Smith, H.; Wilkinson, M.; Gregory, C.; Roulstone, V.; Coffey, M.; Harrington, K.J. Enhanced cytotoxicity of reovirus and radiotherapy in melanoma cells is mediated through increased viral replication and mitochondrial apoptotic signalling. Oncotarget 2016, 7, 48517–48532. [Google Scholar] [CrossRef][Green Version]
- Mezhir, J.J.; Advani, S.J.; Smith, K.D.; Darga, T.E.; Poon, A.P.W.; Schmidt, H.; Posner, M.C.; Roizman, B.; Weichselbaum, R.R. Ionizing radiation activates late herpes simplex virus 1 promoters via the p38 pathway in tumors treated with oncolytic viruses. Cancer Res. 2005, 65, 9479–9484. [Google Scholar] [CrossRef][Green Version]
- Walz, C.; Schlehofer, J.R.; Flentje, M.; Rudat, V.; Zur Hausen, H. Adeno-associated virus sensitizes HeLa cell tumors to gamma rays. J. Virol. 1992, 66, 5651–5657. [Google Scholar]
- Alexander, I.E.; Russell, D.W.; Spence, A.M.; Miller, A.D. Effects of Gamma Irradiation on the Transduction of Dividing and Nondividing Cells in Brain and Muscle of Rats by Adeno-Associated Virus Vectors. Hum. Gene Ther. 1996, 7, 841–850. [Google Scholar] [CrossRef]
- Levine, S. The effect of X-irradiation on the yield of poliovirus. Virology 1960, 10, 257–267. [Google Scholar] [CrossRef]
- Murphy, B.; Glasgow, L. Factors modifying host resistance to viral infection. 3. Effect of whole body x-irradiation on experimental encephalomyocarditis virus infection in mice. J. Exp. Med. 1968, 127, 1035–1052. [Google Scholar] [CrossRef] [PubMed]
- Bradish, C.J.; Titmuss, D.; Fitzgeorge, R. The sensitivity to γ-irradiation of the phases of the virus-host interaction: Studies with strains of Semliki forest virus in mice. J. Gen. Virol. 1980, 48, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, H.; Kadowaki, S.E.; Takahashi, H.; Iwasaki, T.; Tamura, S.I.; Kurata, T. Protection against influenza virus infection by nasal vaccination in advance of sublethal irradiation. Vaccine 2000, 18, 2560–2565. [Google Scholar] [CrossRef]
- Hollingworth, R.; Grand, R.J. Modulation of DNA damage and repair pathways by human tumour viruses. Viruses 2015, 7, 2542–2591. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Qiu, J. Parvovirus infection-induced DNA damage response. Future Virol. 2013, 8, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Jernfors, T.; Kesäniemi, J.; Lavrinienko, A.; Mappes, T.; Milinevsky, G.; Møller, A.P.; Mousseau, T.A.; Tukalenko, E.; Watts, P.C. Transcriptional Upregulation of DNA Damage Response Genes in Bank Voles (Myodes glareolus) Inhabiting the Chernobyl Exclusion Zone. Front. Environ. Sci. 2018, 5, 59. [Google Scholar] [CrossRef]
- Mustonen, V.; Kesäniemi, J.; Lavrinienko, A.; Tukalenko, E.; Mappes, T.; Watts, P.C.; Jurvansuu, J. Fibroblasts from bank voles inhabiting Chernobyl have increased resistance against oxidative and DNA stresses. BMC Cell Biol. 2018, 19, 17. [Google Scholar] [CrossRef]
- Kesäniemi, J.; Jernfors, T.; Lavrinienko, A.; Kivisaari, K.; Kiljunen, M.; Mappes, T.; Watts, P.C. Exposure to environmental radionuclides is associated with altered metabolic and immunity pathways in a wild rodent. Mol. Ecol. 2019, 28, 4620–4635. [Google Scholar] [CrossRef]
- Chesser, R.K.; Bondarkov, M.; Baker, R.J.; Wickliffe, J.K.; Rodgers, B.E. Reconstruction of radioactive plume characteristics along Chernobyl’s Western Trace. J. Environ. Radioact. 2004, 71, 147–157. [Google Scholar] [CrossRef]
- Lavrinienko, A.; Mappes, T.; Tukalenko, E.; Mousseau, T.A.; Møller, A.P.; Knight, R.; Morton, J.T.; Thompson, L.R.; Watts, P.C. Environmental radiation alters the gut microbiome of the bank vole Myodes glareolus. ISME J. 2018, 12, 2801–2806. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Trinity: Reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 2013, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Madan, A. CAP3: A DNA sequence assembly program. Genome Res. 1999, 9, 868–877. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2016, 45, e18. [Google Scholar]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef]
- Zimmermann, L.; Stephens, A.; Nam, S.Z.; Rau, D.; Kübler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at its Core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef]
- Markham, N.R. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets Brief communication. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Mietzsch, M.; Pénzes, J.J.; Agbandje-Mckenna, M. Twenty-five years of structural parvovirology. Viruses 2019, 11, 362. [Google Scholar] [CrossRef]
- Williams, S.H.; Che, X.; Garcia, J.A.; Klena, J.D.; Lee, B.; Muller, D.; Ulrich, W.; Corrigan, R.M.; Nichol, S.; Jain, K.; et al. Viral diversity of house mice in New York City. MBio 2018, 9, e01354-17. [Google Scholar] [CrossRef]
- Qiu, J.; Cheng, F.; Pintel, D. Molecular characterization of caprine adeno-associated virus (AAV-Go.1) reveals striking similarity to human AAV5. Virology 2006, 356, 208–216. [Google Scholar] [CrossRef]
- Snijder, E.J.; Kikkert, M.; Fang, Y. Arterivirus molecular biology and pathogenesis. J. Gen. Virol. 2013, 94, 2141–2163. [Google Scholar] [CrossRef]
- Smith, D.B.; Becher, P.; Bukh, J.; Gould, E.A.; Meyers, G.; Monath, T.; Muerhoff, A.S.; Pletnev, A.; Rico-Hesse, R.; Stapleton, J.T.; et al. Proposed update to the taxonomy of the genera Hepacivirus and Pegivirus within the Flaviviridae family. J. Gen. Virol. 2016, 97, 2894–2907. [Google Scholar] [CrossRef]
- Reuter, G.; Boros, Á.; Kiss, T.; Delwart, E.; Pankovics, P. Complete genome characterization of mosavirus (family Picornaviridae) identified in droppings of a European roller (Coracias garrulus) in Hungary. Arch. Virol. 2014, 159, 2723–2729. [Google Scholar] [CrossRef]
- Luo, X.L.; Lu, S.; Jin, D.; Yang, J.; Wu, S.S.; Xu, J. Marmota himalayana in the Qinghai-Tibetan plateau as a special host for bi-segmented and unsegmented picobirnaviruses article. Emerg. Microbes Infect. 2018, 7, 20. [Google Scholar] [CrossRef]
- Geoffroy, M.-C.; Salvetti, A. Helper Functions Required for Wild Type and Recombinant Adeno- Associated Virus Growth. Curr. Gene Ther. 2005, 5, 265–267. [Google Scholar] [CrossRef]
- Schulte-Hostedde, A.I.; Millar, J.S.; Hickling, G.J. Evaluating body condition in small mammals. Can. J. Zool. 2001, 79, 1021–1029. [Google Scholar] [CrossRef]
- Anthony, S.J.; Epstein, J.H.; Murray, K.A.; Navarrete-Macias, I.; Zambrana-Torrelio, C.M.; Solovyov, A.; Ojeda-Flores, R.; Arrigo, N.C.; Islam, A.; Khan, S.A.; et al. A strategy to estimate unknown viral diversity in mammals. MBio 2013, 4, e00598-13. [Google Scholar] [CrossRef] [PubMed]
- Guy, C.; Thiagavel, J.; Mideo, N.; Ratcliffe, J.M. Phylogeny matters: Revisiting “a comparison of bats and rodents as reservoirs of zoonotic viruses”. R. Soc. Open Sci. 2019, 6, 181182. [Google Scholar] [CrossRef]
- Nikitina, E.; Larionova, I.; Choinzonov, E.; Kzhyshkowska, J. Monocytes and Macrophages as Viral Targets and Reservoirs. Int. J. Mol. Sci. 2018, 19, 2821. [Google Scholar] [CrossRef]
- Sands, M.S. AAV-mediated liver-directed gene therapy. Methods Mol. Biol. 2011, 807, 141–157. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | Genbank ID | Length (nt) | Reads Kyiv West | Reads Kyiv East | Reads CEZ | All Reads | Average Coverage |
|---|---|---|---|---|---|---|---|
| MgAAV1 | MN242366 | 4927 | 1626 | 131 | 0 | 1757 | 35.66 |
| MgAAV2 | MN242367 | 4517 | 0 | 1108 | 102 | 1210 | 26.79 |
| MgAV1 | MN242368 | 15,397 | 0 | 43,566 | 0 | 43,566 | 282.95 |
| MgHV2 | MN242369 | 8900 | 272,994 | 0 | 0 | 272,994 | 3067.35 |
| MgHV3 | MN242370 | 8838 | 143,029 | 414,151 | 7,165,765 | 7,722,945 | 87,383.40 |
| MgHV4 | MN242371 | 8838 | 961 | 1033 | 2,312,332 | 2,314,326 | 26,186.08 |
| MgHV5 | MN242372 | 8828 | 0 | 196,742 | 0 | 196,742 | 2228.61 |
| MgMV1 | MN242373 | 9762 | 4363 | 5379 | 248 | 9990 | 102.34 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kesäniemi, J.; Lavrinienko, A.; Tukalenko, E.; Mappes, T.; Watts, P.C.; Jurvansuu, J. Infection Load and Prevalence of Novel Viruses Identified from the Bank Vole Do Not Associate with Exposure to Environmental Radioactivity. Viruses 2020, 12, 44. https://doi.org/10.3390/v12010044
Kesäniemi J, Lavrinienko A, Tukalenko E, Mappes T, Watts PC, Jurvansuu J. Infection Load and Prevalence of Novel Viruses Identified from the Bank Vole Do Not Associate with Exposure to Environmental Radioactivity. Viruses. 2020; 12(1):44. https://doi.org/10.3390/v12010044
Chicago/Turabian StyleKesäniemi, Jenni, Anton Lavrinienko, Eugene Tukalenko, Tapio Mappes, Phillip C. Watts, and Jaana Jurvansuu. 2020. "Infection Load and Prevalence of Novel Viruses Identified from the Bank Vole Do Not Associate with Exposure to Environmental Radioactivity" Viruses 12, no. 1: 44. https://doi.org/10.3390/v12010044
APA StyleKesäniemi, J., Lavrinienko, A., Tukalenko, E., Mappes, T., Watts, P. C., & Jurvansuu, J. (2020). Infection Load and Prevalence of Novel Viruses Identified from the Bank Vole Do Not Associate with Exposure to Environmental Radioactivity. Viruses, 12(1), 44. https://doi.org/10.3390/v12010044