Detailed Characteristics of Tonsillar Tumors with Extrachromosomal or Integrated Form of Human Papillomavirus

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Clinical Samples
2.2. Processing of Samples and Detection of an Active Infection
2.3. HPV Genome Status Determination
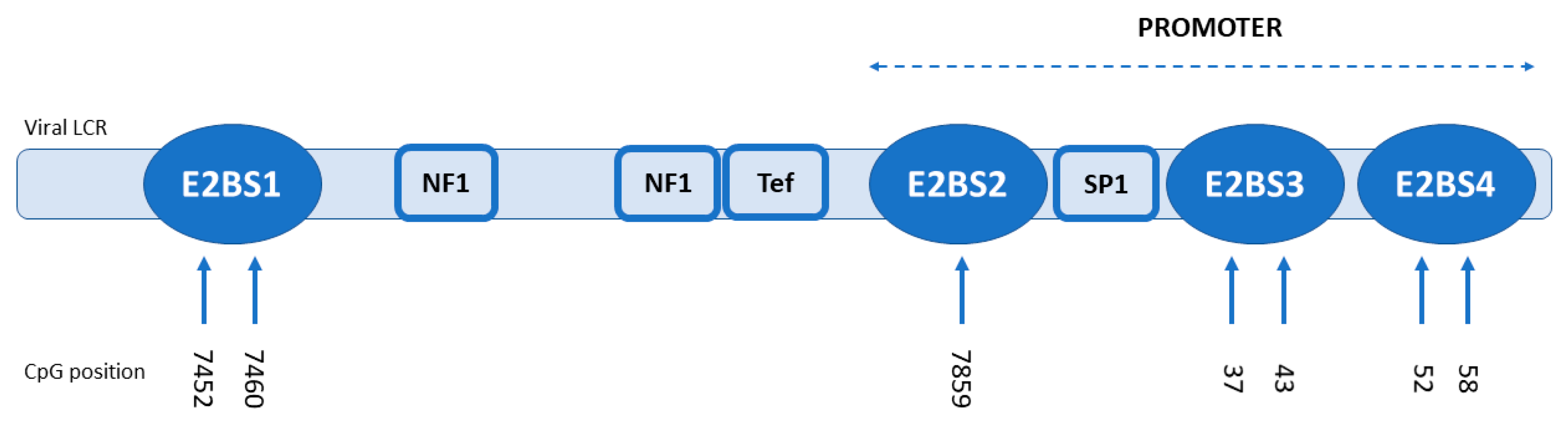
2.4. Methylation Analysis
2.5. HPV16 DNA Load
2.6. HPV16 Oncogene E6/E7 mRNAs Quantification
2.7. LCR and E2 Variant Analysis
2.8. Statistical Analysis
3. Results
3.1. HPV E2 mRNA Determination
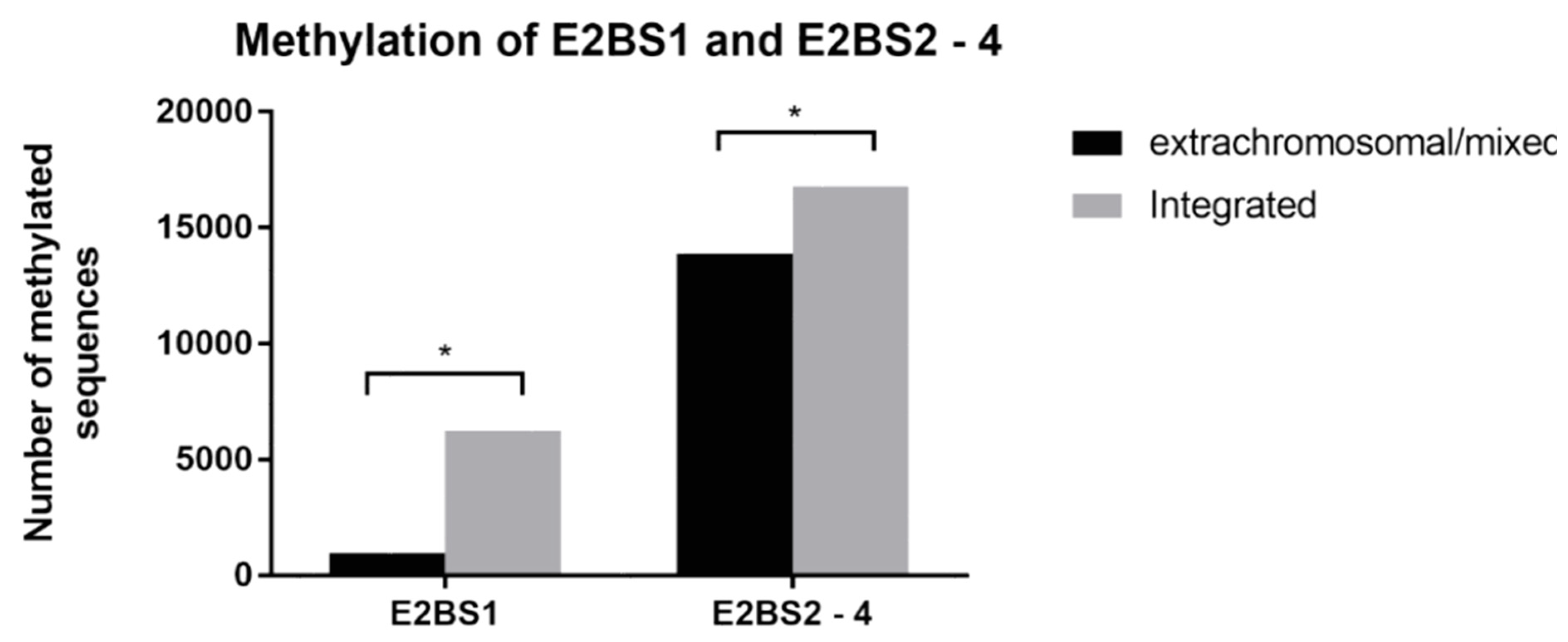
3.2. Methylation Analysis
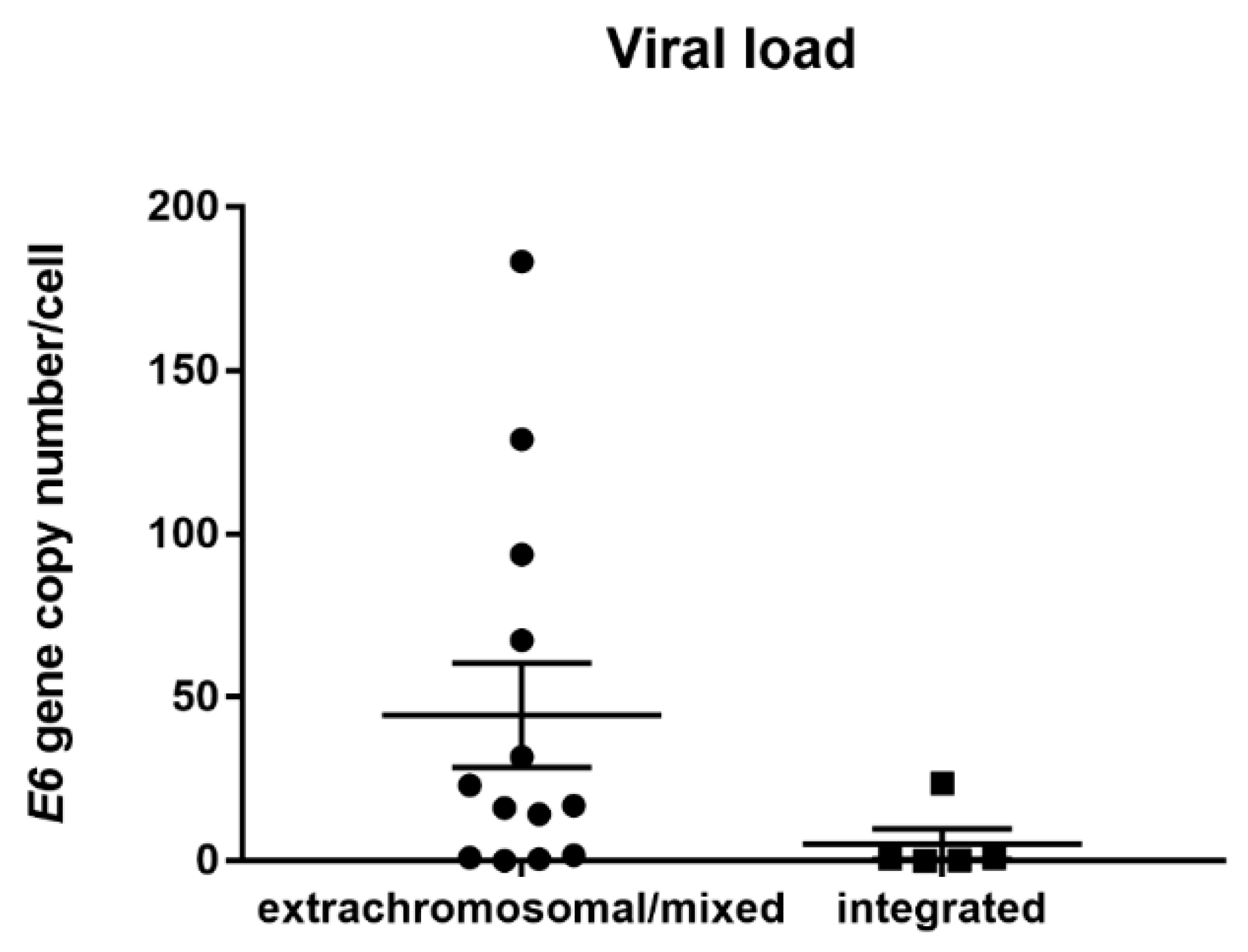
3.3. HPV Viral Load Analysis
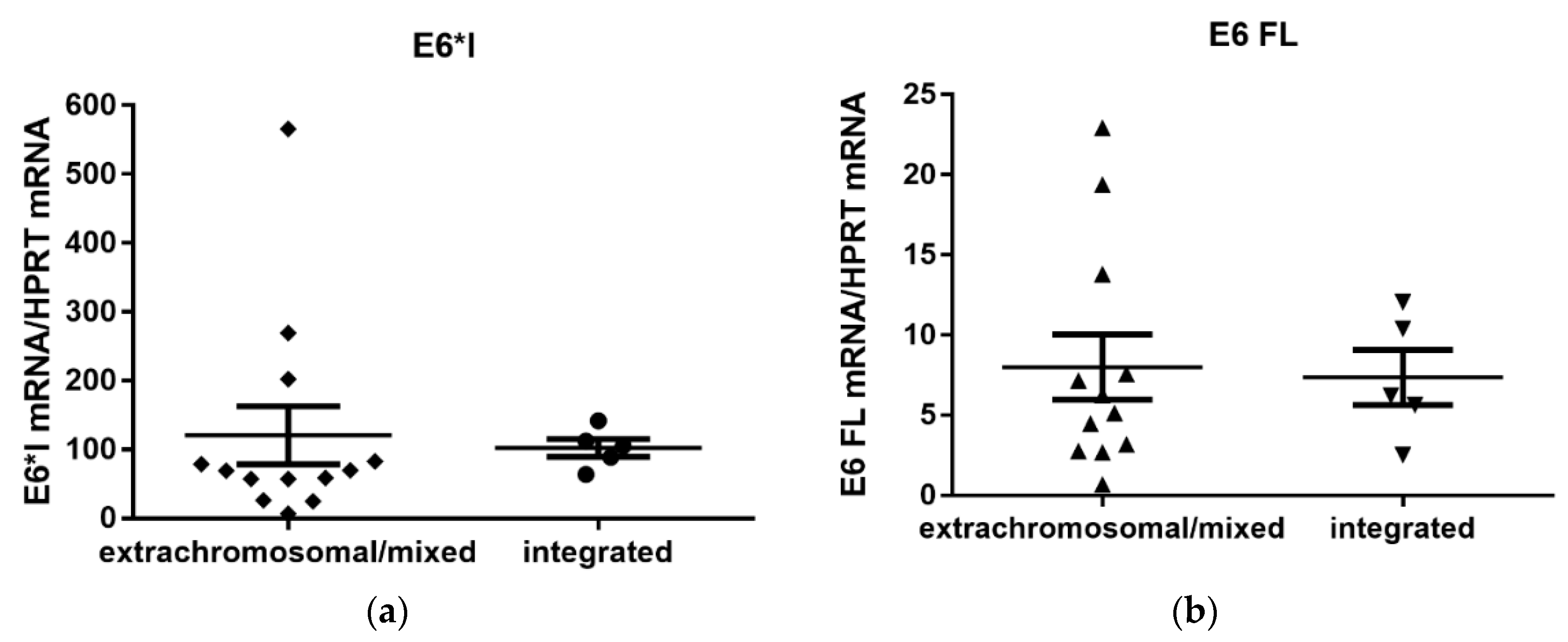
3.4. Quantification of Viral E6/E7 Oncogene Transcripts
3.5. LCR and E2 Variants Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gupta, B.; Johnson, N.W.; Kumar, N. Global Epidemiology of Head and Neck Cancers: A Continuing Challenge. Oncology 2016, 91, 13–23. [Google Scholar] [CrossRef]
- Gillison, M.L.; Chaturvedi, A.K.; Anderson, W.F.; Fakhry, C. Epidemiology of Human Papillomavirus-Positive Head and Neck Squamous Cell Carcinoma. J. Clin. Oncol. 2015, 33, 3235–3242. [Google Scholar] [CrossRef]
- Haeggblom, L.; Ramqvist, T.; Tommasino, M.; Dalianis, T.; Nasman, A. Time to change perspectives on HPV in oropharyngeal cancer. A systematic review of HPV prevalence per oropharyngeal sub-site the last 3 years. Papillomavirus Res. 2017, 4, 1–11. [Google Scholar] [CrossRef]
- Chaturvedi, A.K.; Engels, E.A.; Pfeiffer, R.M.; Hernandez, B.Y.; Xiao, W.; Kim, E.; Jiang, B.; Goodman, M.T.; Sibug-Saber, M.; Cozen, W.; et al. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J. Clin. Oncol. 2011, 29, 4294–4301. [Google Scholar] [CrossRef]
- Stein, A.P.; Saha, S.; Kraninger, J.L.; Swick, A.D.; Yu, M.; Lambertg, P.F.; Kimple, R. Prevalence of Human Papillomavirus in Oropharyngeal Cancer: A Systematic Review. Cancer J. 2015, 21, 138–146. [Google Scholar] [CrossRef]
- Dayyani, F.; Etzel, C.J.; Liu, M.; Ho, C.H.; Lippman, S.M.; Tsao, A.S. Meta-analysis of the impact of human papillomavirus (HPV) on cancer risk and overall survival in head and neck squamous cell carcinomas (HNSCC). Head Neck Oncol. 2010, 2, 15. [Google Scholar] [CrossRef]
- Moody, C.A.; Laimins, L.A. Human papillomavirus oncoproteins: Pathways to transformation. Nat. Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef]
- McBride, A.A.; Warburton, A. The role of integration in oncogenic progression of HPV-associated cancers. PLoS Pathog. 2017, 13, e1006211. [Google Scholar] [CrossRef] [PubMed]
- Doeberitz, M.; Vinokurova, S. Host factors in HPV-related carcinogenesis: Cellular mechanisms controlling HPV infections. Arch. Med. Res. 2009, 40, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.; Allen-Hoffmann, B.L.; Lambert, P.F. Integration of human papillomavirus type 16 into the human genome correlates with a selective growth advantage of cells. J. Virol. 1995, 69, 2989–2997. [Google Scholar] [PubMed]
- Parfenov, M.; Pedamallu, C.S.; Gehlenborg, N.; Freeman, S.S.; Danilova, L.; Bristow, C.A.; Lee, S.; Hadjipanayis, A.G.; Ivanova, E.V.; Wilkerson, M.D. Characterization of HPV and host genome interactions in primary head and neck cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 15544–15549. [Google Scholar] [CrossRef] [PubMed]
- Vinokurova, S.; Wentzensen, N.; Kraus, I.; Klaes, R.; Driesch, C.; Melsheimer, P.; Kisseljov, F.; Dürst, M.; Schneider, A.; von Knebel Doeberitz, M. Type-dependent integration frequency of human papillomavirus genomes in cervical lesions. Cancer Res. 2008, 68, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Joo, J.; Shin, H.J.; Park, B.; Park, S.Y.; Yoo, C.W.; Yoon, K.A.; Kong, S.Y.; Kim, Y.J.; Kim, S.S.; Kim, J.Y. Integration Pattern of Human Papillomavirus Is a Strong Prognostic Factor for Disease-Free Survival After Radiation Therapy in Cervical Cancer Patients. Int. J Radiat. Oncol. Biol. Phys. 2017, 98, 654–661. [Google Scholar] [CrossRef] [PubMed]
- Hudelist, G.; Manavi, M.; Pischinger, K.I.; Watkins-Riedel, T.; Singer, C.F.; Kubista, E.; Czerwenka, K.F. Physical state and expression of HPV DNA in benign and dysplastic cervical tissue: Different levels of viral integration are correlated with lesion grade. Gynecol. Oncol. 2004, 92, 873–880. [Google Scholar] [CrossRef]
- Shin, H.J.; Joo, J.; Yoon, J.H.; Yoo, C.W.; Kim, J.Y. Physical status of human papillomavirus integration in cervical cancer is associated with treatment outcome of the patients treated with radiotherapy. PLoS ONE 2014, 9, e78995. [Google Scholar] [CrossRef]
- Anayannis, N.V.; Schlecht, N.F.; Ben-Dayan, M.; Smith, R.V.; Belbin, T.J.; Ow, T.J.; Blakaj, D.M.; Burk, R.D.; Leonard, S.M.; Woodman, C.B. Association of an intact E2 gene with higher HPV viral load, higher viral oncogene expression, and improved clinical outcome in HPV16 positive head and neck squamous cell carcinoma. PLoS ONE 2018, 13, e0191581. [Google Scholar] [CrossRef]
- Nulton, T.J.; Kim, N.K.; DiNardo, L.J.; Morgan, I.M.; Windle, B. Patients with integrated HPV16 in head and neck cancer show poor survival. Oral Oncol. 2018, 80, 52–55. [Google Scholar] [CrossRef]
- Vojtechova, Z.; Sabol, I.; Salakova, M.; Turek, L.; Grega, M.; Smahelova, J.; Vencalek, O.; Lukesova, E.; Klozar, J.; Tachezy, R. Analysis of the integration of human papillomaviruses in head and neck tumours in relation to patients’ prognosis. Int. J. Cancer. 2016, 138, 386–395. [Google Scholar] [CrossRef]
- Cheung, J.L.; Cheung, T.H.; Yu, M.Y.; Chan, P.K. Virological characteristics of cervical cancers carrying pure episomal form of HPV16 genome. Gynecol. Oncol. 2013, 131, 374–379. [Google Scholar] [CrossRef]
- Faust, H.; Eldenhed Alwan, E.; Roslin, A.; Wennerberg, J.; Forslund, O. Prevalence of human papillomavirus types, viral load and physical status of HPV16 in head and neck squamous cell carcinoma from the South Swedish Health Care Region. J. Gen. Virol. 2016, 97, 2949–2956. [Google Scholar] [CrossRef][Green Version]
- Schwarz, E.; Freese, U.K.; Gissmann, L.; Mayer, W.; Roggenbuck, B.; Stremlau, A.; Zur Hausen, H. Structure and transcription of human papillomavirus sequences in cervical carcinoma cells. Nature 1985, 314, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Smotkin, D.; Wettstein, F.O. Transcription of human papillomavirus type 16 early genes in a cervical cancer and a cancer-derived cell line and identification of the E7 protein. Proc. Natl. Acad. Sci. USA 1986, 83, 4680–4684. [Google Scholar] [CrossRef] [PubMed]
- Olthof, N.C.; Huebbers, C.U.; Kolligs, J.; Henfling, M.; Ramaekers, F.C.; Cornet, I.; van Lent-Albrechts, J.A.; Stegmann, A.P.; Silling, S.; Wieland, U.; et al. Viral load, gene expression and mapping of viral integration sites in HPV16-associated HNSCC cell lines. Int. J. Cancer 2015, 136, E207–E218. [Google Scholar] [CrossRef] [PubMed]
- Häfner, N.; Driesch, C.; Gajda, M.; Jansen, L.; Kirchmayr, R.; Runnebaum, I.B.; Dürst, M. Integration of the HPV16 genome does not invariably result in high levels of viral oncogene transcripts. Oncogene 2008, 27, 1610–1617. [Google Scholar] [CrossRef]
- Sun, Z.; Zhang, R.; Liu, Z.; Liu, C.; Li, X.; Zhou, W.; Yang, L.; Ruan, Q.; Zhang, X. Development of a fluorescence-based multiplex genotyping method for simultaneous determination of human papillomavirus infections and viral loads. BMC Cancer 2015, 15, 860. [Google Scholar] [CrossRef]
- Jung, A.C.; Briolat, J.; Millon, R.; De Reyniès, A.; Rickman, D.; Thomas, E.; Abecassis, J.; Clavel, C.; Wasylyk, B. Biological and clinical relevance of transcriptionally active human papillomavirus (HPV) infection in oropharynx squamous cell carcinoma. Int. J. Cancer 2010, 126, 1882–1894. [Google Scholar] [CrossRef]
- Olthof, N.C.; Speel, E.J.M.; Kolligs, J.; Haesevoets, A.; Henfling, M.; Ramaekers, F.C.; Preuss, S.F.; Drebber, U.; Wieland, U.; Silling, S.; et al. Comprehensive analysis of HPV16 integration in OSCC reveals no significant impact of physical status on viral oncogene and virally disrupted human gene expression. PLoS ONE 2014, 9, e88718. [Google Scholar] [CrossRef]
- Chaiwongkot, A.; Vinokurova, S.; Pientong, C.; Ekalaksananan, T.; Kongyingyoes, B.; Kleebkaow, P.; Chumworathayi, B.; Patarapadungkit, N.; Reuschenbach, M.; von Knebel Doeberitz, M. Differential methylation of E2 binding sites in episomal and integrated HPV 16 genomes in preinvasive and invasive cervical lesions. Int. J. Cancer 2013, 132, 2087–2094. [Google Scholar] [CrossRef]
- Thain, A.; Jenkins, O.; Clarke, A.R.; Gaston, K. CpG methylation directly inhibits binding of the human papillomavirus type 16 E2 protein to specific DNA sequences. J. Virol. 1996, 70, 7233–7235. [Google Scholar]
- Vinokurova, S.; von Knebel Doeberitz, M. Differential methylation of the HPV 16 upstream regulatory region during epithelial differentiation and neoplastic transformation. PLoS ONE 2011, 6, e24451. [Google Scholar] [CrossRef]
- Reuschenbach, M.; Huebbers, C.U.; Prigge, E.S.; Bermejo, J.L.; Kalteis, M.S.; Preuss, S.F.; Seuthe, I.M.; Kolligs, J.; Speel, E.J.M.; Olthof, N.; et al. Methylation status of HPV16 E2-binding sites classifies subtypes of HPV-associated oropharyngeal cancers. Cancer 2015, 121, 1966–1976. [Google Scholar] [CrossRef] [PubMed]
- Rotnáglová, E.; Tachezy, R.; Saláková, M.; Procházka, B.; Košlabová, E.; Veselá, E.; Ludvíková, V.; Hamšíková, E.; Klozar, J. HPV involvement in tonsillar cancer: Prognostic significance and clinically relevant markers. Int. J. Cancer 2011, 129, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Tachezy, R.; Smahelova, J.; Kaspirkova, J.; Salakova, M. Human papillomavirus type-specific prevalence in the cervical cancer screening population of Czech women. PLoS ONE 2013, 8, e79156. [Google Scholar] [CrossRef] [PubMed]
- Gravitt, P.E.; Peyton, C.; Wheeler, C.; Apple, R.; Higuchi, R.; Shah, K.V. Reproducibility of HPV 16 and HPV 18 viral load quantitation using TaqMan real-time PCR assays. J. Virol. Methods 2003, 112, 23–33. [Google Scholar] [CrossRef]
- Salakova, M.; Koslabova, E.; Vojtechova, Z.; Tachezy, R.; Sroller, V. Detection of human polyomaviruses MCPyV, HPyV6, and HPyV7 in malignant and non-malignant tonsillar tissues. J. Med. Virol. 2016, 88, 695–702. [Google Scholar] [CrossRef]
- Tachezy, R.; Mikyšková, I.; Ludvikova, V.; Rob, L.; Kučera, T.; Slavik, V.; Bekova, A.; Robova, H.; Pluta, M.; Hamšíková, E. Longitudinal study of patients after surgical treatment for cervical lesions: Detection of HPV DNA and prevalence of HPV-specific antibodies. Eur. J. Clin. Microbiol. Infect. Dis. 2006, 25, 492–500. [Google Scholar] [CrossRef]
- Xi, L.F.; Demers, W.; Kiviat, N.B.; Kuypers, J.; Beckmann, A.M.; Galloway, D.A. Sequence variation in the noncoding region of human papillomavirus type 16 detected by single-strand conformation polymorphism analysis. J. Infect. Dis. 1993, 168, 610–617. [Google Scholar] [CrossRef]
- Collins, S.I.; Constandinou-Williams, C.; Wen, K.; Young, L.S.; Roberts, S.; Murray, P.G.; Woodman, C.B. Disruption of the E2 gene is a common and early event in the natural history of cervical human papillomavirus infection: A longitudinal cohort study. Cancer Res. 2009, 69, 3828–3832. [Google Scholar] [CrossRef]
- Lace, M.J.; Anson, J.R.; Klussmann, J.P.; Wang, D.H.; Smith, E.M.; Haugen, T.H.; Turek, L.P. Human papillomavirus type 16 (HPV-16) genomes integrated in head and neck cancers and in HPV-16-immortalized human keratinocyte clones express chimeric virus-cell mRNAs similar to those found in cervical cancers. J. Virol. 2011, 85, 1645–1654. [Google Scholar] [CrossRef]
- Akagi, K.; Li, J.; Broutian, T.R.; Padilla-Nash, H.; Xiao, W.; Jiang, B.; Rocco, J.W.; Teknos, T.N.; Kumar, B.; Wangsa, D.; et al. Genome-wide analysis of HPV integration in human cancers reveals recurrent, focal genomic instability. Genome Res. 2014, 24, 185–199. [Google Scholar] [CrossRef]
- Park, I.S.; Chang, X.; Loyo, M.; Wu, G.; Chuang, A.; Kim, M.S.; Chae, Y.K.; Lyford-Pike, S.; Westra, W.H.; Saunders, J.R.; et al. Characterization of the methylation patterns in human papillomavirus type 16 viral DNA in head and neck cancers. Cancer Prev. Res. 2011, 4, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Hatano, T.; Sano, D.; Takahashi, H.; Hyakusoku, H.; Isono, Y.; Shimada, S.; Sawakuma, K.; Takada, K.; Oikawa, R.; Watanabe, Y.; et al. Identification of human papillomavirus (HPV) 16 DNA integration and the ensuing patterns of methylation in HPV-associated head and neck squamous cell carcinoma cell lines. Int. J. Cancer 2017, 140, 1571–1580. [Google Scholar] [CrossRef] [PubMed]
- Lace, M.J.; Yamakawa, Y.; Ushikai, M.; Anson, J.R.; Haugen, T.H.; Turek, L.P. Cellular factor YY1 downregulates the human papillomavirus 16 E6/E7 promoter, P97, in vivo and in vitro from a negative element overlapping the transcription-initiation site. J. Gen. Virol. 2009, 90, 2402–2412. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Kedzia, W.; Gozdzicka-Jozefiak, A. Intratype HPV16 sequence variation within LCR of isolates from asymptomatic carriers and cervical cancers. J. Clin. Virol. 2001, 23, 65–77. [Google Scholar] [CrossRef]
- Das, P.; Thomas, A.; Kannan, S.; Deodhar, K.; Shrivastava, S.K.; Mahantshetty, U.; Mulherkar, R. Human papillomavirus (HPV) genome status & cervical cancer outcome--A retrospective study. Indian J. Med. Res. 2015, 142, 525–532. [Google Scholar] [PubMed]
- Deng, Z.; Hasegawa, M.; Kiyuna, A.; Matayoshi, S.; Uehara, T.; Agena, S.; Yamashita, Y.; Ogawa, K.; Maeda, H.; Suzuki, M. Viral load, physical status, and E6/E7 mRNA expression of human papillomavirus in head and neck squamous cell carcinoma. Head Neck 2013, 35, 800–808. [Google Scholar] [CrossRef]
- Nulton, T.J.; Olex, A.L.; Dozmorov, M.; Morgan, I.M.; Windle, B. Analysis of The Cancer Genome Atlas sequencing data reveals novel properties of the human papillomavirus 16 genome in head and neck squamous cell carcinoma. Oncotarget 2017, 8, 17684–17699. [Google Scholar] [CrossRef]
- Schmitt, M.; Dalstein, V.; Waterboer, T.; Clavel, C.; Gissmann, L.; Pawlita, M. Diagnosing cervical cancer and high-grade precursors by HPV16 transcription patterns. Cancer Res. 2010, 70, 249–256. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| APOT Assay | E2 Breakpoint Mapping | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample | Status | Chromosome | Gene Code | Region in Genome | E6 mRNA | E7 mRNA | E7-E4 mRNA | E2 mRNA | E2/E5 mRNA | E5 mRNA | E2 DNA |
| ORL 111 | Integrated | 1q32 | − | Intergenic area | + | + | − | − | − | − | − |
| ORL 125 | Integrated | 20p12.1 | MACROD2 | Intron | + | + | + | + | − | − | + |
| ORL 128 | Integrated | − | − | − | + | + | − | − | − | − | + |
| ORL 133 | Integrated | 8p23.2 | CSMD1 | Intron | + | + | − | − | − | − | + |
| ORL 160 | Integrated | 17q23.1 | TUBD1 | Exon | + | + | − | − | − | − | − |
| ORL 126 | Mixed | 5q35 | CANX | 3′UTR | + | + | + | + | + | + | + |
| ORL 187 | Mixed | 13q14 | TSC22D1 | Intron | + | + | + | + | + | + | + |
| ORL 243 | Mixed | 3q28 | TP63 | Exon | + | + | + | + | + | + | + |
| ORL 244 | Mixed | 19p13.2 | CDC37 | 5′UTR | + | + | + | + | + | + | + |
| ORL 104 | Extrachromosomal | − | − | − | + | + | + | + | + | + | + |
| ORL 116 | Extrachromosomal | − | − | − | + | + | + | + | + | + | + |
| ORL 137 | Extrachromosomal | − | − | − | + | + | + | + | + | + | + |
| ORL 155 | Extrachromosomal | − | − | − | + | + | + | + | + | + | + |
| ORL 161 | Extrachromosomal | − | − | − | + | + | + | + | + | + | + |
| ORL 181 | Extrachromosomal | − | − | − | + | + | + | + | + | + | + |
| ORL 257 | Extrachromosomal | − | − | − | + | + | + | + | + | + | + |
| ORL 265 | Extrachromosomal | − | − | − | + | + | + | + | + | + | + |
| ORL 280 | Extrachromosomal | − | − | − | + | + | + | + | + | + | + |
| E2BS1 | E2BS2 | E2BS3 | E2BS4 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HPV Genome | Position | 7452 | 7460 | 7859 | 37 | 43 | 52 | 58 | |||||||
| Samples | M | U | M | U | M | U | M | U | M | U | M | U | M | U | |
| Integrated | ORL 111 | 13 | 2283 | 12 | 2284 | 1 | 2591 | 2582 | 17 | 2583 | 12 | 2586 | 14 | 2590 | 11 |
| ORL 125 | 17 | 3166 | 10 | 3173 | 1 | 97 | 1 | 100 | 1 | 100 | 1 | 100 | 1 | 100 | |
| ORL 128 | 37 | 3081 | 34 | 3083 | 1 | 2332 | 5 | 2341 | 74 | 2272 | 6 | 2338 | 13 | 2331 | |
| ORL 133 | 668 | 2051 | 642 | 2077 | 159 | 2499 | 1505 | 1177 | 1650 | 1032 | 1596 | 1086 | 1426 | 1256 | |
| ORL 160 | 2397 | 3 | 2400 | 2 | 1 | 1950 | 1 | 1958 | 2 | 1957 | 6 | 1593 | 1 | 1958 | |
| Mixed | ORL 126 | 36 | 2624 | 30 | 2630 | 1 | 2936 | 33 | 2920 | 25 | 2926 | 43 | 2910 | 9 | 2944 |
| ORL 187 | 1 | 0 | 1 | 0 | 222 | 1054 | 1181 | 121 | 1185 | 117 | 1179 | 123 | 1185 | 117 | |
| ORL 243 | 26 | 2486 | 21 | 2491 | 219 | 49 | 1 | 271 | 2 | 270 | 1 | 271 | 1 | 271 | |
| ORL 244 | 94 | 2154 | 92 | 2157 | 1 | 2671 | 2246 | 445 | 2274 | 417 | 312 | 2378 | 131 | 2560 | |
| Extrachromosomal | ORL 104 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
| ORL 116 | 123 | 1106 | 102 | 1127 | 22 | 1241 | 791 | 484 | 805 | 469 | 797 | 477 | 850 | 425 | |
| ORL 137 | 150 | 3120 | 56 | 3214 | 1 | 1403 | 14 | 2402 | 16 | 2399 | 14 | 2402 | 1 | 2415 | |
| ORL 155 | 10 | 2495 | 9 | 2496 | 1 | 2399 | 3 | 2413 | 6 | 2409 | 1 | 2415 | 2 | 2414 | |
| ORL 161 | 15 | 2530 | 11 | 2534 | 1 | 2281 | 9 | 2286 | 6 | 2289 | 10 | 2285 | 4 | 2291 | |
| ORL 181 | 52 | 1284 | 32 | 1305 | 7 | 1588 | 14 | 1588 | 1 | 1602 | 34 | 1569 | 27 | 1576 | |
| ORL 257 | 14 | 2035 | 13 | 2036 | 1 | 1933 | 2 | 1945 | 2 | 1945 | 4 | 1943 | 5 | 1942 | |
| ORL 265 | 25 | 2217 | 7 | 2235 | 2 | 1773 | 1 | 1788 | 4 | 1785 | 17 | 1772 | 1 | 1788 | |
| ORL 280 | 28 | 2462 | 22 | 2468 | 2 | 3247 | 20 | 3234 | 23 | 3230 | 22 | 3232 | 106 | 3148 | |
| Nucleotide Position | 7482 | 7486 | 7518 | 7629 | 7666 | 7686 | 7712 | 7726 | 7740 | 7761 | 7783 | 7853 | 7883 | 7888 | 12 | 13 | 24 | 28 | 109 | 131 | 145 | 188 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Integrated | ORL 111 | ||||||||||||||||||||||
| ORL 125 | A | C | A | G | |||||||||||||||||||
| ORL 128 | A | G | |||||||||||||||||||||
| ORL 133 | A | ||||||||||||||||||||||
| ORL 160 | A | ||||||||||||||||||||||
| Mixed | ORL 126 | A | |||||||||||||||||||||
| ORL 187 | A | ||||||||||||||||||||||
| ORL 243 | T | C | |||||||||||||||||||||
| ORL 244 | A | ||||||||||||||||||||||
| Extrachromosomal | ORL 116 | ||||||||||||||||||||||
| ORL 137 | A | ||||||||||||||||||||||
| ORL 155 | A | ||||||||||||||||||||||
| ORL 161 | A | ||||||||||||||||||||||
| ORL 181 | A | A | |||||||||||||||||||||
| ORL 257 | A | ||||||||||||||||||||||
| ORL 265 | A | G | C | ||||||||||||||||||||
| ORL 280 | A | ||||||||||||||||||||||
| ORL 104 | C | A | A | T | A | C | G | T | T | T | T | T | |||||||||||
| AF402678 | C | A | A | T | A | C | G | T | T | T | A | T | |||||||||||
| K02718.1 | A | G | G | C | C | C | T | A | T | C | C | G | C | T | C | C | G | T | A | G | G | ||
| TF | YY1 | Tef1 | NF1 | YY1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pokrývková, B.; Saláková, M.; Šmahelová, J.; Vojtěchová, Z.; Novosadová, V.; Tachezy, R. Detailed Characteristics of Tonsillar Tumors with Extrachromosomal or Integrated Form of Human Papillomavirus. Viruses 2020, 12, 42. https://doi.org/10.3390/v12010042
Pokrývková B, Saláková M, Šmahelová J, Vojtěchová Z, Novosadová V, Tachezy R. Detailed Characteristics of Tonsillar Tumors with Extrachromosomal or Integrated Form of Human Papillomavirus. Viruses. 2020; 12(1):42. https://doi.org/10.3390/v12010042
Chicago/Turabian StylePokrývková, Barbora, Martina Saláková, Jana Šmahelová, Zuzana Vojtěchová, Vendula Novosadová, and Ruth Tachezy. 2020. "Detailed Characteristics of Tonsillar Tumors with Extrachromosomal or Integrated Form of Human Papillomavirus" Viruses 12, no. 1: 42. https://doi.org/10.3390/v12010042
APA StylePokrývková, B., Saláková, M., Šmahelová, J., Vojtěchová, Z., Novosadová, V., & Tachezy, R. (2020). Detailed Characteristics of Tonsillar Tumors with Extrachromosomal or Integrated Form of Human Papillomavirus. Viruses, 12(1), 42. https://doi.org/10.3390/v12010042