miR-26a Inhibits Feline Herpesvirus 1 Replication by Targeting SOCS5 and Promoting Type I Interferon Signaling

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Virus
2.2. siRNAs, miRNAs and Transfection
2.3. Antibodies and Western Blot
2.4. microRNA Target Gene Prediction and Plasmid Construction
2.5. Quantitative Reverse Transcription-PCR (qRT-PCR)
2.6. Dual-Luciferase Reporter Assays
2.7. Plaque Assays
2.8. Statistical Analysis
3. Results
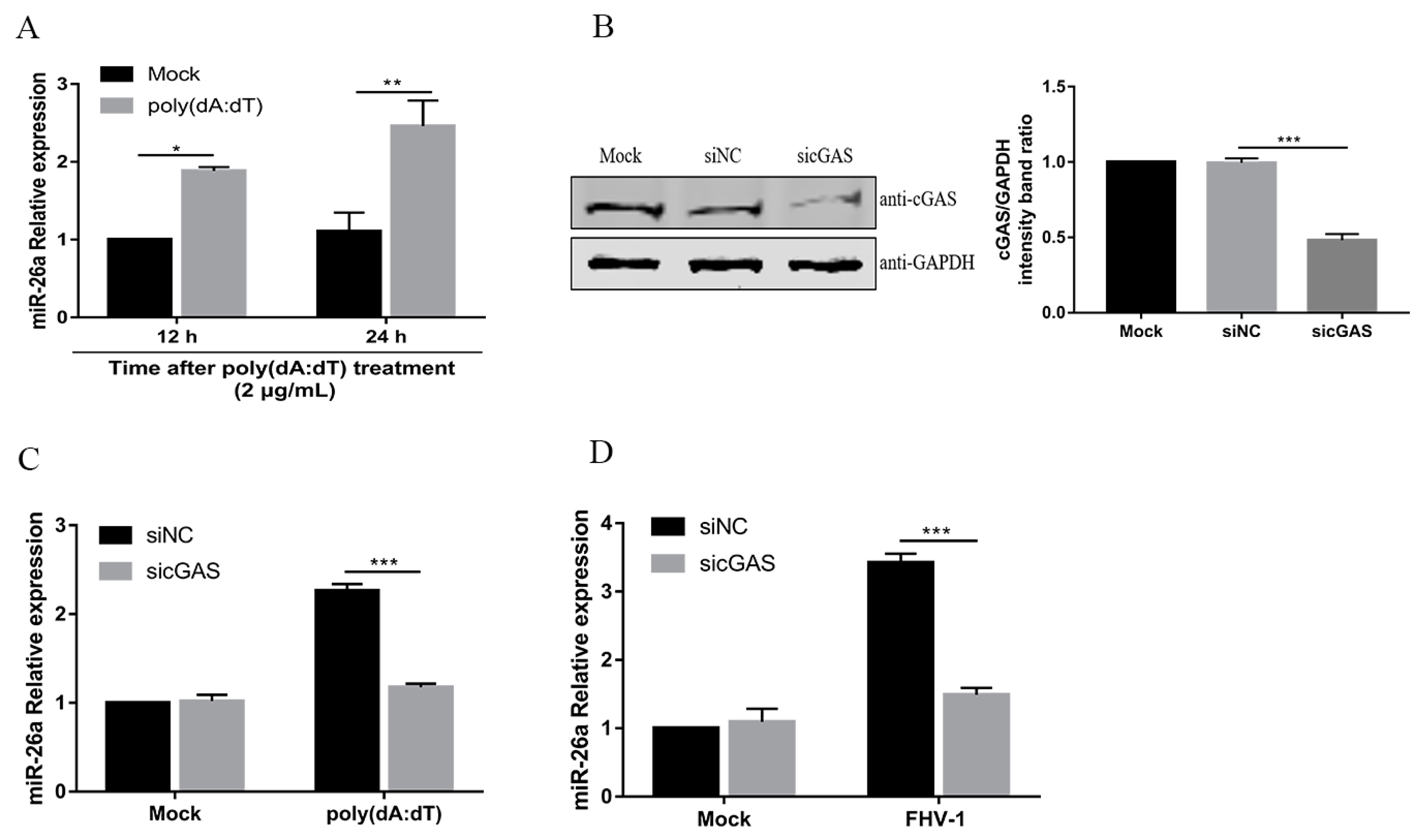
3.1. FHV-1 Infection Increases the Expression of miR-26a
3.2. FHV-1 Infection Upregulates the Level of miR-26a via the cGAS-Mediated Signalling Pathway
3.3. miR-26a Inhibits FHV-1 Replication
3.4. miR-26a Directly Targets SOCS5
3.5. miR-26a Enhance JAK-STAT Antiviral Signalling
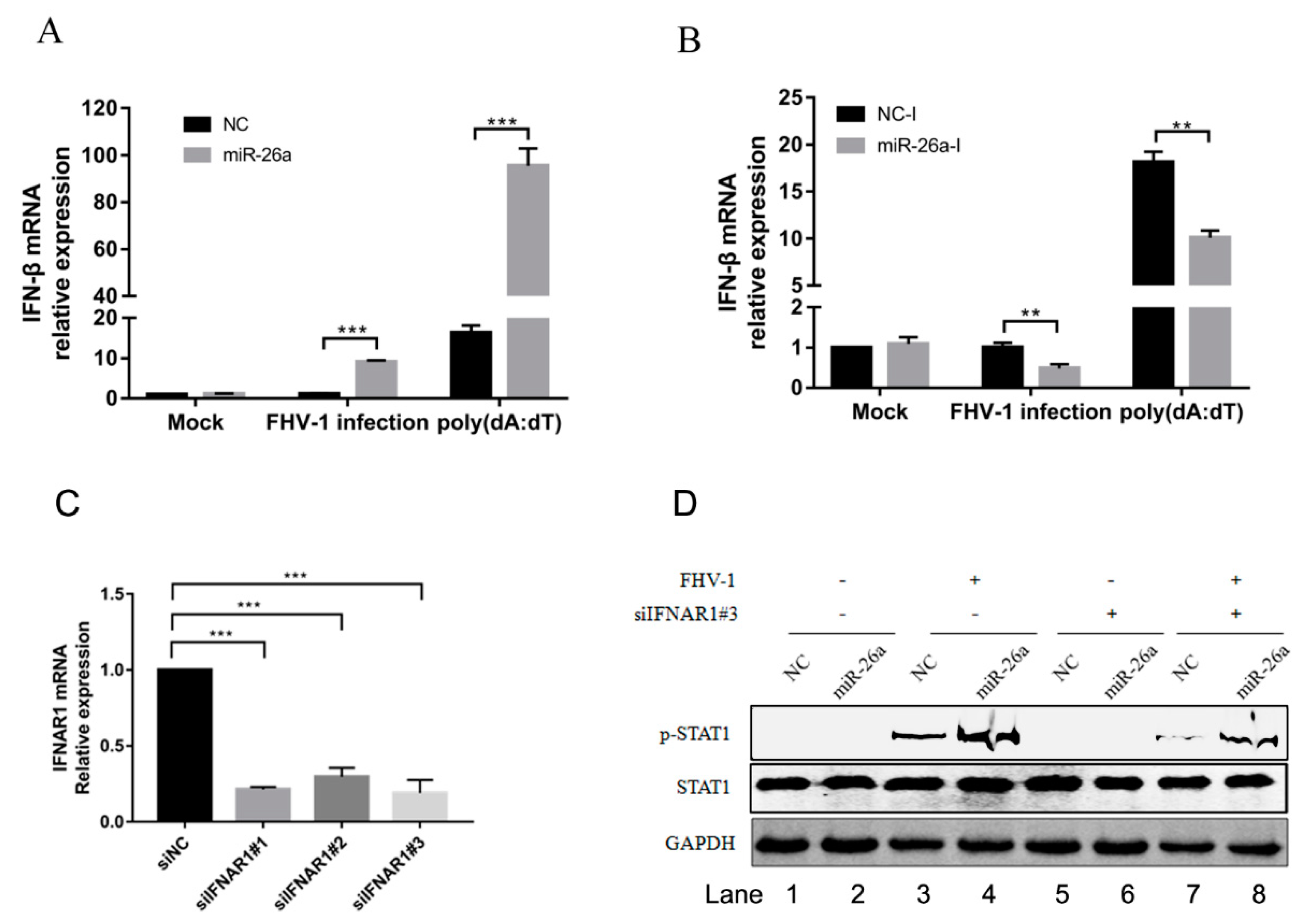
3.6. miR-26a Is Involved in Type I Interferon Production
3.7. SOCS5 Can Inhibit Type I Antiviral Signalling and Facilitate FHV-1 Replication
3.8. miR-26a Enhances IFN Antiviral Signalling Through Regulating Downstream SOCS5
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Davison, A.J.; Eberle, R.; Ehlers, B.; Hayward, G.S.; McGeoch, D.J.; Minson, A.C.; Pellett, P.E.; Roizman, B.; Studdert, M.J.; Thiry, E. The order Herpesvirales. Arch. Virol. 2009, 154, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Stanley, J.; Bisaro, D.; Briddon, R.; Brown, J.; Fauquet, C.; Harrison, B.; Rybicki, E.; Stenger, D.C. Geminivridae: Eighth Report of the ICTV on Virus Taxonomy. In Virus Taxonomy-Eighth Report of the International Committee on Taxonomy of Viruses; Elsevier: San Diego, CA, USA, 2005; pp. 301–306. [Google Scholar]
- Daniels, M.J.; Golder, M.C.; Jarrett, O.; MacDonald, D.W. MacDonald, Feline viruses in wildcats from Scotland. J. Wildl. Dis. 1999, 35, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Hofmann-Lehmann, R.; Fehr, D.; Grob, M.; Elgizoli, M.; Packer, C.; Martenson, J.S.; O’Brien, S.J.; Lutz, H. Prevalence of antibodies to feline parvovirus, calicivirus, herpesvirus, coronavirus, and immunodeficiency virus and of feline leukemia virus antigen and the interrelationship of these viral infections in free-ranging lions in east Africa. Clin. Diagn. Lab. Immunol. 1996, 3, 554–562. [Google Scholar] [PubMed]
- Munson, L.; Wack, R.; Duncan, M.; Montali, R.J.; Boon, D.; Stalis, I.; Crawshaw, G.J.; Cameron, K.N.; Mortenson, J.; Citino, S.; et al. Chronic eosinophilic dermatitis associated with persistent feline herpes virus infection in cheetahs (Acinonyx jubatus). Vet. Pathol. 2004, 41, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Ostrowski, S.; van Vuuren, M.; Lenain, D.M.; Durand, A. A serologic survey of wild felids from central west Saudi Arabia. J. Wildl. Dis. 2003, 39, 696–701. [Google Scholar] [CrossRef]
- Paul-Murphy, J.; Work, T.; Hunter, D.; McFie, E.; Fjelline, D. Serologic survey and serum biochemical reference ranges of the free-ranging mountain lion (Felis concolor) in California. J. Wildl. Dis. 1994, 30, 205–215. [Google Scholar] [CrossRef][Green Version]
- Van Vuuren, M.; Goosen, T.; Rogers, P. Feline herpesvirus infection in a group of semi-captive cheetahs. J. S. Afr. Vet. Assoc. 1999, 70, 132–134. [Google Scholar]
- Thiry, E.; Addie, D.; Belák, S.; Boucraut-Baralon, C.; Egberink, H.; Frymus, T.; Gruffydd-Jones, T.; Hartmann, K.; Hosie, M.J.; Lloret, A.; et al. Feline herpesvirus infection. ABCD guidelines on prevention and management. J. Feline Med. 2009, 11, 547–555. [Google Scholar] [CrossRef]
- Townsend, W.M.; Jacobi, S.; Tai, S.H.; Kiupel, M.; Wise, A.G.; Maes, R.K. Ocular and neural distribution of feline herpesvirus-1 during active and latent experimental infection in cats. BMC Vet. Res. 2013, 9, 185. [Google Scholar] [CrossRef]
- Albà, M.M.; Das, R.; Orengo, C.A.; Kellam, P. Genomewide function conservation and phylogeny in the Herpesviridae. Genome Res. 2001, 11, 43–54. [Google Scholar] [CrossRef]
- Tai, S.S.; Niikura, M.; Cheng, H.H.; Kruger, J.M.; Wise, A.G.; Maes, R.K. Complete genomic sequence and an infectious BAC clone of feline herpesvirus-1 (FHV-1). Virology 2010, 401, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Liu, Y.; Liu, X.; Sun, X.; Zhang, J.; Qu, L. Feline Herpesvirus 1 US3 Blocks the Type I Interferon Signal Pathway by Targeting Interferon Regulatory Factor 3 Dimerization in a Kinase-Independent Manner. J. Virol. 2018, 92, e00047-18. [Google Scholar] [CrossRef] [PubMed]
- Kloosterman, W.P.; Plasterk, R.H. The diverse functions of microRNAs in animal development and disease. Dev. Cell 2006, 11, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Bushati, N.; Cohen, S.M. microRNA functions. Annu. Rev. Cell Dev. Biol. 2007, 23, 175–205. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.C.; Doudna, J.A. Molecular mechanisms of RNA interference. Annu. Rev. Biophys. 2013, 42, 217–239. [Google Scholar] [CrossRef]
- Kim, Y.K.; Heo, I.; Kim, V.N. Modifications of small RNAs and their associated proteins. Cell 2010, 143, 703–709. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Zhu, B.; Ye, J.; Nie, Y.; Ashraf, U.; Zohaib, A.; Duan, X.; Fu, Z.F.; Song, Y.; Chen, H.; Cao, S. MicroRNA-15b Modulates Japanese Encephalitis Virus-Mediated Inflammation via Targeting RNF125. J. Immunol. 2015, 195, 2251–2262. [Google Scholar] [CrossRef]
- Yin, Y.; Liu, W.; Dai, Y. SOCS3 and its role in associated diseases. Hum. Immunol. 2015, 76, 775–780. [Google Scholar] [CrossRef]
- Wang, P.; Hou, J.; Lin, L.; Wang, C.; Liu, X.; Li, D.; Ma, F.; Wang, Z.; Cao, X. Inducible microRNA-155 feedback promotes type I IFN signaling in antiviral innate immunity by targeting suppressor of cytokine signaling 1. J. Immunol. 2010, 185, 6226–6233. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, C.; Xue, M.; Fu, F.; Zhang, X.; Li, L.; Yin, L.; Xu, W.; Feng, L.; Liu, P. The Coronavirus Transmissible Gastroenteritis Virus Evades the Type I Interferon Response through IRE1alpha-Mediated Manipulation of the MicroRNA miR-30a-5p/SOCS1/3 Axis. J. Virol. 2018, 92, e00728-18. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Wang, B.; Chen, X.; He, Z.; Wang, Y.; Li, X.; Cao, H.; Zheng, S.J. MicroRNA gga-miR-130b Suppresses Infectious Bursal Disease Virus Replication via Targeting of the Viral Genome and Cellular Suppressors of Cytokine Signaling 5. J. Virol. 2018, 92, e01646-17. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Kumawat, K.L.; Rastogi, M.; Basu, A.; Singh, S.K. Japanese Encephalitis Virus exploits the microRNA-432 to regulate the expression of Suppressor of Cytokine Signaling (SOCS) 5. Sci. Rep. 2016, 6, 27685. [Google Scholar] [CrossRef] [PubMed]
- Haldipur, B.; Bhukya, P.L.; Arankalle, V.; Lole, K. Positive Regulation of Hepatitis E Virus Replication by MicroRNA-122. J. Virol. 2018, 92, e01999-17. [Google Scholar] [CrossRef]
- Zhang, Q.; Guo, X.K.; Gao, L.; Huang, C.; Li, N.; Jia, X.; Liu, W.; Feng, W.H. MicroRNA-23 inhibits PRRSV replication by directly targeting PRRSV RNA and possibly by upregulating type I interferons. Virology 2014, 450, 182–195. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, Z. Identification and Analysis of the Profile of Viral and Host microRNAs in Feline Herpesvirus 1-Infected CRFK cells. Virol. J. 2020. (submitted). [Google Scholar]
- Sun, J.Z.; Wang, J.; Wang, S.; Yuan, D.; Birame, B.M.; Li, Z.; Yi, B.; Liu, W. MicroRNA profile analysis of a feline kidney cell line before and after infection with mink enteritis virus. Gene 2014, 539, 224–229. [Google Scholar] [CrossRef]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef]
- Linossi, E.M.; Calleja, D.J.; Nicholson, S.E. Understanding SOCS protein specificity. Growth Factors 2018, 36, 1–14. [Google Scholar] [CrossRef]
- Hilton, D.J.; Richardson, R.T.; Alexander, W.S.; Viney, E.M.; Willson, T.A.; Sprigg, N.S.; Starr, R.; Nicholson, S.E.; Metcalf, D.; Nicola, N.A. Twenty proteins containing a C-terminal SOCS box form five structural classes. Proc. Natl. Acad. Sci. USA 1998, 95, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, A.; Ito, M.; Chikuma, S.; Akanuma, T.; Nakatsukasa, H. Negative Regulation of Cytokine Signaling in Immunity. Cold Spring Harb. Perspect. Biol. 2018, 10, a028571. [Google Scholar] [CrossRef] [PubMed]
- Linossi, E.M.; Nicholson, S.E. Kinase inhibition, competitive binding and proteasomal degradation: Resolving the molecular function of the suppressor of cytokine signaling (SOCS) proteins. Immunol. Rev. 2015, 266, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.J.; Chang, B.L.; Yu, H.P.; Liao, C.L.; Lin, Y.L. Blocking of interferon-induced Jak-Stat signaling by Japanese encephalitis virus NS5 through a protein tyrosine phosphatase-mediated mechanism. J. Virol. 2006, 80, 5908–5918. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Koshizuka, T.; Ishibashi, K.; Hashimoto, K.; Ishioka, K.; Ikuta, K.; Yokota, S.I.; Fujii, N.; Suzutani, T. Involvement of herpes simplex virus type 1 UL13 protein kinase in induction of SOCS genes, the negative regulators of cytokine signaling. Microbiol. Immunol. 2017, 61, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Yokota, S.; Yokosawa, N.; Okabayashi, T.; Suzutani, T.; Fujii, N. Induction of suppressor of cytokine signaling-3 by herpes simplex virus type 1 confers efficient viral replication. Virology 2005, 338, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Yokota, S.; Yokosawa, N.; Okabayashi, T.; Suzutani, T.; Miura, S.; Jimbow, K.; Fujii, N. Induction of suppressor of cytokine signaling-3 by herpes simplex virus type 1 contributes to inhibition of the interferon signaling pathway. J. Virol. 2004, 78, 6282–6286. [Google Scholar] [CrossRef]
- Frey, K.G.; Ahmed, C.M.; Dabelic, R.; Jager, L.D.; Noon-Song, E.N.; Haider, S.M.; Johnson, H.M.; Bigley, N.J. HSV-1-induced SOCS-1 expression in keratinocytes: Use of a SOCS-1 antagonist to block a novel mechanism of viral immune evasion. J. Immunol. 2009, 183, 1253–1262. [Google Scholar] [CrossRef]
- Linossi, E.M.; Chandrashekaran, I.R.; Kolesnik, T.B.; Murphy, J.M.; Webb, A.I.; Willson, T.A.; Kedzierski, L.; Bullock, A.N.; Babon, J.J.; Norton, R.S.; et al. Suppressor of Cytokine Signaling (SOCS) 5 utilises distinct domains for regulation of JAK1 and interaction with the adaptor protein Shc-1. PLoS ONE 2013, 8, e70536. [Google Scholar] [CrossRef]
- Bruscella, P.; Bottini, S.; Baudesson, C.; Pawlotsky, J.M.; Feray, C.; Trabucchi, M. Viruses and miRNAs: More Friends than Foes. Front. Microbiol. 2017, 8, 824. [Google Scholar] [CrossRef]
- Piedade, D.; Azevedo-Pereira, J.M. The Role of microRNAs in the Pathogenesis of Herpesvirus Infection. Viruses 2016, 8, 156. [Google Scholar] [CrossRef] [PubMed]
- Frappier, L. Regulation of herpesvirus reactivation by host microRNAs. J. Virol. 2015, 89, 2456–2458. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Forster, S.C.; Tate, M.D.; Hertzog, P.J. MicroRNA as Type I Interferon-Regulated Transcripts and Modulators of the Innate Immune Response. Front. Immunol. 2015, 6, 334. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; He, L.; Li, Y.; Wang, T.; Feng, L.; Jiang, L.; Zhang, P.; Huang, X. miR-146a facilitates replication of dengue virus by dampening interferon induction by targeting TRAF6. J. Infect. 2013, 67, 329–341. [Google Scholar] [CrossRef]
- Wang, B.; Fu, M.; Liu, Y.; Wang, Y.; Li, X.; Cao, H.; Zheng, S.J. gga-miR-155 Enhances Type I Interferon Expression and Suppresses Infectious Burse Disease Virus Replication via Targeting SOCS1 and TANK. Front. Cell. Infect. Microbiol. 2018, 8, 55. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, L.; Wang, H.; Zhang, G.; Sun, X. Respiratory syncytial virus non-structural protein 1 facilitates virus replication through miR-29a-mediated inhibition of interferon-alpha receptor. Biochem. Biophys. Res. Commun. 2016, 478, 1436–1441. [Google Scholar] [CrossRef]
- Xie, Y.; He, S.; Wang, J. MicroRNA-373 facilitates HSV-1 replication through suppression of type I IFN response by targeting IRF1. Biomed. Pharmacother. 2018, 97, 1409–1416. [Google Scholar] [CrossRef]
- Zheng, C. Evasion of Cytosolic DNA-Stimulated Innate Immune Responses by Herpes Simplex Virus 1. J. Virol. 2018, 92, e00099-17. [Google Scholar] [CrossRef]
- Melroe, G.T.; DeLuca, N.A.; Knipe, D.M. Herpes simplex virus 1 has multiple mechanisms for blocking virus-induced interferon production. J. Virol. 2004, 78, 8411–8420. [Google Scholar] [CrossRef]
- Lin, R.; Noyce, R.S.; Collins, S.E.; Everett, R.D.; Mossman, K.L. The herpes simplex virus ICP0 RING finger domain inhibits IRF3- and IRF7-mediated activation of interferon-stimulated genes. J. Virol. 2004, 78, 1675–1684. [Google Scholar] [CrossRef]
- Xing, J.; Wang, S.; Lin, R.; Mossman, K.L.; Zheng, C. Herpes simplex virus 1 tegument protein US11 downmodulates the RLR signaling pathway via direct interaction with RIG-I and MDA-5. J. Virol. 2012, 86, 3528–3540. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, K.; Li, J.; Zheng, C. Herpes simplex virus 1 ubiquitin-specific protease UL36 inhibits beta interferon production by deubiquitinating TRAF3. J. Virol. 2013, 87, 11851–11860. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, S.; Wang, K.; Zheng, C. Herpes simplex virus 1 DNA polymerase processivity factor UL42 inhibits TNF-alpha-induced NF-kappaB activation by interacting with p65/RelA and p50/NF-kappaB1. Med. Microbiol. Immunol. 2013, 202, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.; Ni, L.; Wang, S.; Wang, K.; Lin, R.; Zheng, C. Herpes simplex virus 1-encoded tegument protein VP16 abrogates the production of beta interferon (IFN) by inhibiting NF-kappaB activation and blocking IFN regulatory factor 3 to recruit its coactivator CBP. J. Virol. 2013, 87, 9788–9801. [Google Scholar] [CrossRef]
- Zhang, D.; Su, C.; Zheng, C. Herpes Simplex Virus 1 Serine Protease VP24 Blocks the DNA-Sensing Signal Pathway by Abrogating Activation of Interferon Regulatory Factor 3. J. Virol. 2016, 90, 5824–5829. [Google Scholar] [CrossRef]
- Wang, S.; Wang, K.; Lin, R.; Zheng, C. Herpes simplex virus 1 serine/threonine kinase US3 hyperphosphorylates IRF3 and inhibits beta interferon production. J. Virol. 2013, 87, 12814–12827. [Google Scholar] [CrossRef] [PubMed]
- You, H.; Zheng, S.; Huang, Z.; Lin, Y.; Shen, Q.; Zheng, C. Herpes Simplex Virus 1 Tegument Protein UL46 Inhibits TANK-Binding Kinase 1-Mediated Signaling. MBio 2019, 10, e00919-19. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Li, Z.; Huang, J.; Yin, H.; Tian, J.; Qu, L. miR-26a Inhibits Feline Herpesvirus 1 Replication by Targeting SOCS5 and Promoting Type I Interferon Signaling. Viruses 2020, 12, 2. https://doi.org/10.3390/v12010002
Zhang J, Li Z, Huang J, Yin H, Tian J, Qu L. miR-26a Inhibits Feline Herpesvirus 1 Replication by Targeting SOCS5 and Promoting Type I Interferon Signaling. Viruses. 2020; 12(1):2. https://doi.org/10.3390/v12010002
Chicago/Turabian StyleZhang, Jikai, Zhijie Li, Jiapei Huang, Hang Yin, Jin Tian, and Liandong Qu. 2020. "miR-26a Inhibits Feline Herpesvirus 1 Replication by Targeting SOCS5 and Promoting Type I Interferon Signaling" Viruses 12, no. 1: 2. https://doi.org/10.3390/v12010002
APA StyleZhang, J., Li, Z., Huang, J., Yin, H., Tian, J., & Qu, L. (2020). miR-26a Inhibits Feline Herpesvirus 1 Replication by Targeting SOCS5 and Promoting Type I Interferon Signaling. Viruses, 12(1), 2. https://doi.org/10.3390/v12010002