Bacterial Virus Lambda Gpd-Fusions to Cathelicidins, α- and β-Defensins, and Disease-Specific Epitopes Evaluated for Antimicrobial Toxicity and Ability to Support Phage Display


Abstract
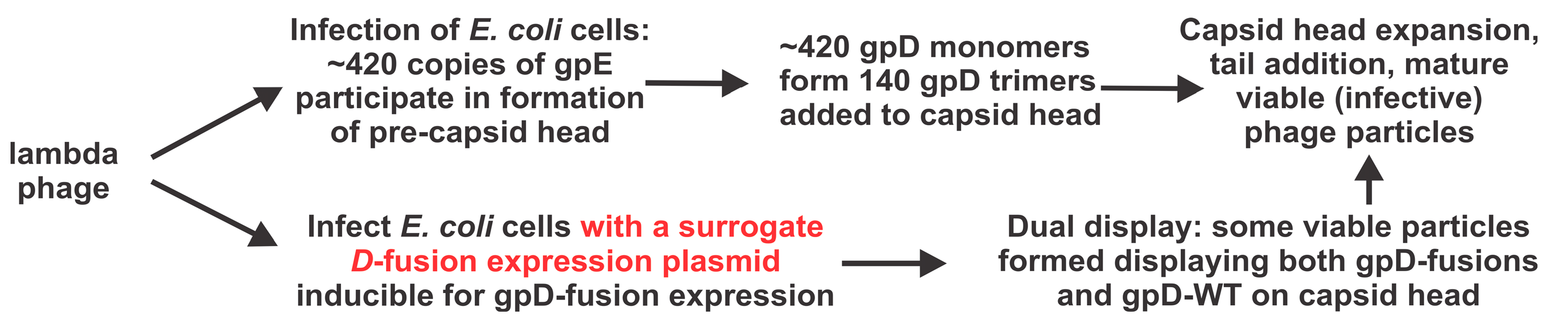
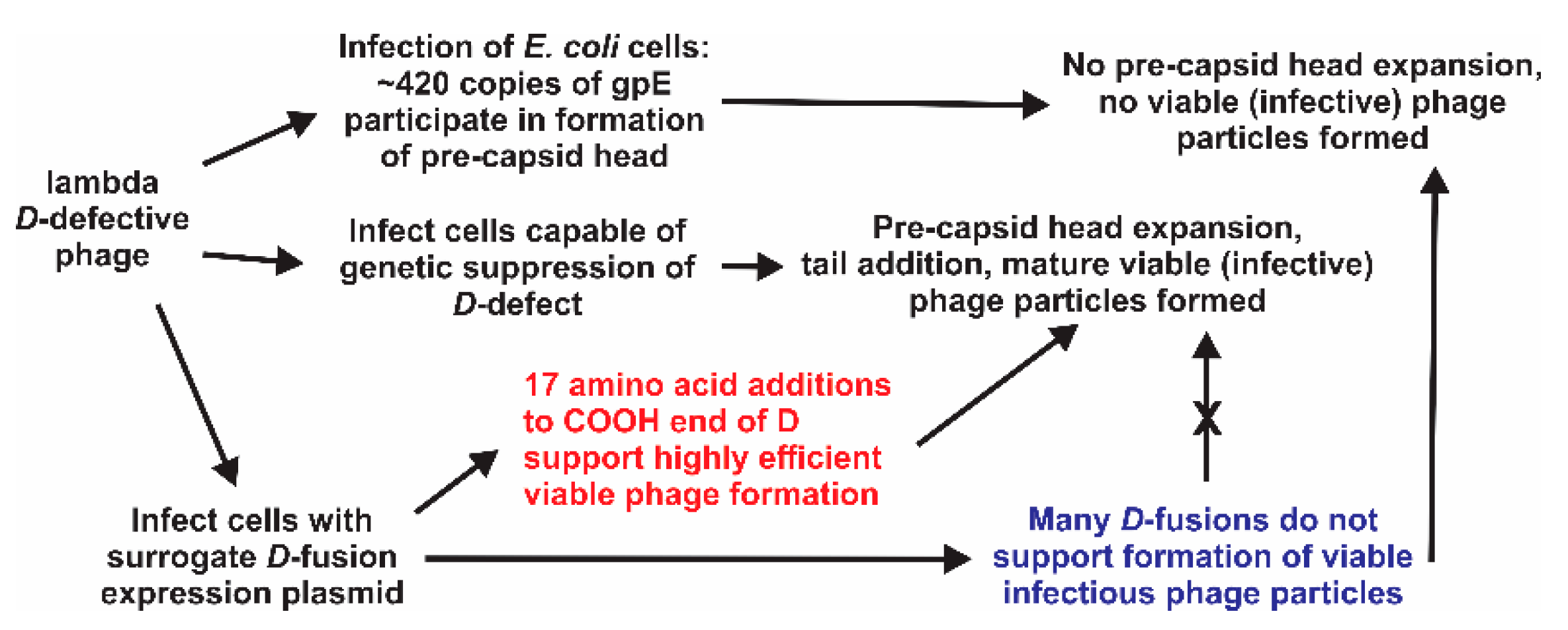
1. Introduction
2. Materials and Methods
2.1. Strains
2.2. Growth Medium and General Dilution “Buffer”
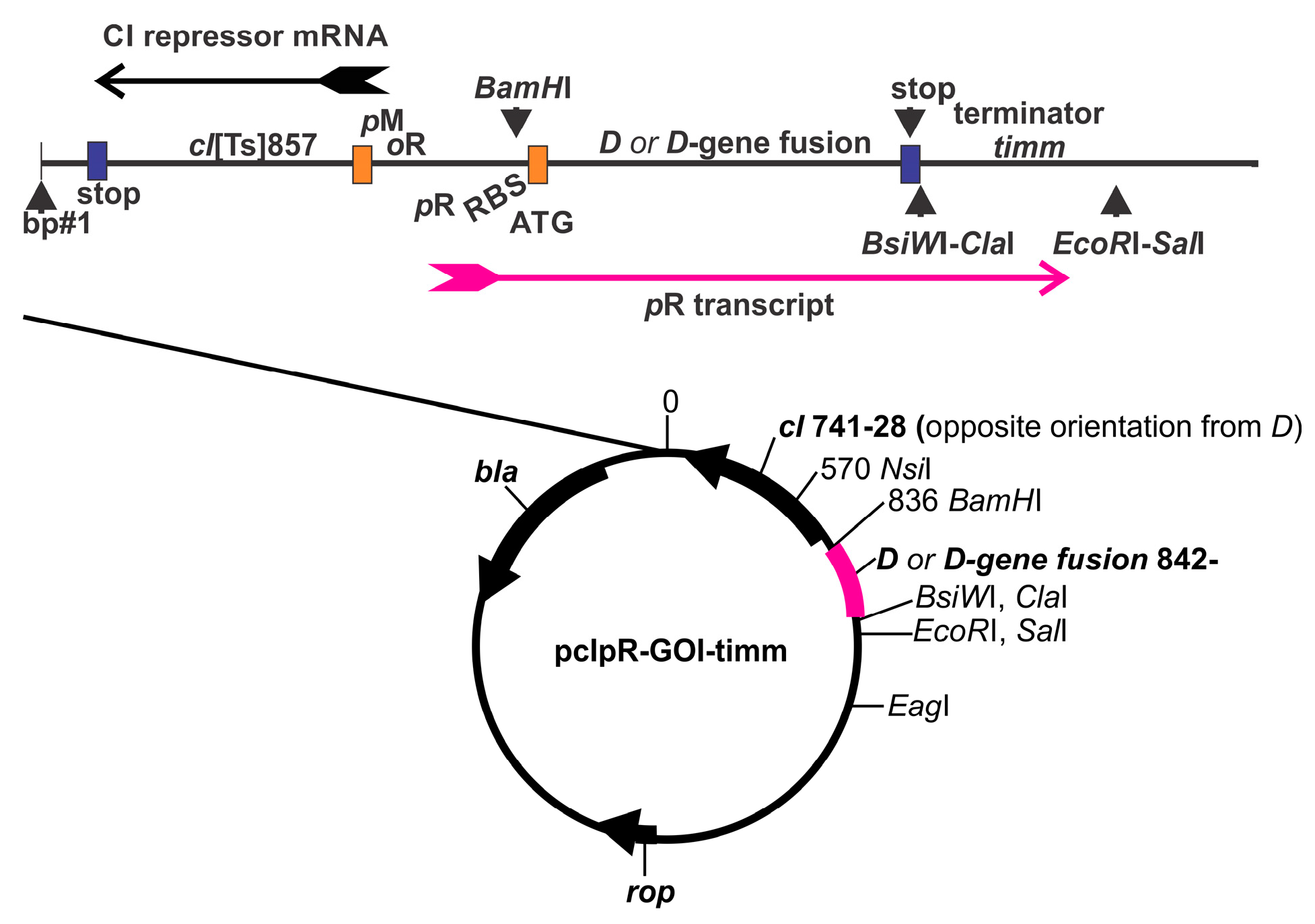
2.3. Plasmid Construction; D-Fusion Plasmids and Encoded Amino Acids
2.4. Single-Burst LDP-Vaccine and SEV Production
2.5. Infection-Complementation Using Transformed Cells
2.6. Complementation Methodology
2.7. Complementation Versus Reversion, Versus Marker rescue
2.7.1. D-Amber Reversion
2.7.2. D-Amber Marker Rescue
2.7.3. Complementation Temperature
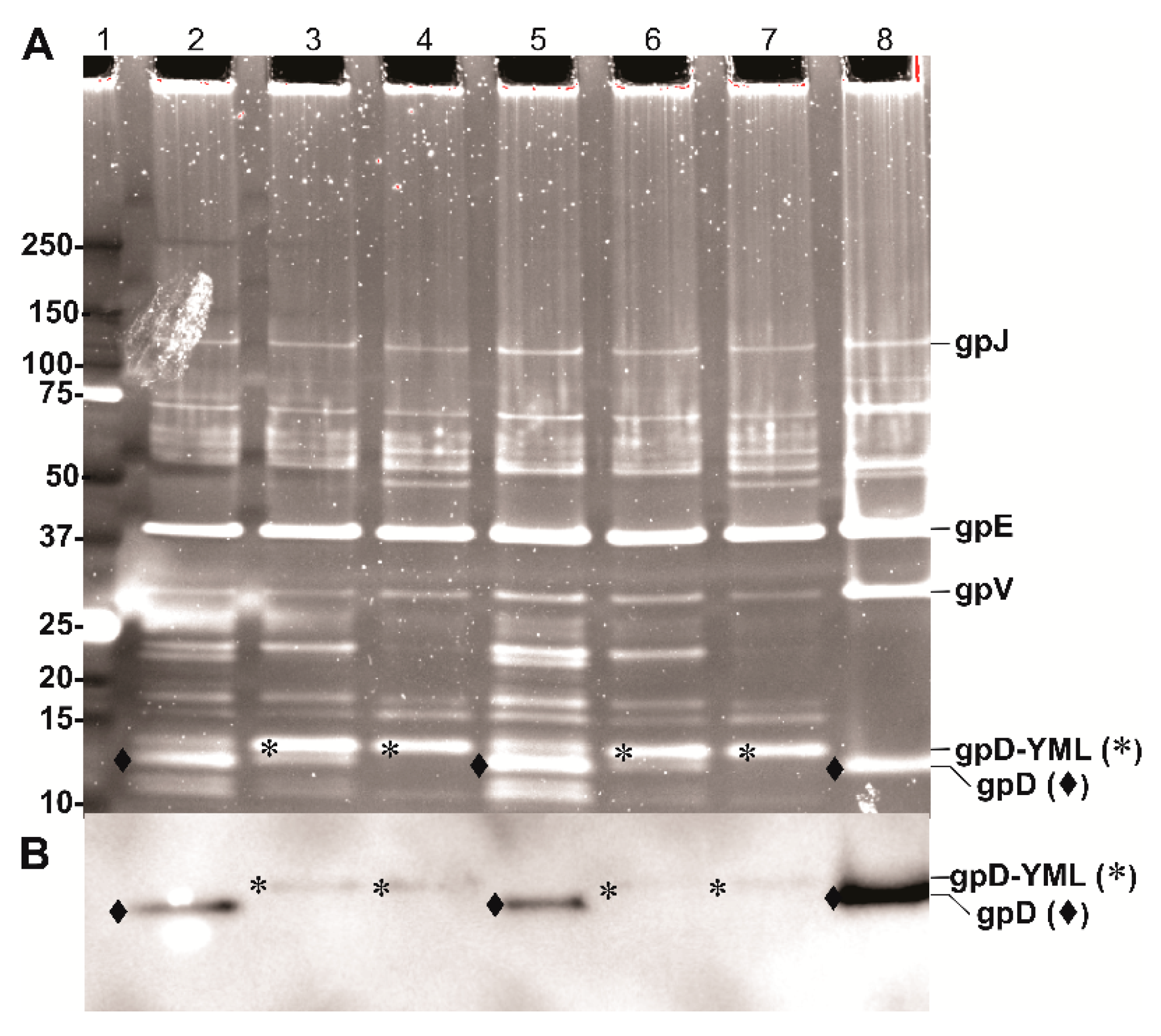
2.8. Protein Gels and Western Blots
3. Results and Discussion
3.1. Temperature Dependent Gene Expression from Plasmid pcIpR-GOI-Timm
3.2. Evaluating D-Fusion Cathelicidins for Cellular Toxicity
3.3. Temperature-Dependent Complementation for Phage Growth in D-Defective Phage mutant by Plasmids Expressing gpD or gpD-Fusions
3.4. Evaluating the Influence of D-fusion Size and Toxicity on LDP Production
4. Further Considerations and Summary
Supplementary Materials
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Casjens, S.R.; Hendrix, R.W. Locations and amounts of major structural proteins in bacteriophage lambda. J. Mol. Biol. 1974, 88, 535–545. [Google Scholar] [CrossRef]
- Imber, R.; Tsugita, A.; Wurtz, M.; Hohn, T. Outer surface protein of bacteriophage lambda. J. Mol. Biol. 1980, 139, 277–295. [Google Scholar] [CrossRef]
- Georgopoulos, C.; Tilly, K.; Casjens, S. Lambdoid phage head assembly. In Lambda ii; Hendrix, R.W., Roberts, J.R., Stahl, F.S., Weisberg, R.A., Eds.; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 1983; pp. 279–304. [Google Scholar]
- Sternberg, N.; Hoess, R.H. Display of peptides and proteins on the surface of bacteriophage lambda. Proc. Natl. Acad. Sci. USA 1995, 92, 1609–1613. [Google Scholar] [CrossRef]
- Sternberg, N.; Weisberg, R. Packaging of coliphage lambda DNA. Ii. The role of the gene d protein. J. Mol. Biol. 1977, 117, 733–759. [Google Scholar] [CrossRef]
- Ansuini, H.; Cicchini, C.; Nicosia, A.; Tripodi, M.; Cortese, R.; Luzzago, A. Biotin-tagged cdna expression libraries displayed on lambda phage: A new tool for the selection of natural protein ligands. Nucleic Acids Res. 2002, 30, e78. [Google Scholar] [CrossRef]
- Cicchini, C.; Ansuini, H.; Amicone, L.; Alonzi, T.; Nicosia, A.; Cortese, R.; Tripodi, M.; Luzzago, A. Searching for DNA-protein interactions by lambda phage display. J. Mol. Biol. 2002, 322, 697–706. [Google Scholar] [CrossRef]
- Cortese, R.; Monaci, P.; Luzzago, A.; Santini, C.; Bartoli, F.; Cortese, I.; Fortugno, P.; Galfre, G.; Nicosia, A.; Felici, F. Selection of biologically active peptides by phage display of random peptide libraries. Curr. Opin. Biotechnol. 1996, 7, 616–621. [Google Scholar] [CrossRef]
- Kong, B.; Ma, W.J. Display of aggregation-prone ligand binding domain of human ppar gamma on surface of bacteriophage lambda. Acta Pharmacol. Sin. 2006, 27, 91–99. [Google Scholar] [CrossRef]
- Maruyama, I.N.; Maruyama, H.I.; Brenner, S. Lambda foo: A lambda phage vector for the expression of foreign proteins. Proc. Natl. Acad. Sci. USA 1994, 91, 8273–8277. [Google Scholar] [CrossRef]
- Mikawa, Y.G.; Maruyama, I.N.; Brenner, S. Surface display of proteins on bacteriophage lambda heads. J. Mol. Biol. 1996, 262, 21–30. [Google Scholar] [CrossRef]
- Niwa, M.; Maruyama, H.; Fujimoto, T.; Dohi, K.; Maruyama, I.N. Affinity selection of cdna libraries by lambda phage surface display. Gene 2000, 256, 229–236. [Google Scholar] [CrossRef]
- Santi, E.; Capone, S.; Mennuni, C.; Lahm, A.; Tramontano, A.; Luzzago, A.; Nicosia, A. Bacteriophage lambda display of complex cdna libraries: A new approach to functional genomics. J. Mol. Biol 2000, 296, 497–508. [Google Scholar] [CrossRef]
- Santini, C.; Brennan, D.; Mennuni, C.; Hoess, R.H.; Nicosia, A.; Cortese, R.; Luzzago, A. Efficient display of an hcv cdna expression library as c-terminal fusion to the capsid protein d of bacteriophage lambda. J. Mol. Biol. 1998, 282, 125–135. [Google Scholar] [CrossRef]
- Vilchez, S.; Jacoby, J.; Ellar, D.J. Display of biologically functional insecticidal toxin on the surface of lambda phage. Appl. Environ. Microbiol. 2004, 70, 6587–6594. [Google Scholar] [CrossRef]
- Zucconi, A.; Dente, L.; Santonico, E.; Castagnoli, L.; Cesareni, G. Selection of ligands by panning of domain libraries displayed on phage lambda reveals new potential partners of synaptojanin 1. J. Mol. Biol. 2001, 307, 1329–1339. [Google Scholar] [CrossRef]
- Gupta, A.; Onda, M.; Pastan, I.; Adhya, S.; Chaudhary, V.K. High-density functional display of proteins on bacteriophage lambda. J. Mol. Biol. 2003, 334, 241–254. [Google Scholar] [CrossRef]
- Zanghi, C.N.; Lankes, H.A.; Bradel-Tretheway, B.; Wegman, J.; Dewhurst, S. A simple method for displaying recalcitrant proteins on the surface of bacteriophage lambda. Nucleic Acids Res. 2005, 33, e160. [Google Scholar] [CrossRef]
- Zanghi, C.N.; Sapinoro, R.; Bradel-Tretheway, B.; Dewhurst, S. A tractable method for simultaneous modifications to the head and tail of bacteriophage lambda and its application to enhancing phage-mediated gene delivery. Nucleic Acids Res. 2007, 35, e59. [Google Scholar] [CrossRef]
- Gao, J.; Wang, Y.; Liu, Z. Phage display an its application in vaccine design. Ann. Microbiol. 2010, 60, 13–19. [Google Scholar] [CrossRef]
- Ren, Z.J.; Lewis, G.K.; Wingfield, P.T.; Locke, E.G.; Steven, A.C.; Black, L.W. Phage display of intact domains at high copy number: A system based on soc, the small outer capsid protein of bacteriophage t4. Protein Sci. 1996, 5, 1833–1843. [Google Scholar] [CrossRef]
- Ren, Z.J.; Tian, C.J.; Zhu, Q.S.; Zhao, M.Y.; Xin, A.G.; Nie, W.X.; Ling, S.R.; Zhu, M.W.; Wu, J.Y.; Lan, H.Y.; et al. Orally delivered foot-and-mouth disease virus capsid protomer vaccine displayed on t4 bacteriophage surface: 100% protection from potency challenge in mice. Vaccine 2008, 26, 1471–1481. [Google Scholar] [CrossRef]
- Sathaliyawala, T.; Rao, M.; Maclean, D.M.; Birx, D.L.; Alving, C.R.; Rao, V.B. Assembly of human immunodeficiency virus (hiv) antigens on bacteriophage t4: A novel in vitro approach to construct multicomponent hiv vaccines. J. Virol. 2006, 80, 7688–7698. [Google Scholar] [CrossRef]
- Shivachandra, S.B.; Rao, M.; Janosi, L.; Sathaliyawala, T.; Matyas, G.R.; Alving, C.R.; Leppla, S.H.; Rao, V.B. In vitro binding of anthrax protective antigen on bacteriophage t4 capsid surface through hoc-capsid interactions: A strategy for efficient display of large full-length proteins. Virology 2006, 345, 190–198. [Google Scholar] [CrossRef]
- Wu, J.; Tu, C.; Yu, X.; Zhang, M.; Zhang, N.; Zhao, M.; Nie, W.; Ren, Z. Bacteriophage t4 nanoparticle capsid surface soc and hoc bipartite display with enhanced classical swine fever virus immunogenicity: A powerful immunological approach. J. Virol. Methods 2007, 139, 50–60. [Google Scholar] [CrossRef]
- Hayes, S.; Gamage, L.N.; Hayes, C. Dual expression system for assembling phage lambda display particle (ldp) vaccine to porcine circovirus 2 (pcv2). Vaccine 2010, 28, 6789–6799. [Google Scholar] [CrossRef]
- Gamage, L.N.; Ellis, J.; Hayes, S. Immunogenicity of bacteriophage lambda particles displaying porcine circovirus 2 (pcv2) capsid protein epitopes. Vaccine 2009, 27, 6595–6604. [Google Scholar] [CrossRef]
- Gonzalez-Cano, P.; Gamage, L.N.A.; Marciniuk, K.; Hayes, C.; Napper, S.; Hayes, S.; Griebel, P.J. Lambda display phage as a mucosal vaccine delivery vehicle for peptide antigens. Vaccine 2017, 35, 7256–7263. [Google Scholar] [CrossRef]
- Hayes, S.; Erker, C.; Horbay, M.A.; Marciniuk, K.; Wang, W.; Hayes, C. Phage lambda p protein: Trans-activation, inhibition phenotypes and their suppression. Viruses 2013, 5, 619–653. [Google Scholar] [CrossRef]
- Hayes, S.; Rajamanickam, K.; Hayes, C. Complementation studies of bacteriophage lambda o amber mutants by allelic forms of o expressed from plasmid, and o-p interaction phenotypes. Antibiotics 2018, 7, 31. [Google Scholar] [CrossRef]
- Rajamanickam, K.; Hayes, S. The bacteriophage lambda cii phenotypes for complementation, cellular toxicity and replication inhibition are suppressed in cii-oop constructs expressing the small rna oop. Viruses 2018, 10, 115. [Google Scholar] [CrossRef]
- Braff, M.H.; Hawkins, M.A.; Di Nardo, A.; Lopez-Garcia, B.; Howell, M.D.; Wong, C.; Lin, K.; Streib, J.E.; Dorschner, R.; Leung, D.Y.; et al. Structure-function relationships among human cathelicidin peptides: Dissociation of antimicrobial properties from host immunostimulatory activities. J. Immunol. 2005, 174, 4271–4278. [Google Scholar] [CrossRef]
- Vandamme, D.; Landuyt, B.; Luyten, W.; Schoofs, L. A comprehensive summary of ll-37, the factotum human cathelicidin peptide. Cell Immunol. 2012, 280, 22–35. [Google Scholar] [CrossRef]
- Yang, D.; Chertov, O.; Oppenheim, J.J. Participation of mammalian defensins and cathelicidins in anti-microbial immunity: Receptors and activities of human defensins and cathelicidin (ll-37). J. Leukoc. Biol. 2001, 69, 691–697. [Google Scholar]
- Zanetti, M. Cathelicidins, multifunctional peptides of the innate immunity. J. Leukoc. Biol. 2004, 75, 39–48. [Google Scholar] [CrossRef]
- Sang, Y.; Blecha, F. Porcine host defense peptides: Expanding repertoire and functions. Dev. Comp. Immunol. 2009, 33, 334–343. [Google Scholar] [CrossRef]
- Selsted, M.E.; Harwig, S.S.; Ganz, T.; Schilling, J.W.; Lehrer, R.I. Primary structures of three human neutrophil defensins. J. Clin. Investig. 1985, 76, 1436–1439. [Google Scholar] [CrossRef]
- Schneider, J.J.; Unholzer, A.; Schaller, M.; Schafer-Korting, M.; Korting, H.C. Human defensins. J. Mol. Med. 2005, 83, 587–595. [Google Scholar] [CrossRef]
- Tollner, T.L.; Bevins, C.L.; Cherr, G.N. Multifunctional glycoprotein defb126—A curious story of defensin-clad spermatozoa. Nat. Rev. Urol. 2012, 9, 365–375. [Google Scholar] [CrossRef]
- Tollner, T.L.; Venners, S.A.; Hollox, E.J.; Yudin, A.I.; Liu, X.; Tang, G.; Xing, H.; Kays, R.J.; Lau, T.; Overstreet, J.W.; et al. A common mutation in the defensin defb126 causes impaired sperm function and subfertility. Sci. Transl. Med. 2011, 3, 92ra65. [Google Scholar] [CrossRef]
- Bessette, P.H.; Aslund, F.; Beckwith, J.; Georgiou, G. Efficient folding of proteins with multiple disulfide bonds in the escherichia coli cytoplasm. Proc. Natl. Acad. Sci. USA 1999, 96, 13703–13708. [Google Scholar] [CrossRef]
- Lehrer, R.I.; Barton, A.; Daher, K.A.; Harwig, S.S.; Ganz, T.; Selsted, M.E. Interaction of human defensins with escherichia coli. Mechanism of bactericidal activity. J. Clin. Investig. 1989, 84, 553–561. [Google Scholar] [CrossRef]
- Schroeder, B.O.; Wu, Z.; Nuding, S.; Groscurth, S.; Marcinowski, M.; Beisner, J.; Buchner, J.; Schaller, M.; Stange, E.F.; Wehkamp, J. Reduction of disulphide bonds unmasks potent antimicrobial activity of human beta-defensin 1. Nature 2011, 469, 419–423. [Google Scholar] [CrossRef]
- Hendrix, R.W.; Duda, R.L. Bacteriophage lambda papa: Not the mother of all lambda phages. Science 1992, 258, 1145–1148. [Google Scholar] [CrossRef]
- Lemon, D.J.; Kay, M.K.; Titus, J.K.; Ford, A.A.; Chen, W.; Hamlin, N.J.; Hwang, Y.Y. Construction of a genetically modified T7 select phage system to express the antimicrobial peptide 1018. J. Microbiol. 2019, 57, 532–538. [Google Scholar] [CrossRef]
- Gordon, Y.J.; Romanowski, E.G.; McDermott, A.M. A review of antimicrobial peptides and their therapeutic potential as anti-infective drugs. Curr. Eye Res. 2005, 30, 505–515. [Google Scholar] [CrossRef]
- Knappe, D.; Henklein, P.; Hoffmann, R.; Hilpert, K. Easy strategy to protect antimicrobial peptides from fast degradation in serum. Antimicrob. Agents Chemother. 2010, 54, 4003–4005. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| D-Fusion Name (Plasmid Number) | |
|---|---|
| COOH-terminal additions: a | NH2-terminal additions: a |
| His-TAGZ-Dcoe-TEV-LL37 (p619) b | LL37-TEV-Dcoe-His (p620) |
| Dcoe-LL37 (p627) c | LL37-Dcoe (p617) c |
| Dcoe-PR39 (p625) c | PR39-Dcoe (p623) c |
| Dcoe-DEFB126ΔC (p618) d | DEFB126ΔC-Dcoe (p622) d |
| Dcoe-HBD3 (p616) | HBD3-Dcoe (p624) |
| Dcoe-HD5 (p628) | HD5-Dcoe (p621) |
| Dcoe-HNP1 (p615) [see text] | HNP1-Dcoe (p626) [see text] |
| Dcoe-YML (p674) | YML-Dcoe (p676) |
| D-YML (p675) | His-Dcoe (p614) |
| Dcoe-5EV2 (p521,p629) e | His-Dcoe- (p614*) f |
| Plasmid In E. coli, Encoded D or D-Fusion Protein | Non-Induces (0 Time) | Non-Induced, 140 min Cell Growth at 30 °C | Induced, 140 min Cell Growth after Shifting Culture to 42 °C |
|---|---|---|---|
| p613, gpDcoe | 0.10 | 0.44 | 0.79 |
| p627, gpD-PR39 | 0.14 | 0.51 | 0.70 |
| p625, gpD-LL37 | 0.15 | 0.71 | 0.70 |
| Strain 594[pcIpR-D-fusion-timm] Plasmid Construct a | Relative Culture Viability at 42 °C (D-fusion Toxicity) b | EOP c of λimm434 Dam123 at 42 °C on Assay Strain d |
|---|---|---|
| p613: Dcoe | 0.65 | 0.56 |
| p613*: Dcoe- (67 bp deletion within D) e | 0.79 | <0.0001 |
| p614: His-Dcoe (11AA N-terminal) | 0.09 | <0.0001 f |
| p614*: His- Dcoe-:*(Pro48Glu mutation in D) | 0.87 | <0.0001 |
| p615: Dcoe-HNP1 (35AA C-term.) | 0.83 | <0.0001 f |
| p626: HNP1- Dcoe (36AA N-term.) | 1.00 | <0.0001 f |
| p616: Dcoe-HBD3 (50AA C-term.) | <0.0001 | <0.0005 |
| p624: HBD3- Dcoe (53AA N-term.) | 0.78 | <0.0001 f |
| p627: Dcoe-LL37 (45AA C-term.) | 0.0002 | g |
| p617: LL37-Dcoe (46AA N-term.) | 0.93 | <0.0001 f |
| p618: Dcoe-DEFB126-Δ (84AA C-term.) | 0.0001 | g |
| p622: DEFB126-ΔC- Dcoe (85AA N-term.) | 0.85 | <0.0001 f |
| p619: His-TAGZ-Dcoe-TEV-LL37 (11AA N-, 55AA C-term.) | 0.0003 | <0.000 1f |
| p620: LL37-TEV- Dcoe-His (50AA N-, 12AA C-term.) | 0.87 | <0.0001 f |
| p628: Dcoe-HD5 (40AA C-term.) | 0.0034 | <0.0001 f |
| p621: HD5- Dcoe (41AA N-term.) | 0.77 | <0.0001 f |
| p625: Dcoe-PR39 (48AA C-term.) | <0.0001 | g |
| p623: PR39-Dcoe (49AA N-term.) | 0.97 | <0.0001 f |
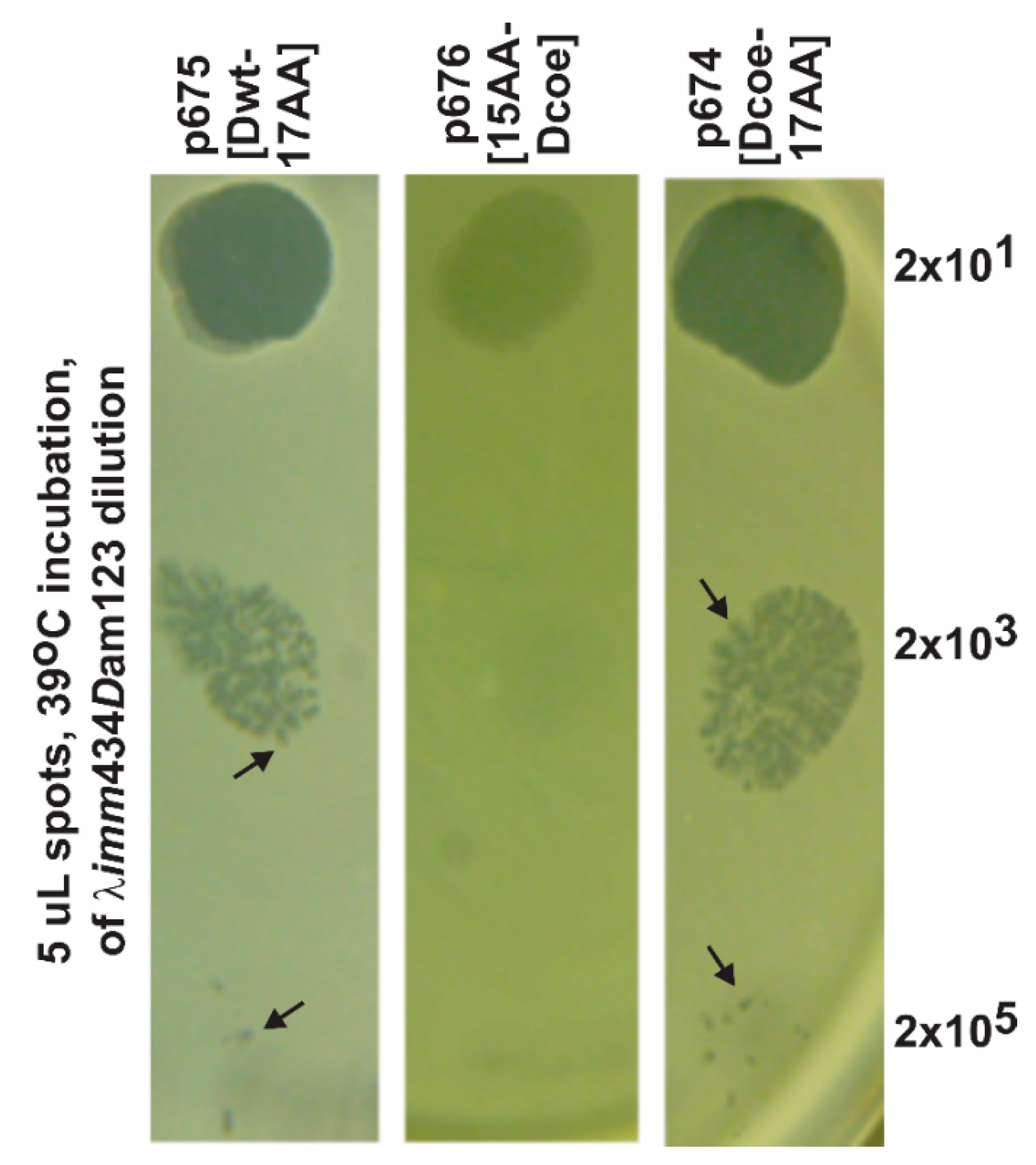
| p674: Dcoe-YML (17AA C-term.) | 0.05 | ≥0.90 h |
| p675: D(wild type)-YML (17AA C-term.) | 0.88 | 0.93 |
| p676: YML-Dcoe (15AA N-term.) | 1.00 | 0.06 |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hayes, S. Bacterial Virus Lambda Gpd-Fusions to Cathelicidins, α- and β-Defensins, and Disease-Specific Epitopes Evaluated for Antimicrobial Toxicity and Ability to Support Phage Display. Viruses 2019, 11, 869. https://doi.org/10.3390/v11090869
Hayes S. Bacterial Virus Lambda Gpd-Fusions to Cathelicidins, α- and β-Defensins, and Disease-Specific Epitopes Evaluated for Antimicrobial Toxicity and Ability to Support Phage Display. Viruses. 2019; 11(9):869. https://doi.org/10.3390/v11090869
Chicago/Turabian StyleHayes, Sidney. 2019. "Bacterial Virus Lambda Gpd-Fusions to Cathelicidins, α- and β-Defensins, and Disease-Specific Epitopes Evaluated for Antimicrobial Toxicity and Ability to Support Phage Display" Viruses 11, no. 9: 869. https://doi.org/10.3390/v11090869
APA StyleHayes, S. (2019). Bacterial Virus Lambda Gpd-Fusions to Cathelicidins, α- and β-Defensins, and Disease-Specific Epitopes Evaluated for Antimicrobial Toxicity and Ability to Support Phage Display. Viruses, 11(9), 869. https://doi.org/10.3390/v11090869