An Update on African Swine Fever Virology

 ,
,  , , , and
, , , and {kind=link}
{kind=link}
Abstract
1. Introduction
2. Proteomics on ASFV
3. Cell Entry of ASFV
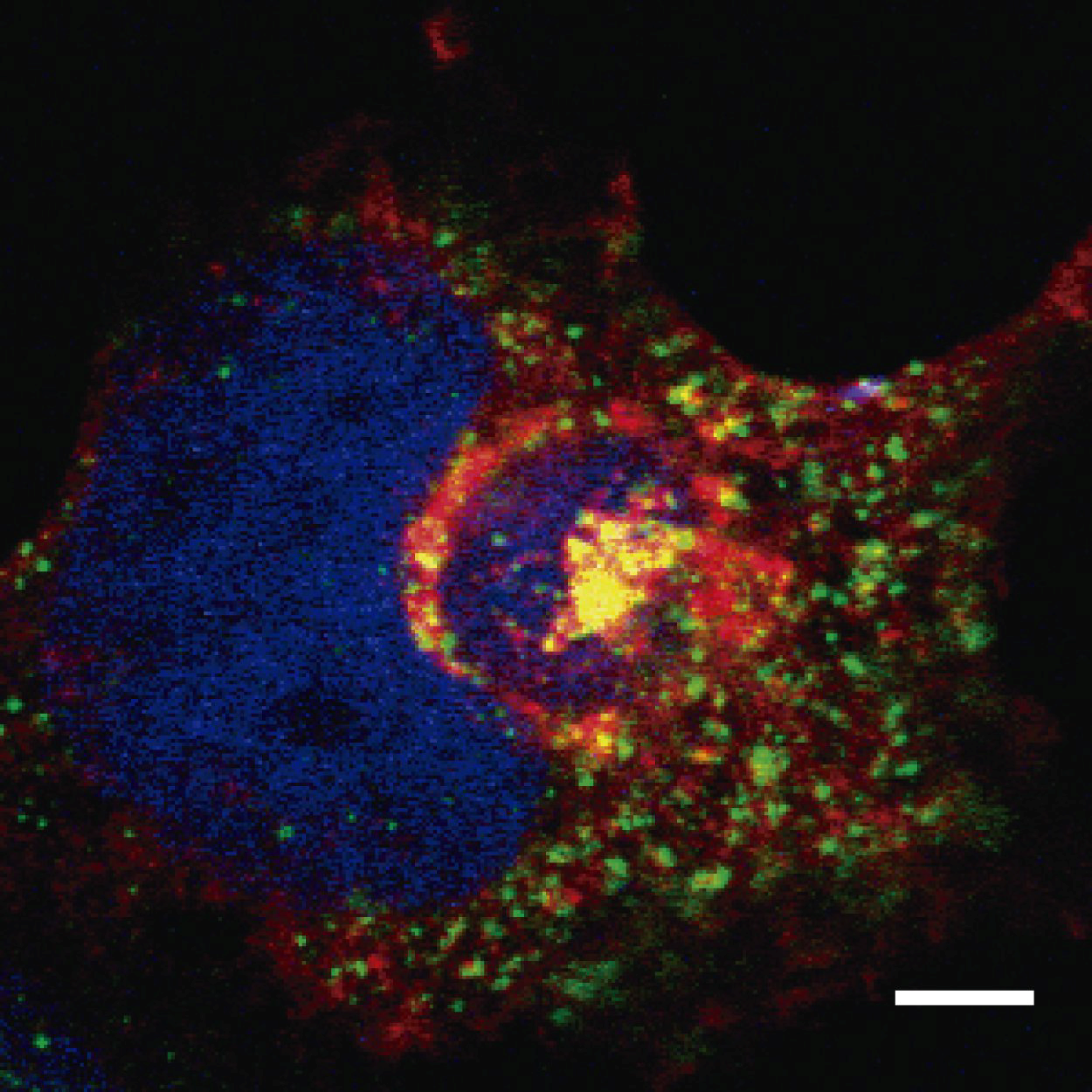
4. Endosomal Traffic of ASFV
5. CD2v Interaction with Adaptor Protein 1 (AP1)
6. ASFV Genes Involved in Cycle Progression and Viral-Host Interactions
7. I215L—E2 Ubiquitin-Conjugating Enzyme
8. A104R—Histone-Like Protein
9. QP509L and Q706L RNA Helicases
10. P1192R—Topoisomerase II
11. Summary
Author Contributions
Funding
Conflicts of Interest
References
- VanderWaal, K.; Deen, J. Global trends in infectious diseases of swine. Proc. Natl. Acad. Sci. USA 2018, 115, 11495–11500. [Google Scholar] [CrossRef] [PubMed]
- Mulumba-Mfumu, L.K.; Saegerman, C.; Dixon, L.K.; Madimba, K.C.; Kazadi, E.; Mukalakata, N.T.; Oura, C.A.L.; Chenais, E.; Masembe, C.; Stahl, K.; et al. African swine fever: Update on Eastern, Central and Southern Africa. Transbound. Emerg. Dis. 2019, 10, 1111. [Google Scholar] [CrossRef] [PubMed]
- Cwynar, P.; Stojkov, J.; Wlazlak, K. African swine fever status in Europe. Viruses 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.; Li, J.; Fan, X.; Liu, F.; Li, L.; Wang, Q.; Ren, W.; Bao, J.; Liu, C.; Wang, H.; et al. Molecular characterization of African swine fever virus, China, 2018. Emerg. Infect. Dis. 2018, 24, 2131–2133. [Google Scholar] [CrossRef] [PubMed]
- Alonso, C.; Borca, M.; Dixon, L.; Revilla, Y.; Rodriguez, F.; Escribano, J.M.; Ictv Report, C. ICTV virus taxonomy profile Asfarviridae. J. Gen. Virol. 2018, 99, 613–614. [Google Scholar] [CrossRef] [PubMed]
- Dixon, L.K.; Sun, H.; Roberts, H. African swine fever. Antiviral. Res. 2019, 165, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Dixon, L.K.; Islam, M.; Nash, R.; Reis, A.L. African swine fever virus evasion of host defences. Virus Res. 2019, 266, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Revilla, Y.; Perez-Nunez, D.; Richt, J.A. African swine fever virus biology and vaccine approaches. Adv. Virus Res. 2018, 100, 41–74. [Google Scholar] [CrossRef] [PubMed]
- Alcaraz, C.; Brun, A.; Ruiz-Gonzalvo, F.; Escribano, J.M. Cell culture propagation modifies the African swine fever virus replication phenotype in macrophages and generates viral subpopulations differing in protein p54. Virus Res. 1992, 23, 173–182. [Google Scholar] [CrossRef]
- Alfonso, P.; Rivera, J.; Hernaez, B.; Alonso, C.; Escribano, J.M. Identification of cellular proteins modified in response to African swine fever virus infection by proteomics. Proteomics 2004, 4, 2037–2046. [Google Scholar] [CrossRef] [PubMed]
- Esteves, A.; Marques, M.I.; Costa, J.V. Two-dimensional analysis of African swine fever virus proteins and proteins induced in infected cells. Virology 1986, 152, 192–206. [Google Scholar] [CrossRef]
- Herrera-Uribe, J.; Jimenez-Marin, A.; Lacasta, A.; Monteagudo, P.L.; Pina-Pedrero, S.; Rodriguez, F.; Moreno, A.; Garrido, J.J. Comparative proteomic analysis reveals different responses in porcine lymph nodes to virulent and attenuated homologous African swine fever virus strains. Vet. Res. 2018, 49, 90. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.M.; Salas, M.L.; Santaren, J.F. African swine fever virus-induced polypeptides in porcine alveolar macrophages and in Vero cells: Two-dimensional gel analysis. Proteomics 2001, 1, 1447–1456. [Google Scholar] [CrossRef]
- Alejo, A.; Matamoros, T.; Guerra, M.; Andres, G. A proteomic atlas of the African swine fever Virus particle. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Kessler, C.; Forth, J.H.; Keil, G.M.; Mettenleiter, T.C.; Blome, S.; Karger, A. The intracellular proteome of African swine fever virus. Sci. Rep. 2018, 8, 14714. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, E.G.; Perez-Nunez, D.; Revilla, Y. Mechanisms of entry and endosomal pathway of African swine fever virus. Vaccines 2017, 5, 42. [Google Scholar] [CrossRef] [PubMed]
- Alcami, A.; Carrascosa, A.L.; Vinuela, E. The entry of African swine fever virus into Vero cells. Virology 1989, 171, 68–75. [Google Scholar] [CrossRef]
- Valdeira, M.L.; Bernardes, C.; Cruz, B.; Geraldes, A. Entry of African swine fever virus into Vero cells and uncoating. Vet. Microbiol. 1998, 60, 131–140. [Google Scholar] [CrossRef]
- Galindo, I.; Cuesta-Geijo, M.A.; Hlavova, K.; Munoz-Moreno, R.; Barrado-Gil, L.; Dominguez, J.; Alonso, C. African swine fever virus infects macrophages, the natural host cells, via clathrin and cholesterol-dependent endocytosis. Virus Res. 2015, 200, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Hernaez, B.; Alonso, C. Dynamin- and clathrin-dependent endocytosis in African swine fever virus entry. J. Virol. 2010, 84, 2100–2109. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, E.G.; Quintas, A.; Perez-Nunez, D.; Nogal, M.; Barroso, S.; Carrascosa, A.L.; Revilla, Y. African swine fever virus uses macropinocytosis to enter host cells. PLoS Pathog. 2012, 8, e1002754. [Google Scholar] [CrossRef] [PubMed]
- Netherton, C.L.; Wileman, T.E. African swine fever virus organelle rearrangements. Virus Res. 2013, 173, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Hernaez, B.; Guerra, M.; Salas, M.L.; Andres, G. African swine fever virus undergoes outer envelope disruption, capsid disassembly and inner envelope fusion before core release from multivesicular endosomes. PLoS Pathog. 2016, 12, e1005595. [Google Scholar] [CrossRef] [PubMed]
- Cuesta-Geijo, M.A.; Galindo, I.; Hernaez, B.; Quetglas, J.I.; Dalmau-Mena, I.; Alonso, C. Endosomal maturation, Rab7 GTPase and phosphoinositides in African swine fever virus entry. PLoS ONE 2012, 7, e48853. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.J.; Lampe, M.; Merrifield, C.J. A feedback loop between dynamin and actin recruitment during clathrin-mediated endocytosis. PLoS Biol. 2012, 10, e1001302. [Google Scholar] [CrossRef] [PubMed]
- Alonso, C.; Galindo, I.; Cuesta-Geijo, M.A.; Cabezas, M.; Hernaez, B.; Munoz-Moreno, R. African swine fever virus-cell interactions: From virus entry to cell survival. Virus Res. 2013, 173, 42–57. [Google Scholar] [CrossRef] [PubMed]
- Quetglas, J.I.; Hernaez, B.; Galindo, I.; Munoz-Moreno, R.; Cuesta-Geijo, M.A.; Alonso, C. Small RHO GTPases and cholesterol biosynthetic pathway intermediates in African swine fever virus infection. J. Virol. 2012, 86, 1758–1767. [Google Scholar] [CrossRef] [PubMed]
- Bernardes, C.; Antonio, A.; Pedroso de Lima, M.C.; Valdeira, M.L. Cholesterol affects African swine fever virus infection. Biochimica Biophysica Acta 1998, 1393, 19–25. [Google Scholar] [CrossRef]
- Andres, G. African swine fever virus gets undressed: New insights on the entry pathway. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Taylor, M.J.; Perrais, D.; Merrifield, C.J. A high precision survey of the molecular dynamics of mammalian clathrin-mediated endocytosis. PLoS Biol. 2011, 9, e1000604. [Google Scholar] [CrossRef]
- Cuesta-Geijo, M.A.; Chiappi, M.; Galindo, I.; Barrado-Gil, L.; Munoz-Moreno, R.; Carrascosa, J.L.; Alonso, C. Cholesterol flux is required for endosomal progression of African swine fever virions during the initial establishment of infection. J. Virol. 2016, 90, 1534–1543. [Google Scholar] [CrossRef] [PubMed]
- Barrado-Gil, L.; Galindo, I.; Martinez-Alonso, D.; Viedma, S.; Alonso, C. The ubiquitin-proteasome system is required for African swine fever replication. PLoS ONE 2017, 12, e0189741. [Google Scholar] [CrossRef] [PubMed]
- Cuesta-Geijo, M.A.; Barrado-Gil, L.; Galindo, I.; Munoz-Moreno, R.; Alonso, C. Redistribution of endosomal membranes to the African swine fever virus replication site. Viruses 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Galindo, I.; Cuesta-Geijo, M.A.; Del Puerto, A.; Soriano, E.; Alonso, C. Lipid exchange factors at membrane contact sites in African swine fever virus infection. Viruses 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Raychaudhuri, S.; Prinz, W.A. The diverse functions of oxysterol-binding proteins. Annu. Rev. Cell Dev. Biol. 2010, 26, 157–177. [Google Scholar] [CrossRef]
- Weber-Boyvat, M.; Zhong, W.; Yan, D.; Olkkonen, V.M. Oxysterol-binding proteins: Functions in cell regulation beyond lipid metabolism. Biochem. Pharmacol. 2013, 86, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Strating, J.R.; van der Linden, L.; Albulescu, L.; Bigay, J.; Arita, M.; Delang, L.; Leyssen, P.; van der Schaar, H.M.; Lanke, K.H.; Thibaut, H.J.; et al. Itraconazole inhibits enterovirus replication by targeting the oxysterol-binding protein. Cell Rep. 2015, 10, 600–615. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.M.; Yanez, R.J.; Almazan, F.; Vinuela, E.; Rodriguez, J.F. African swine fever virus encodes a CD2 homolog responsible for the adhesion of erythrocytes to infected cells. J. Virol. 1993, 67, 5312–5320. [Google Scholar]
- Borca, M.V.; Carrillo, C.; Zsak, L.; Laegreid, W.W.; Kutish, G.F.; Neilan, J.G.; Burrage, T.G.; Rock, D.L. Deletion of a CD2-like gene, 8-DR, from African swine fever virus affects viral infection in domestic swine. J. Virol. 1998, 72, 2881–2889. [Google Scholar]
- Chapman, D.A.; Tcherepanov, V.; Upton, C.; Dixon, L.K. Comparison of the genome sequences of non-pathogenic and pathogenic African swine fever virus isolates. J. Gen. Virol. 2008, 89, 397–408. [Google Scholar] [CrossRef]
- Rowlands, R.J.; Duarte, M.M.; Boinas, F.; Hutchings, G.; Dixon, L.K. The CD2v protein enhances African swine fever virus replication in the tick vector, Ornithodoros erraticus. Virology 2009, 393, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Goatley, L.C.; Dixon, L.K. Processing and localization of the african swine fever virus CD2v transmembrane protein. J. Virol. 2011, 85, 3294–3305. [Google Scholar] [CrossRef] [PubMed]
- Kay-Jackson, P.C.; Goatley, L.C.; Cox, L.; Miskin, J.E.; Parkhouse, R.M.; Wienands, J.; Dixon, L.K. The CD2v protein of African swine fever virus interacts with the actin-binding adaptor protein SH3P7. J. Gen. Virol. 2004, 85, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Nakatsu, F.; Ohno, H. Adaptor protein complexes as the key regulators of protein sorting in the post-Golgi network. Cell Struct. Funct. 2003, 28, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Canagarajah, B.J.; Ren, X.; Bonifacino, J.S.; Hurley, J.H. The clathrin adaptor complexes as a paradigm for membrane-associated allostery. Protein Sci. 2013, 22, 517–529. [Google Scholar] [CrossRef] [PubMed]
- Stamnes, M.A.; Rothman, J.E. The binding of AP-1 clathrin adaptor particles to Golgi membranes requires ADP-ribosylation factor, a small GTP-binding protein. Cell 1993, 73, 999–1005. [Google Scholar] [CrossRef]
- Donaldson, J.G.; Honda, A. Localization and function of Arf family GTPases. Biochem. Soc. Trans. 2005, 33, 639–642. [Google Scholar] [CrossRef]
- Robinson, M.S.; Kreis, T.E. Recruitment of coat proteins onto Golgi membranes in intact and permeabilized cells: Effects of brefeldin A and G protein activators. Cell 1992, 69, 129–138. [Google Scholar] [CrossRef]
- Bresnahan, P.A.; Yonemoto, W.; Ferrell, S.; Williams-Herman, D.; Geleziunas, R.; Greene, W.C. A dileucine motif in HIV-1 Nef acts as an internalization signal for CD4 downregulation and binds the AP-1 clathrin adaptor. Curr. Biol. 1998, 8, 1235–1238. [Google Scholar] [CrossRef]
- Laguette, N.; Bregnard, C.; Benichou, S.; Basmaciogullari, S. Human immunodeficiency virus (HIV) type-1, HIV-2 and simian immunodeficiency virus Nef proteins. Mol. Aspects Med. 2010, 31, 418–433. [Google Scholar] [CrossRef]
- Janvier, K.; Craig, H.; Hitchin, D.; Madrid, R.; Sol-Foulon, N.; Renault, L.; Cherfils, J.; Cassel, D.; Benichou, S.; Guatelli, J. HIV-1 Nef stabilizes the association of adaptor protein complexes with membranes. J. Biol. Chem. 2003, 278, 8725–8732. [Google Scholar] [CrossRef]
- Madrid, R.; Janvier, K.; Hitchin, D.; Day, J.; Coleman, S.; Noviello, C.; Bouchet, J.; Benmerah, A.; Guatelli, J.; Benichou, S. Nef-induced alteration of the early/recycling endosomal compartment correlates with enhancement of HIV-1 infectivity. J. Biol. Chem. 2005, 280, 5032–5044. [Google Scholar] [CrossRef]
- Tong, X.; Boll, W.; Kirchhausen, T.; Howley, P.M. Interaction of the bovine papillomavirus E6 protein with the clathrin adaptor complex AP-1. J. Virol. 1998, 72, 476–482. [Google Scholar]
- Johnson, D.C.; Webb, M.; Wisner, T.W.; Brunetti, C. Herpes simplex virus gE/gI sorts nascent virions to epithelial cell junctions, promoting virus spread. J. Virol. 2001, 75, 821–833. [Google Scholar] [CrossRef]
- Mori, Y.; Koike, M.; Moriishi, E.; Kawabata, A.; Tang, H.; Oyaizu, H.; Uchiyama, Y.; Yamanishi, K. Human herpesvirus-6 induces MVB formation, and virus egress occurs by an exosomal release pathway. Traffic 2008, 9, 1728–1742. [Google Scholar] [CrossRef]
- Wonderlich, E.R.; Williams, M.; Collins, K.L. The tyrosine binding pocket in the adaptor protein 1 (AP-1) mu1 subunit is necessary for Nef to recruit AP-1 to the major histocompatibility complex class I cytoplasmic tail. J. Biol. Chem. 2008, 283, 3011–3022. [Google Scholar] [CrossRef]
- Perez-Nunez, D.; Garcia-Urdiales, E.; Martinez-Bonet, M.; Nogal, M.L.; Barroso, S.; Revilla, Y.; Madrid, R. CD2v Interacts with adaptor protein AP-1 during African swine fever infection. PLoS ONE 2015, 10, e0123714. [Google Scholar] [CrossRef]
- Dinkel, H.; Van Roey, K.; Michael, S.; Davey, N.E.; Weatheritt, R.J.; Born, D.; Speck, T.; Kruger, D.; Grebnev, G.; Kuban, M.; et al. The eukaryotic linear motif resource ELM: 10 years and counting. Nucleic Acids Res. 2014, 42, D259–D266. [Google Scholar] [CrossRef]
- Netherton, C.L.; Connell, S.; Benfield, C.T.O.; Dixon, L.K. The genetics of life and death: Virus-host interactions underpinning resistance to African swine fever, a viral hemorrhagic disease. Front. Genet. 2019, 10, 402. [Google Scholar] [CrossRef]
- Hingamp, P.M.; Arnold, J.E.; Mayer, R.J.; Dixon, L.K. A ubiquitin conjugating enzyme encoded by African swine fever virus. EMBO J. 1992, 11, 361–366. [Google Scholar] [CrossRef]
- Hingamp, P.M.; Leyland, M.L.; Webb, J.; Twigger, S.; Mayer, R.J.; Dixon, L.K. Characterization of a ubiquitinated protein which is externally located in African swine fever virions. J. Virol. 1995, 69, 1785–1793. [Google Scholar]
- Gonzalez-Santamaria, J.; Campagna, M.; Garcia, M.A.; Marcos-Villar, L.; Gonzalez, D.; Gallego, P.; Lopitz-Otsoa, F.; Guerra, S.; Rodriguez, M.S.; Esteban, M.; et al. Regulation of vaccinia virus E3 protein by small ubiquitin-like modifier proteins. J. Virol. 2011, 85, 12890–12900. [Google Scholar] [CrossRef]
- Gustin, J.K.; Moses, A.V.; Fruh, K.; Douglas, J.L. Viral takeover of the host ubiquitin system. Front. Microbiol. 2011, 2, 161. [Google Scholar] [CrossRef]
- Isaacson, M.K.; Ploegh, H.L. Ubiquitination, ubiquitin-like modifiers, and deubiquitination in viral infection. Cell Host Microbe 2009, 5, 559–570. [Google Scholar] [CrossRef]
- Randow, F.; Lehner, P.J. Viral avoidance and exploitation of the ubiquitin system. Nat. Cell Biol. 2009, 11, 527–534. [Google Scholar] [CrossRef]
- Freitas, F.B.; Frouco, G.; Martins, C.; Ferreira, F. African swine fever virus encodes for an E2-ubiquitin conjugating enzyme that is mono- and di-ubiquitinated and required for viral replication cycle. Sci. Rep. 2018, 8, 3471. [Google Scholar] [CrossRef]
- Sojka, D.; Franta, Z.; Horn, M.; Caffrey, C.R.; Mares, M.; Kopacek, P. New insights into the machinery of blood digestion by ticks. Trends Parasitol. 2013, 29, 276–285. [Google Scholar] [CrossRef]
- Fukuyo, Y.; Horikoshi, N.; Ishov, A.M.; Silverstein, S.J.; Nakajima, T. The herpes simplex virus immediate-early ubiquitin ligase ICP0 induces degradation of the ICP0 repressor protein E2FBP1. J. Virol. 2011, 85, 3356–3366. [Google Scholar] [CrossRef]
- Borca, M.V.; Irusta, P.M.; Kutish, G.F.; Carillo, C.; Afonso, C.L.; Burrage, A.T.; Neilan, J.G.; Rock, D.L. A structural DNA binding protein of African swine fever virus with similarity to bacterial histone-like proteins. Arch. Virol. 1996, 141, 301–313. [Google Scholar] [CrossRef]
- Neilan, J.G.; Lu, Z.; Kutish, G.F.; Sussman, M.D.; Roberts, P.C.; Yozawa, T.; Rock, D.L. An African swine fever virus gene with similarity to bacterial DNA binding proteins, bacterial integration host factors, and the bacillus phage SPO1 transcription factor, TF1. Nucleic Acids Res. 1993, 21, 1496. [Google Scholar] [CrossRef][Green Version]
- Frouco, G.; Freitas, F.B.; Coelho, J.; Leitao, A.; Martins, C.; Ferreira, F. DNA-binding properties of African swine fever virus pA104R, a histone-like protein involved in viral replication and transcription. J. Virol. 2017, 91, e02498-16. [Google Scholar] [CrossRef] [PubMed]
- Loregian, A.; Sinigalia, E.; Mercorelli, B.; Palu, G.; Coen, D.M. Binding parameters and thermodynamics of the interaction of the human cytomegalovirus DNA polymerase accessory protein, UL44, with DNA: Implications for the processivity mechanism. Nucleic Acids Res. 2007, 35, 4779–4791. [Google Scholar] [CrossRef] [PubMed]
- Rochester, S.C.; Traktman, P. Characterization of the single-stranded DNA binding protein encoded by the vaccinia virus I3 gene. J. Virol. 1998, 72, 2917–2926. [Google Scholar] [PubMed]
- Coelho, J.; Ferreira, F.; Martins, C.; Leitao, A. Functional characterization and inhibition of the type II DNA topoisomerase coded by African swine fever virus. Virology 2016, 493, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Freitas, F.B.; Frouco, G.; Martins, C.; Leitao, A.; Ferreira, F. In vitro inhibition of African swine fever virus-topoisomerase II disrupts viral replication. Antiviral. Res. 2016, 134, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Bensaid, A.; Almeida, A.; Drlica, K.; Rouviere-Yaniv, J. Cross-talk between topoisomerase I and HU in Escherichia coli. J. Mol. Biol. 1996, 256, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Mallick, B.; Nagaraja, V. Direct regulation of topoisomerase activity by a nucleoid-associated protein. Nucleic Acids Res. 2014, 42, 11156–11165. [Google Scholar] [CrossRef] [PubMed]
- Bogner, E.; Radsak, K.; Stinski, M.F. The gene product of human cytomegalovirus open reading frame UL56 binds the pac motif and has specific nuclease activity. J. Virol. 1998, 72, 2259–2264. [Google Scholar] [PubMed]
- Thoma, C.; Borst, E.; Messerle, M.; Rieger, M.; Hwang, J.S.; Bogner, E. Identification of the interaction domain of the small terminase subunit pUL89 with the large subunit pUL56 of human cytomegalovirus. Biochemistry 2006, 45, 8855–8863. [Google Scholar] [CrossRef]
- Salas, M.L.; Andres, G. African swine fever virus morphogenesis. Virus Res. 2013, 173, 29–41. [Google Scholar] [CrossRef]
- Simoes, M.; Rino, J.; Pinheiro, I.; Martins, C.; Ferreira, F. Alterations of nuclear architecture and epigenetic signatures during African swine fever virus infection. Viruses 2015, 7, 4978–4996. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.M.; Salas, M.L. African swine fever virus transcription. Virus Res. 2013, 173, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Freije, J.M.; Lain, S.; Vinuela, E.; Lopez-Otin, C. Nucleotide sequence of a nucleoside triphosphate phosphohydrolase gene from African swine fever virus. Virus Res. 1993, 30, 63–72. [Google Scholar] [CrossRef]
- Linder, P.; Jankowsky, E. From unwinding to clamping—The DEAD box RNA helicase family. Nat. Rev. Mol. Cell Biol. 2011, 12, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Frick, D.N.; Lam, A.M. Understanding helicases as a means of virus control. Curr. Pharm. Des. 2006, 12, 1315–1338. [Google Scholar] [CrossRef] [PubMed]
- Ranji, A.; Boris-Lawrie, K. RNA helicases: Emerging roles in viral replication and the host innate response. RNA Biol. 2010, 7, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Freitas, F.B.; Frouco, G.; Martins, C.; Ferreira, F. The QP509L and Q706L superfamily II RNA helicases of African swine fever virus are required for viral replication, having non-redundant activities. Emerg. Microbes Infect. 2019, 8, 291–302. [Google Scholar] [CrossRef]
- Baylis, S.A.; Twigg, S.R.; Vydelingum, S.; Dixon, L.K.; Smith, G.L. Three African swine fever virus genes encoding proteins with homology to putative helicases of vaccinia virus. J. Gen. Virol. 1993, 74, 1969–1974. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S.; Shackelton, L.A.; Holmes, E.C. Rates of evolutionary change in viruses: Patterns and determinants. Nat. Rev. Genet. 2008, 9, 267–276. [Google Scholar] [CrossRef]
- Roberts, P.C.; Lu, Z.; Kutish, G.F.; Rock, D.L. Three adjacent genes of African swine fever virus with similarity to essential poxvirus genes. Arch. Virol. 1993, 132, 331–342. [Google Scholar] [CrossRef]
- Yanez, R.J.; Rodriguez, J.M.; Boursnell, M.; Rodriguez, J.F.; Vinuela, E. Two putative African swine fever virus helicases similar to yeast ‘DEAH’ pre-mRNA processing proteins and vaccinia virus ATPases D11L and D6R. Gene 1993, 134, 161–174. [Google Scholar] [CrossRef]
- Garcia-Beato, R.; Salas, M.L.; Vinuela, E.; Salas, J. Role of the host cell nucleus in the replication of African swine fever virus DNA. Virology 1992, 188, 637–649. [Google Scholar] [CrossRef]
- Dumont, S.; Cheng, W.; Serebrov, V.; Beran, R.K.; Tinoco, I., Jr.; Pyle, A.M.; Bustamante, C. RNA translocation and unwinding mechanism of HCV NS3 helicase and its coordination by ATP. Nature 2006, 439, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Jankowsky, E.; Gross, C.H.; Shuman, S.; Pyle, A.M. The DExH protein NPH-II is a processive and directional motor for unwinding RNA. Nature 2000, 403, 447–451. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Yates, J.; Liang, Y.; Lemon, S.M.; Yi, M. NS3 helicase domains involved in infectious intracellular hepatitis C virus particle assembly. J. Virol. 2008, 82, 7624–7639. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, A.; Guo, H.S.; Saenz, P.; Simon-Buela, L.; Gomez de Cedron, M.; Garcia, J.A. The motif V of plum pox potyvirus CI RNA helicase is involved in NTP hydrolysis and is essential for virus RNA replication. Nucleic Acids Res. 1997, 25, 4474–4480. [Google Scholar] [CrossRef] [PubMed]
- Gross, C.H.; Shuman, S. The nucleoside triphosphatase and helicase activities of vaccinia virus NPH-II are essential for virus replication. J. Virol. 1998, 72, 4729–4736. [Google Scholar]
- Lam, A.M.; Frick, D.N. Hepatitis C virus subgenomic replicon requires an active NS3 RNA helicase. J. Virol. 2006, 80, 404–411. [Google Scholar] [CrossRef]
- Mackintosh, S.G.; Lu, J.Z.; Jordan, J.B.; Harrison, M.K.; Sikora, B.; Sharma, S.D.; Cameron, C.E.; Raney, K.D.; Sakon, J. Structural and biological identification of residues on the surface of NS3 helicase required for optimal replication of the hepatitis C virus. J. Biol. Chem. 2006, 281, 3528–3535. [Google Scholar] [CrossRef]
- Shuman, S. Vaccinia virus RNA helicase: An essential enzyme related to the DE-H family of RNA-dependent NTPases. Proc. Natl. Acad. Sci. USA 1992, 89, 10935–10939. [Google Scholar] [CrossRef]
- Garcia-Beato, R.; Freije, J.M.; Lopez-Otin, C.; Blasco, R.; Vinuela, E.; Salas, M.L. A gene homologous to topoisomerase II in African swine fever virus. Virology 1992, 188, 938–947. [Google Scholar] [CrossRef]
- Champoux, J.J. DNA topoisomerases: Structure, function, and mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. [Google Scholar] [CrossRef]
- De Souza, R.F.; Iyer, L.M.; Aravind, L. Diversity and evolution of chromatin proteins encoded by DNA viruses. Biochimica Biophysica Acta 2010, 1799, 302–318. [Google Scholar] [CrossRef]
- Forterre, P.; Gribaldo, S.; Gadelle, D.; Serre, M.C. Origin and evolution of DNA topoisomerases. Biochimie 2007, 89, 427–446. [Google Scholar] [CrossRef]
- Mottola, C.; Freitas, F.B.; Simoes, M.; Martins, C.; Leitao, A.; Ferreira, F. In vitro antiviral activity of fluoroquinolones against African swine fever virus. Vet. Microbiol. 2013, 165, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Coelho, J.; Martins, C.; Ferreira, F.; Leitao, A. African swine fever virus ORF P1192R codes for a functional type II DNA topoisomerase. Virology 2015, 474, 82–93. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karger, A.; Pérez-Núñez, D.; Urquiza, J.; Hinojar, P.; Alonso, C.; Freitas, F.B.; Revilla, Y.; Le Potier, M.-F.; Montoya, M. An Update on African Swine Fever Virology. Viruses 2019, 11, 864. https://doi.org/10.3390/v11090864
Karger A, Pérez-Núñez D, Urquiza J, Hinojar P, Alonso C, Freitas FB, Revilla Y, Le Potier M-F, Montoya M. An Update on African Swine Fever Virology. Viruses. 2019; 11(9):864. https://doi.org/10.3390/v11090864
Chicago/Turabian StyleKarger, Axel, Daniel Pérez-Núñez, Jesús Urquiza, Patricia Hinojar, Covadonga Alonso, Ferdinando B. Freitas, Yolanda Revilla, Marie-Frédérique Le Potier, and Maria Montoya. 2019. "An Update on African Swine Fever Virology" Viruses 11, no. 9: 864. https://doi.org/10.3390/v11090864
APA StyleKarger, A., Pérez-Núñez, D., Urquiza, J., Hinojar, P., Alonso, C., Freitas, F. B., Revilla, Y., Le Potier, M.-F., & Montoya, M. (2019). An Update on African Swine Fever Virology. Viruses, 11(9), 864. https://doi.org/10.3390/v11090864