Cyprinid herpesvirus 3 Evolves In Vitro through an Assemblage of Haplotypes that Alternatively Become Dominant or Under-Represented

 ,
,  , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. CyHV-3 Propagation Onto CCB Cells and Virus Harvest and Storage
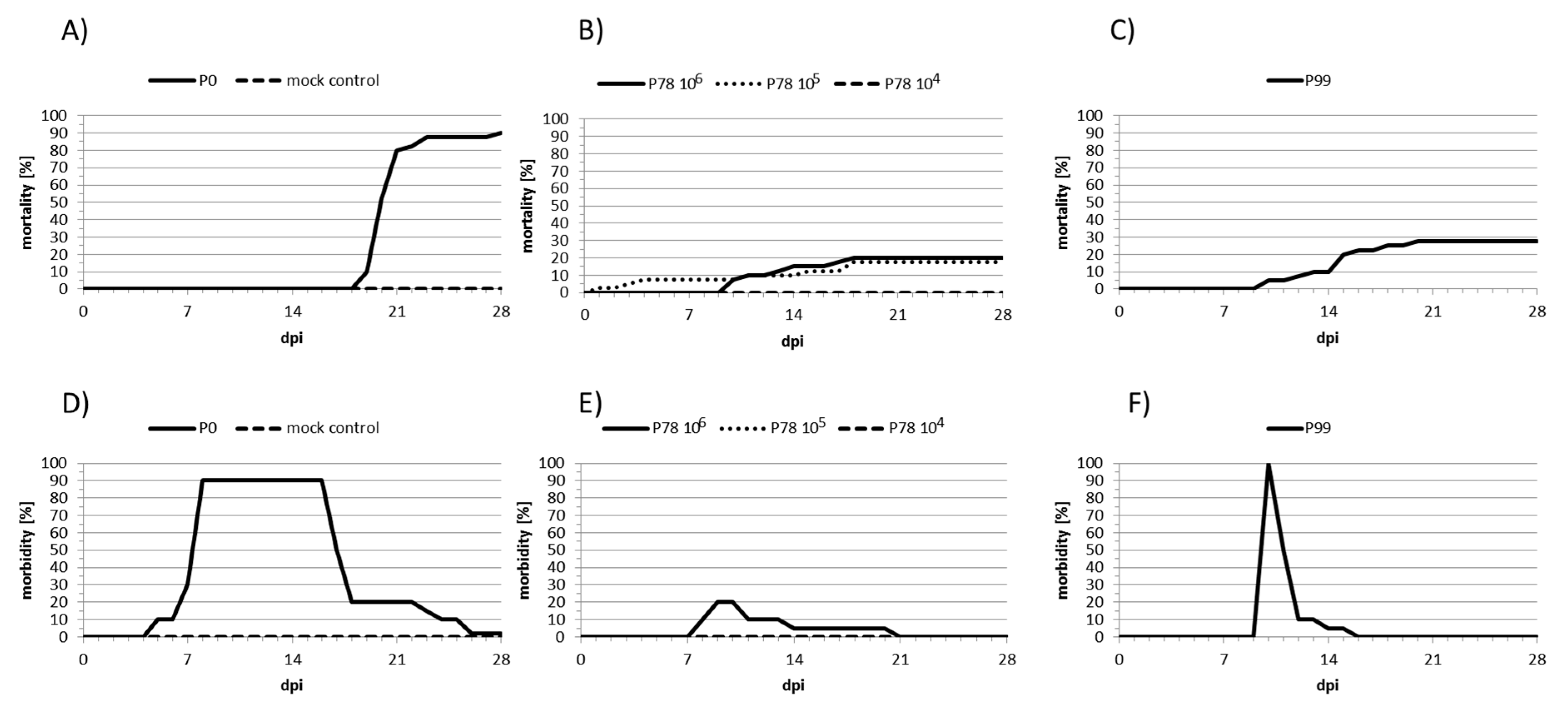
2.2. Experimental Infections of Carp
2.3. Extraction of Viral DNA, Library Preparation, and Genome Sequencing of P78 and P99
2.4. Preparation and Genome Sequencing of KHV-T (P0)
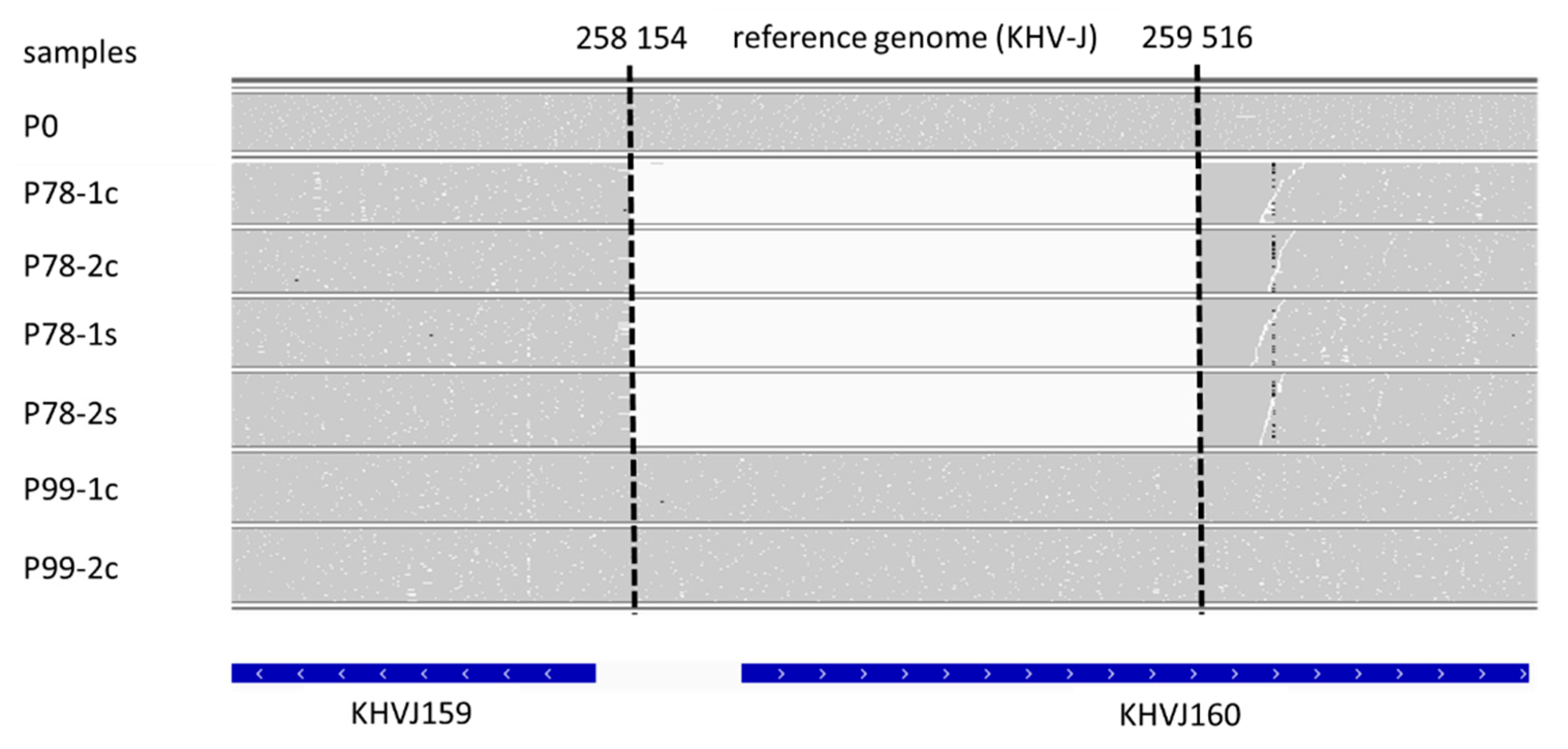
2.5. Genomic Sequence Analysis
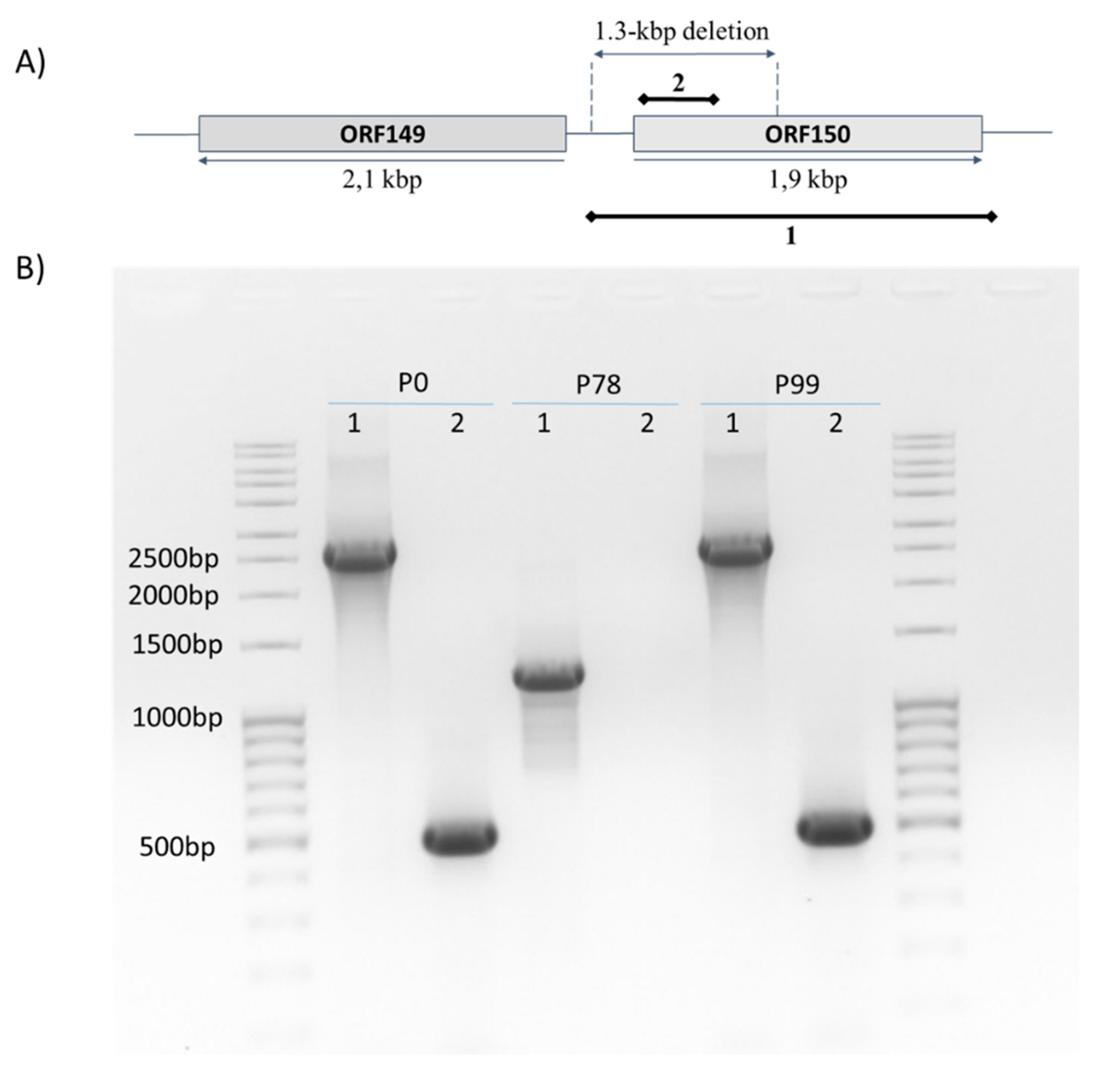
2.6. PCR Assays
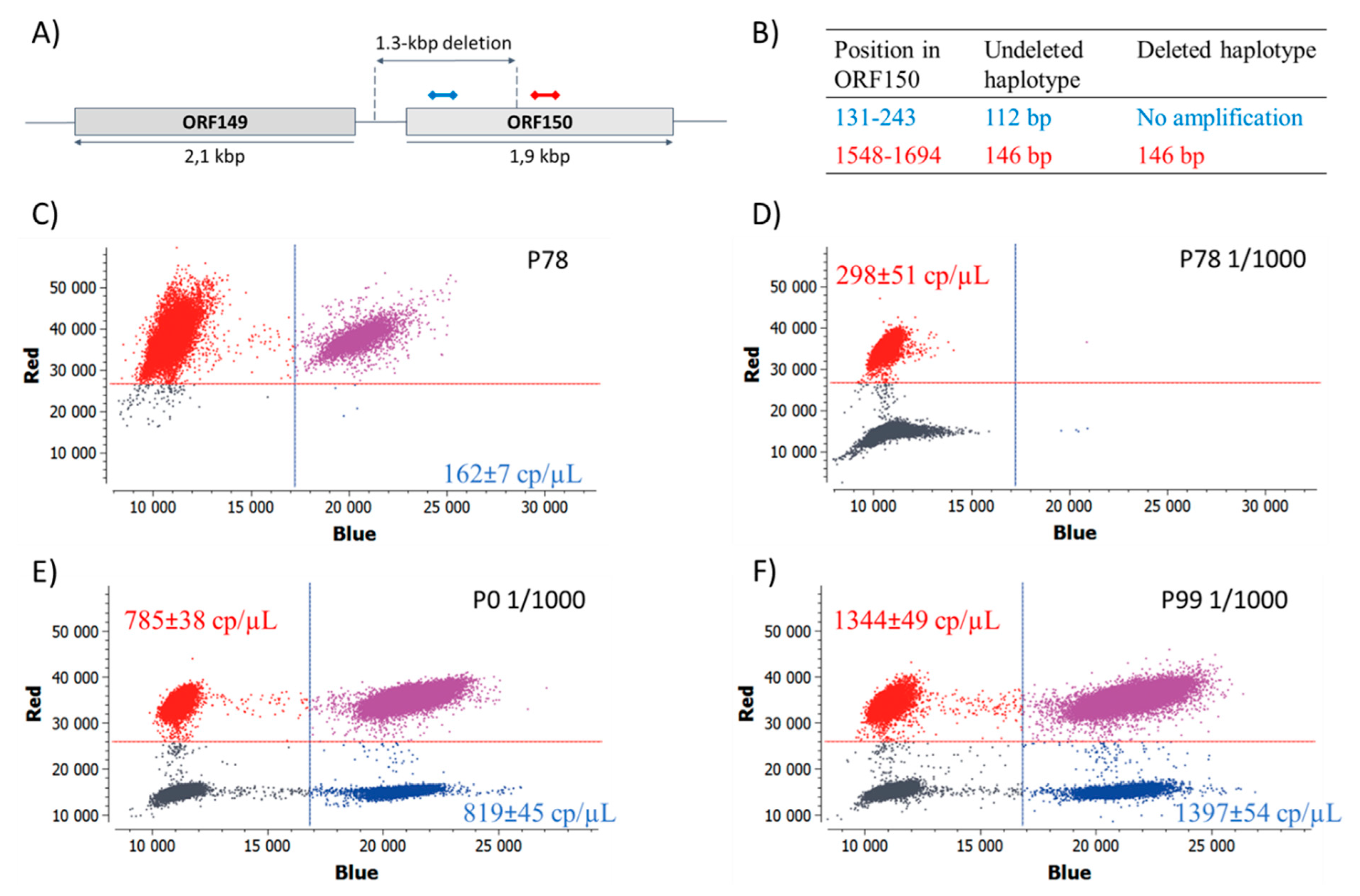
2.7. Digital PCR Assays
3. Results and Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability
References
- Hedrick, R.P.; Gilad, O.; Yun, S.; Spangenberg, J.V.; Marty, G.D.; Nordhausen, R.W.; Kebus, M.J.; Bercovier, H.; Eldar, A. A herpesvirus associated with mass mortality of juvenile and adult koi, a strain of common carp. J. Aquat. Anim. Health 2000, 12, 44–57. [Google Scholar] [CrossRef]
- Pokorova, D.; Vesely, T.; Piackova, V.; Reschova, S.; Hulova, J. Current knowledge on koi herpesvirus (khv): A review. Vet. Med. 2005, 50, 139–147. [Google Scholar] [CrossRef]
- Donohoe, O.H.; Henshilwood, K.; Way, K.; Hakimjavadi, R.; Stone, D.M.; Walls, D. Identification and characterization of cyprinid herpesvirus-3 (cyhv-3) encoded micrornas. PLoS ONE 2015, 10, e0125434. [Google Scholar] [CrossRef]
- Haenen, O.L.M.; Way, K.; Bergmann, S.M.; Ariel, E. The emergence of koi herpesvirus and its significance to european aquaculture. Bull. Eur. Assoc. Fish Pathol. 2004, 24, 293–307. [Google Scholar]
- Waltzek, T.B.; Kelley, G.O.; Alfaro, M.E.; Kurobe, T.; Davison, A.J.; Hedrick, R.P. Phylogenetic relationships in the family alloherpesviridae. Dis. Aquat. Org. 2009, 84, 179–194. [Google Scholar] [CrossRef] [PubMed]
- Bretzinger, A.; Fischer-Scherl, T.; Oumouna, M.; Hoffmann, R.; Truyen, U. Mass mortalities in koi carp, cyprinus carpio, associated with gill and skin disease. Bull. Eur. Assoc. Fish Pathol. 1999, 19, 182–185. [Google Scholar]
- Eide, K.; Miller-Morgan, T.; Heidel, J.; Bildfell, R.; Jin, L. Results of total DNA measurement in koi tissue by koi herpes virus real-time pcr. J. Virol. Methods 2011, 172, 81–84. [Google Scholar] [CrossRef]
- Eide, K.E.; Miller-Morgan, T.; Heidel, J.R.; Kent, M.L.; Bildfell, R.J.; Lapatra, S.; Watson, G.; Jin, L. Investigation of koi herpesvirus latency in koi. J. Virol. 2011, 85, 4954–4962. [Google Scholar] [CrossRef]
- Reed, A.N.; Putman, T.; Sullivan, C.; Jin, L. Application of a nanoflare probe specific to a latency associated transcript for isolation of khv latently infected cells. Virus Res. 2015, 208, 129–135. [Google Scholar] [CrossRef]
- Aoki, T.; Hirono, I.; Kurokawa, K.; Fukuda, H.; Nahary, R.; Eldar, A.; Davison, A.J.; Waltzek, T.B.; Bercovier, H.; Hedrick, R.P. Genome sequences of three koi herpesvirus isolates representing the expanding distribution of an emerging disease threatening koi and common carp worldwide. J. Virol. 2007, 81, 5058–5065. [Google Scholar] [CrossRef]
- Gao, Y.; Suárez, N.M.; Wilkie, G.S.; Dong, C.; Bergmann, S.; Lee, P.-Y.A.; Davison, A.J.; Vanderplasschen, A.F.C.; Boutier, M. Genomic and biologic comparisons of cyprinid herpesvirus 3 strains. Vet. Res. 2018, 49, 40. [Google Scholar] [CrossRef]
- Hammoumi, S.; Vallaeys, T.; Santika, A.; Leleux, P.; Borzym, E.; Klopp, C.; Avarre, J.C. Targeted genomic enrichment and sequencing of cyhv-3 from carp tissues confirms low nucleotide diversity and mixed genotype infections. PeerJ 2016, 4, e2516. [Google Scholar] [CrossRef]
- Li, W.; Lee, X.; Weng, S.; He, J.; Dong, C. Whole-genome sequence of a novel chinese cyprinid herpesvirus 3 isolate reveals the existence of a distinct european genotype in east asia. Vet. Microbiol. 2015, 175, 185–194. [Google Scholar] [CrossRef]
- Davison, A.J.; Kurobe, T.; Gatherer, D.; Cunningham, C.; Korf, I.; Fukuda, H.; Hedrick, R.P.; Waltzek, T.B. Comparative genomics of carp herpesviruses. J. Virol. 2013, 87, 2908–2922. [Google Scholar] [CrossRef]
- Costes, B.; Fournier, G.; Michel, B.; Delforge, C.; Raj, V.S.; Dewals, B.; Gillet, L.; Drion, P.; Body, A.; Schynts, F.; et al. Cloning of the koi herpesvirus genome as an infectious bacterial artificial chromosome demonstrates that disruption of the thymidine kinase locus induces partial attenuation in cyprinus carpio koi. J. Virol. 2008, 82, 4955–4964. [Google Scholar] [CrossRef]
- Boutier, M.; Ronsmans, M.; Ouyang, P.; Fournier, G.; Reschner, A.; Rakus, K.; Wilkie, G.S.; Farnir, F.; Bayrou, C.; Lieffrig, F.; et al. Rational development of an attenuated recombinant cyprinid herpesvirus 3 vaccine using prokaryotic mutagenesis and in vivo bioluminescent imaging. PLoS Pathog. 2015, 11, e1004690. [Google Scholar] [CrossRef]
- Ronen, A.; Perelberg, A.; Abramowitz, J.; Hutoran, M.; Tinman, S.; Bejerano, I.; Steinitz, M.; Kotler, M. Efficient vaccine against the virus causing a lethal disease in cultured cyprinus carpio. Vaccine 2003, 21, 4677–4684. [Google Scholar] [CrossRef]
- Enzmann, P.-J.; Fichtner, D.; Schütze, H.; Walliser, G. Development of vaccines against vhs and ihn: Oral application, molecular marker and discrimination of vaccinated fish from infected populations. J. Appl. Ichthyol. 1998, 14, 179–183. [Google Scholar] [CrossRef]
- Klafack, S.; Wang, Q.; Zeng, W.W.; Wang, Y.Y.; Li, Y.Y.; Zheng, S.C.; Kempter, J.; Lee, P.Y.; Matras, M.; Bergmann, S.M. Genetic variability of koi herpesvirus in vitro-a natural event? Front. Microbiol. 2017, 8, 982. [Google Scholar] [CrossRef]
- Avarre, J.C.; Santika, A.; Bentenni, A.; Zainun, Z.; Madeira, J.P.; Maskur, M.; Bigarré, L.; Caruso, D. Spatio-temporal analysis of cyprinid herpesvirus 3 genetic diversity at a local scale. J. Fish Dis. 2012, 35, 767–774. [Google Scholar] [CrossRef]
- Sunarto, A.; McColl, K.A.; Crane, M.S.; Sumiati, T.; Hyatt, A.D.; Barnes, A.C.; Walker, P.J. Isolation and characterization of koi herpesvirus (khv) from indonesia: Identification of a new genetic lineage. J. Fish Dis. 2011, 34, 87–101. [Google Scholar] [CrossRef]
- Neukirch, M.; Böttcher, K.; Bunnajirakul, S. Isolation of a virus from koi with altered gills. Bull. Eur. Assoc. Fish Pathol. 1999, 19, 221–224. [Google Scholar]
- Bergmann, S.M.; Riechardt, M.; Fichtner, D.; Lee, P.; Kempter, J. Investigation on the diagnostic sensitivity of molecular tools used for detection of koi herpesvirus. J. Virol. Methods 2010, 163, 229–233. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing Subgroup. The sequence alignment/map format and samtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative genomics viewer (igv): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Bryant, S.H. Cd-search: Protein domain annotations on the fly. Nucleic Acids Res. 2004, 32, W327–W331. [Google Scholar] [CrossRef]
- Bergmann, S.M.; Sadowski, J.; Kiełpiński, M.; Bartłomiejczyk, M.; Fichtner, D.; Riebe, R.; Lenk, M.; Kempter, J. Susceptibility of koi×crucian carp and koi×goldfish hybrids to koi herpesvirus (khv) and the development of khv disease (khvd). J. Fish Dis. 2010, 33, 267–272. [Google Scholar] [CrossRef]
- D’Amore, R.; Ijaz, U.Z.; Schirmer, M.; Kenny, J.G.; Gregory, R.; Darby, A.C.; Shakya, M.; Podar, M.; Quince, C.; Hall, N. A comprehensive benchmarking study of protocols and sequencing platforms for 16s rrna community profiling. BMC Genom. 2016, 17, 55. [Google Scholar] [CrossRef]
- MacConaill, L.E.; Burns, R.T.; Nag, A.; Coleman, H.A.; Slevin, M.K.; Giorda, K.; Light, M.; Lai, K.; Jarosz, M.; McNeill, M.S.; et al. Unique, dual-indexed sequencing adapters with umis effectively eliminate index cross-talk and significantly improve sensitivity of massively parallel sequencing. BMC Genom. 2018, 19, 30. [Google Scholar] [CrossRef]
- Wright, E.S.; Vetsigian, K.H. Quality filtering of illumina index reads mitigates sample cross-talk. BMC Genom. 2016, 17, 876. [Google Scholar] [CrossRef]
- Spencer, D.H.; Tyagi, M.; Vallania, F.; Bredemeyer, A.J.; Pfeifer, J.D.; Mitra, R.D.; Duncavage, E.J. Performance of common analysis methods for detecting low-frequency single nucleotide variants in targeted next-generation sequence data. J. Mol. Diagn. 2014, 16, 75–88. [Google Scholar] [CrossRef]
- Xu, C.; Nezami Ranjbar, M.R.; Wu, Z.; DiCarlo, J.; Wang, Y. Detecting very low allele fraction variants using targeted DNA sequencing and a novel molecular barcode-aware variant caller. BMC Genom. 2017, 18, 5. [Google Scholar] [CrossRef]
- Liu, S.-L.; Rodrigo, A.G.; Shankarappa, R.; Learn, G.H.; Hsu, L.; Davidov, O.; Zhao, L.P.; Mullins, J.I. Hiv quasispecies and resampling. Science 1996, 273, 415–416. [Google Scholar] [CrossRef]
- Kebschull, J.M.; Zador, A.M. Sources of pcr-induced distortions in high-throughput sequencing data sets. Nucleic Acids Res. 2015, 43, e143. [Google Scholar] [CrossRef]
- Illingworth, C.J.R.; Roy, S.; Beale, M.A.; Tutill, H.; Williams, R.; Breuer, J. On the effective depth of viral sequence data. Virus Evol. 2017, 3, vex030. [Google Scholar] [CrossRef]
- Armbruster, D.A.; Pry, T. Limit of blank, limit of detection and limit of quantitation. Clin. Biochem. Rev. 2008, 29 (Suppl. 1), S49–S52. [Google Scholar]
- Freemont, P.S. Ubiquitination: Ring for destruction? Curr. Biol. 2000, 10, R84–R87. [Google Scholar] [CrossRef]
- Lorick, K.L.; Jensen, J.P.; Fang, S.; Ong, A.M.; Hatakeyama, S.; Weissman, A.M. Ring fingers mediate ubiquitin-conjugating enzyme (e2)-dependent ubiquitination. Proc. Natl. Acad. Sci. USA 1999, 96, 11364–11369. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, L.-F.; Munir, M.; Qiu, H.-J. Ring-domain e3 ligase-mediated host–virus interactions: Orchestrating immune responses by the host and antagonizing immune defense by viruses. Front. Immunol. 2018, 9, 1083. [Google Scholar] [CrossRef]
- Klafack, S.; Schröder, L. Development of an attenuated vaccine to combat Koi Herpesvirus Disease. Fish Shellfish Immunol. Submitted.
- Nkili-Meyong, A.A.; Bigarré, L.; Labouba, I.; Vallaeys, T.; Avarre, J.C.; Berthet, N. Contribution of next-generation sequencing to aquatic and fish virology. Intervirology 2016, 59, 285–300. [Google Scholar] [CrossRef]
- Posada-Cespedes, S.; Seifert, D.; Beerenwinkel, N. Recent advances in inferring viral diversity from high-throughput sequencing data. Virus Res. 2017, 239, 17–32. [Google Scholar] [CrossRef]
- Xue, K.S.; Moncla, L.H.; Bedford, T.; Bloom, J.D. Within-host evolution of human influenza virus. Trends Microbiol. 2018, 26, 781–793. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer/Probe a | Sequence | 5’ Position b |
|---|---|---|
| Conventional PCR | ||
| ORF150_-363_F | GCGTCGACGGAGCATG | 258055 |
| ORF150_+343_R | CGAAAGAGTAAGCCGTTGCC | 260647 |
| ORF150_11_F | CACAAGAGATGGACGCTCAG | 258428 |
| ORF150_510_R | GTTCTCGCCCAGCACCA | 258927 |
| Digital PCR | ||
| ORF150_131_F | GCTGGACCTGTCACAATTCTAT | 258548 |
| ORF150_195_P (FAM/BHQ1) | TCGCACCGTCGTCAAGCAGT | 258612 |
| ORF150_243_R | TGGTCCAGTAGACGGTTGA | 258659 |
| ORF150_1548_F | GAGCGAGGAACTCTACACAAC | 259965 |
| ORF150_1589_P (Cy5/BHQ1) | TGAGGATGCAGAAGCAGTGGATGT | 260006 |
| ORF150_1694_R | GGTAAGGGTAAAGCAGACCATC | 260110 |
| Sample | Number of Reads a | % Mapped Reads | Mean Coverage [1st-3rd Quartile] | Number of Variants Against KHV-J b | Number of Variants Against P0 b |
|---|---|---|---|---|---|
| P0 | 60,049,308 | 98.62 | 7802 [7868–7932] | 80 | - |
| P78-1c | 1,297,956 | 37.86 | 262 [202–292] | 46 | 21 |
| P78-2c | 2,179,255 | 37.07 | 418 [301–466] | 54 | 26 |
| P78-1s | 3,002,298 | 27.08 | 356 [248–398] | 50 | 25 |
| P78-2s | 3,315,745 | 32.28 | 568 [448–634] | 49 | 23 |
| P99-1c | 3,280,716 | 30.30 | 559 [404–633] | 103 | 58 |
| P99-2c | 2,227,886 | 31.94 | 395 [302–451] | 103 | 57 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klafack, S.; Fiston-Lavier, A.-S.; Bergmann, S.M.; Hammoumi, S.; Schröder, L.; Fuchs, W.; Lusiastuti, A.; Lee, P.-Y.; Heredia, S.V.; Master student consortium; et al. Cyprinid herpesvirus 3 Evolves In Vitro through an Assemblage of Haplotypes that Alternatively Become Dominant or Under-Represented. Viruses 2019, 11, 754. https://doi.org/10.3390/v11080754
Klafack S, Fiston-Lavier A-S, Bergmann SM, Hammoumi S, Schröder L, Fuchs W, Lusiastuti A, Lee P-Y, Heredia SV, Master student consortium, et al. Cyprinid herpesvirus 3 Evolves In Vitro through an Assemblage of Haplotypes that Alternatively Become Dominant or Under-Represented. Viruses. 2019; 11(8):754. https://doi.org/10.3390/v11080754
Chicago/Turabian StyleKlafack, Sandro, Anna-Sophie Fiston-Lavier, Sven M. Bergmann, Saliha Hammoumi, Lars Schröder, Walter Fuchs, Angela Lusiastuti, Pei-Yu Lee, Sarahi Vega Heredia, Master student consortium, and et al. 2019. "Cyprinid herpesvirus 3 Evolves In Vitro through an Assemblage of Haplotypes that Alternatively Become Dominant or Under-Represented" Viruses 11, no. 8: 754. https://doi.org/10.3390/v11080754
APA StyleKlafack, S., Fiston-Lavier, A.-S., Bergmann, S. M., Hammoumi, S., Schröder, L., Fuchs, W., Lusiastuti, A., Lee, P.-Y., Heredia, S. V., Master student consortium, Gosselin-Grenet, A.-S., & Avarre, J.-C. (2019). Cyprinid herpesvirus 3 Evolves In Vitro through an Assemblage of Haplotypes that Alternatively Become Dominant or Under-Represented. Viruses, 11(8), 754. https://doi.org/10.3390/v11080754