Human Virome and Disease: High-Throughput Sequencing for Virus Discovery, Identification of Phage-Bacteria Dysbiosis and Development of Therapeutic Approaches with Emphasis on the Human Gut


Abstract
1. What is the Microbiome?
2. Viruses as Part of the Human Microbiome
2.1. Human Endogenous Retroviruses
2.2. Eukaryotic Viruses
2.3. Bacteriophages
3. Lytic and Lysogenic Cycles in the Control of Bacterial Populations
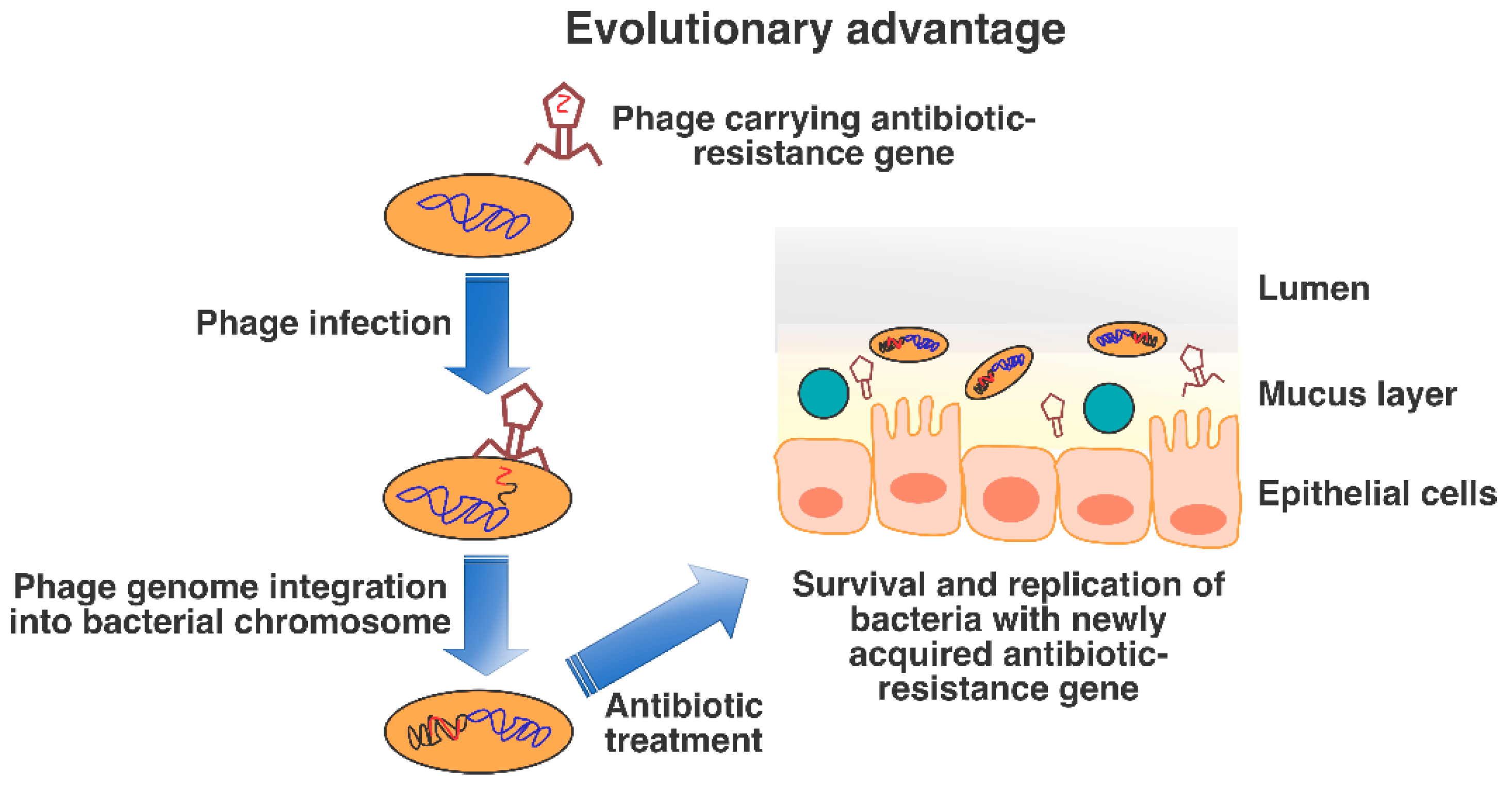
4. Bacterial Adaptation through Lysogenic Conversion
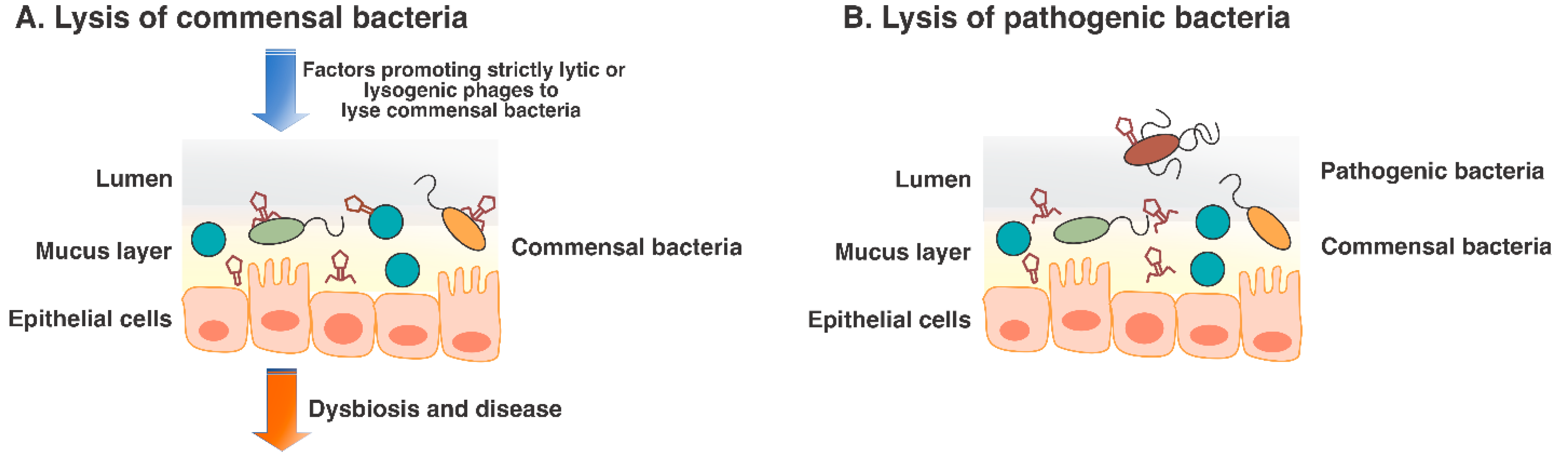
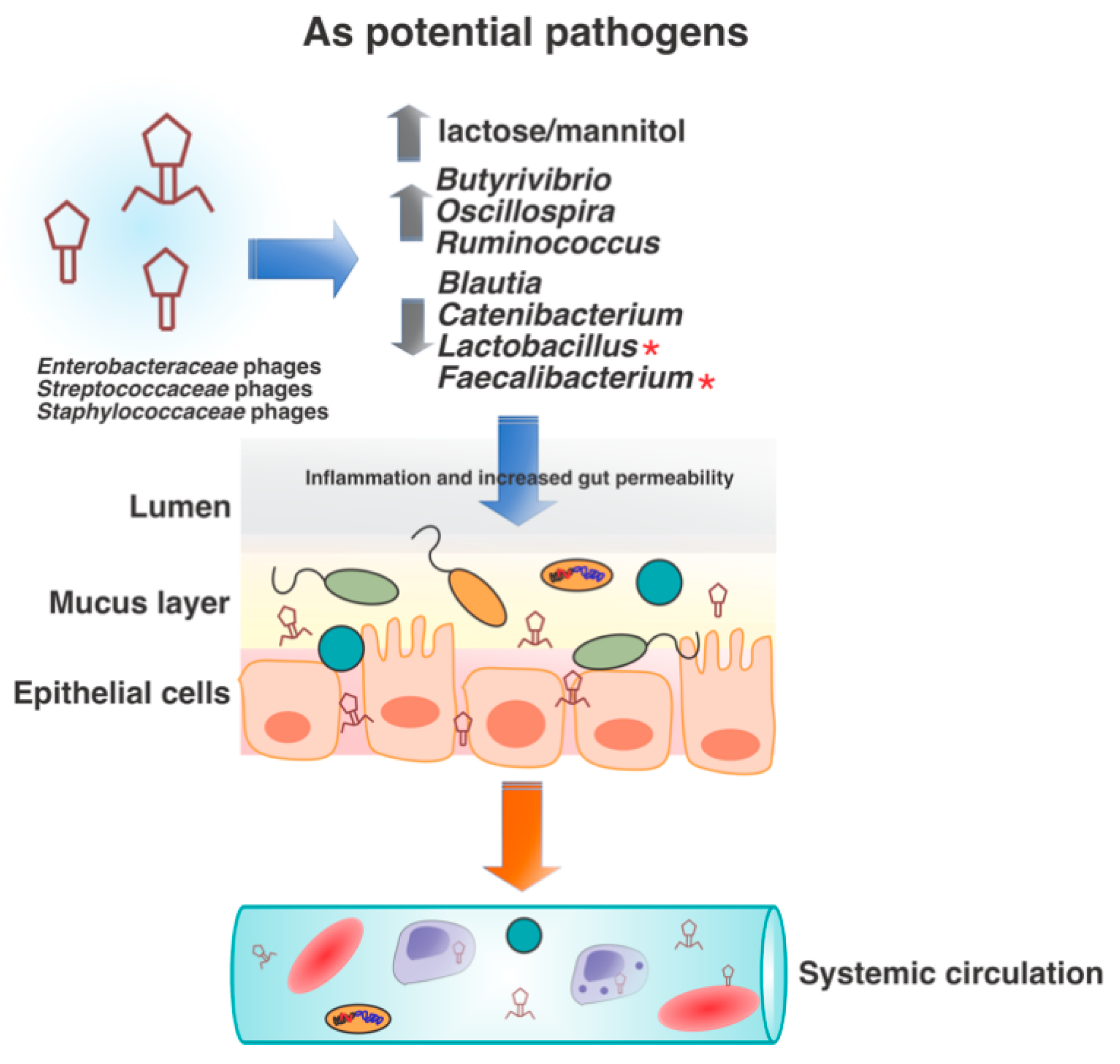
5. Bacteriophages as Human Pathogens?
6. Virome-Associated Dysbiosis
6.1. Type-1 Diabetes
6.2. Type-2 Diabetes
6.3. Inflammatory Bowel Disease (IBD)
6.4. Human Immunodeficiency Virus (HIV) Infection
6.5. Cancer
7. Trans-Kingdom Interactions
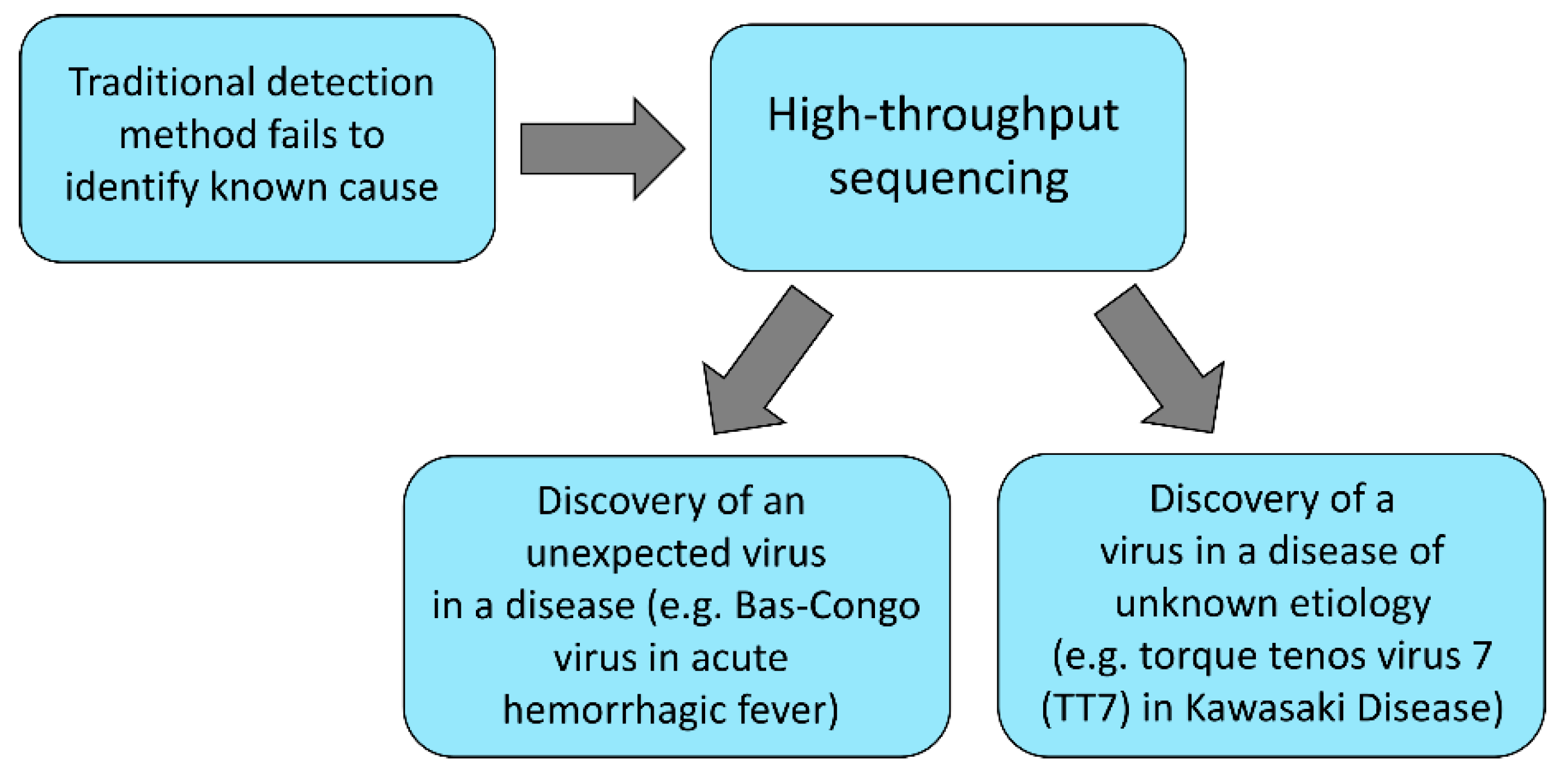
8. Challenges in Virome Research
8.1. Bioinformatic Methods for Virome Analysis
8.2. Viral Mock Communities as Controls in Virome Research
9. Virus- and Virome-Directed Therapeutic Approaches
10. Conclusions and Future Directions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Nicholson, J.K.; Holmes, E.; Kinross, J.; Burcelin, R.; Gibson, G.; Jia, W.; Pettersson, S. Host-gut Metabolic Interactions. Science 2012, 336, 1262–1268. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Bello, M.G.; Costello, E.K.; Contreras, M.; Magris, M.; Hidalgo, G.; Fierer, N.; Knight, R. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc. Natl. Acad. Sci. USA 2010, 107, 11971–11975. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Liu, W.; Piao, M.; Zhu, H. A review of the relationship between the gut microbiota and amino acid metabolism. Amino Acids 2017, 49, 2083–2090. [Google Scholar] [CrossRef] [PubMed]
- Rowland, I.; Gibson, G.; Heinken, A.; Scott, K.; Swann, J.; Thiele, I.; Tuohy, K. Gut microbiota functions: Metabolism of nutrients and other food components. Eur. J. Nutr. 2018, 57, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Koonen, D.; Hofker, M.; Fu, J. Gut microbiome and lipid metabolism: From associations to mechanisms. Curr. Opin. Lipidol. 2016, 27, 216–224. [Google Scholar] [CrossRef]
- Blekhman, R.; Goodrich, J.K.; Huang, K.; Sun, Q.; Bukowski, R.; Bell, J.T.; Spector, T.D.; Keinan, A.; Ley, R.E.; Gevers, D.; et al. Host genetic variation impacts microbiome composition across human body sites. Genome Biol. 2015, 16. [Google Scholar] [CrossRef]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef]
- Clemente, J.C.; Pehrsson, E.C.; Blaser, M.J.; Sandhu, K.; Gao, Z.; Wang, B.; Magris, M.; Hidalgo, G.; Contreras, M.; Noya-Alarcón, Ó.; et al. The microbiome of uncontacted Amerindians. Sci. Adv. 2015, 1, e1500183. [Google Scholar] [CrossRef]
- Nagpal, R.; Newman, T.M.; Wang, S.; Jain, S.; Lovato, J.F.; Yadav, H. Obesity-Linked Gut Microbiome Dysbiosis Associated with Derangements in Gut Permeability and Intestinal Cellular Homeostasis Independent of Diet. J. Diabetes Res. 2018, 2018, 1–9. [Google Scholar] [CrossRef]
- Montassier, E.; Gastinne, T.; Vangay, P.; Al-Ghalith, G.A.; Bruley Des Varannes, S.; Massart, S.; Moreau, P.; Potel, G.; de La Cochetière, M.F.; Batard, E.; et al. Chemotherapy-driven dysbiosis in the intestinal microbiome. Aliment. Pharmacol. Ther. 2015, 42, 515–528. [Google Scholar] [CrossRef]
- Francino, M.P. Antibiotics and the human gut microbiome: Dysbioses and accumulation of resistances. Front. Microbiol. 2016, 6, 1543. [Google Scholar] [CrossRef] [PubMed]
- Pollock, J.; Glendinning, L.; Wisedchanwet, T.; Watson, M. The Madness of Microbiome: Attempting To Find Consensus “Best Practice” for 16S Microbiome Studies. Appl. Environ. Microbiol. 2018, 84. [Google Scholar] [CrossRef] [PubMed]
- Fuks, G.; Elgart, M.; Amir, A.; Zeisel, A.; Turnbaugh, P.J.; Soen, Y.; Shental, N. Combining 16S rRNA gene variable regions enables high-resolution microbial community profiling. Microbiome 2018, 6. [Google Scholar] [CrossRef] [PubMed]
- Bittinger, K.; Koenig, J.E.; Muegge, B.D.; Zaneveld, J.; Kelley, S.T.; Peña, A.G.; Costello, E.K.; Walters, W.A.; Knight, R.; Stombaugh, J.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. QIIME 2: Reproducible, interactive, scalable, and extensible microbiome data science. PeerJ Prepr. 2018. [Google Scholar] [CrossRef]
- Dridi, B.; Raoult, D.; Drancourt, M. Archaea as emerging organisms in complex human microbiomes. Anaerobe 2011, 17, 56–63. [Google Scholar] [CrossRef]
- Wang, Z.K.; Yang, Y.S.; Stefka, A.T.; Sun, G.; Peng, L.H. Fungal microbiota and digestive diseases. Aliment. Pharmacol. Ther. 2014, 39, 751–766. [Google Scholar] [CrossRef] [PubMed]
- Sharpton, T.J. An introduction to the analysis of shotgun metagenomic data. Front. Plant Sci. 2014, 5, 209. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.M.; Pfeiffer, J.K. Viruses and the Microbiota. Annu. Rev. Virol. 2014, 1, 55–69. [Google Scholar] [CrossRef]
- Mukhopadhya, I.; Segal, J.P.; Carding, S.R.; Hart, A.L.; Hold, G.L. The gut virome: The “missing link” between gut bacteria and host immunity? Therap. Adv. Gastroenterol. 2019, 12, 1756284819836620. [Google Scholar] [CrossRef]
- Lodish, H.; Berk, A.; Zipursky, S.L.; Matsudaira, P.; Baltimore, D.; Darnell, J. Viruses: Structure, function, and uses. In Molecular Cell Biology, 4th ed.; Freeman, W.H., Ed.; W.H.Freeman & Co Ltd.: New York, NY, USA, 2000. [Google Scholar]
- Grove, J.; Marsh, M. The cell biology of receptor-mediated virus entry. J. Cell Biol. 2011, 195, 1071–1082. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, D.J. Endogenous retroviruses in the human genome sequence. Genome Biol. 2001, 2, 171. [Google Scholar] [CrossRef] [PubMed]
- Pistello, M.; Antonelli, G. Integration of the viral genome into the host cell genome: A double-edged sword. Clin. Microbiol. Infect. 2016. [Google Scholar] [CrossRef] [PubMed]
- Luzuriaga, K. Introduction to Retroviridae. In Principles and Practice of Pediatric Infectious Diseases, 4th ed.; Long, S.S., Pickering, L.K., Prober, C.G., Eds.; Saunders: Philadelphia, PA, USA, 2012; pp. 1164–1164.e3. ISBN 9781437720594. [Google Scholar]
- Küry, P.; Nath, A.; Créange, A.; Dolei, A.; Marche, P.; Gold, J.; Giovannoni, G.; Hartung, H.P.; Perron, H. Human Endogenous Retroviruses in Neurological Diseases. Trends Mol. Med. 2018, 24, 379–394. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Lee, M.H.; Henderson, L.; Tyagi, R.; Bachani, M.; Steiner, J.; Campanac, E.; Hoffman, D.A.; Geldern, G.V.; Johnson, K.; et al. Human endogenous retrovirus-K contributes to motor neuron disease. Sci. Transl. Med. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Nexø, B.A.; Villesen, P.; Nissen, K.K.; Lindegaard, H.M.; Rossing, P.; Petersen, T.; Tarnow, L.; Hansen, B.; Lorenzen, T.; Hørslev-Petersen, K.; et al. Are human endogenous retroviruses triggers of autoimmune diseases? Unveiling associations of three diseases and viral loci. Immunol. Res. 2016, 64, 55–63. [Google Scholar] [CrossRef]
- Gonzalez-Cao, M.; Iduma, P.; Karachaliou, N.; Santarpia, M.; Blanco, J.; Rosell, R. Human endogenous retroviruses and cancer. Cancer Biol. Med. 2016, 13, 483. [Google Scholar] [CrossRef]
- Lecuit, M.; Eloit, M. Chapter 21—The Viruses of the Gut Microbiota. In The Microbiota in Gastrointestinal Pathophysiology; Floch, M.H., Ringel, Y., Walker, W.A., Ringel, Y., Eds.; Academic Press: Boston, MA, USA, 2017; pp. 179–183. ISBN 978-0-12-804024-9. [Google Scholar]
- Ajami, N.J.; Petrosino, J.F. Chapter 5.1–Enteric Viral Metagenomics. In Viral Gastroenteritis; Svensson, L., Desselberger, U., Greenberg, H.B., Estes, M.K., Eds.; Academic Press: Boston, MA, USA, 2016; pp. 523–533. ISBN 978-0-12-802241-2. [Google Scholar]
- Pannaraj, P.S.; Ly, M.; Cerini, C.; Saavedra, M.; Aldrovandi, G.M.; Saboory, A.A.; Johnson, K.M.; Pride, D.T. Shared and distinct features of human milk and infant stool viromes. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef]
- Lim, E.S.; Zhou, Y.; Zhao, G.; Bauer, I.K.; Droit, L.; Ndao, I.M.; Warner, B.B.; Tarr, P.I.; Wang, D.; Holtz, L.R. Early life dynamics of the human gut virome and bacterial microbiome in infants. Nat. Med. 2015, 21, 1228–1234. [Google Scholar] [CrossRef]
- Reyes, A.; Haynes, M.; Hanson, N.; Angly, F.E.; Heath, A.C.; Rohwer, F.; Gordon, J.I. Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature 2010, 466, 334–338. [Google Scholar] [CrossRef]
- Boone, S.A.; Gerba, C.P. Significance of fomites in the spread of respiratory and enteric viral disease. Appl. Environ. Microbiol. 2007, 73, 1687–1696. [Google Scholar] [CrossRef]
- Rodríguez-Lázaro, D.; Cook, N.; Ruggeri, F.M.; Sellwood, J.; Nasser, A.; Nascimento, M.S.J.; D’Agostino, M.; Santos, R.; Saiz, J.C.; Rzezutka, A.; et al. Virus hazards from food, water and other contaminated environments. FEMS Microbiol. Rev. 2012, 36, 786–814. [Google Scholar] [CrossRef]
- Pallansch, M.A. Enterovirus Infections, Including Poliomyelitis. In Tropical Infectious Diseases; Saunders: Philadelphia, PA, USA, 2011; ISBN 9780702039355. [Google Scholar]
- Bosch, A.; Pintó, R.M.; Guix, S. Human astroviruses. Clin. Microbiol. Rev. 2014, 27, 1048–1074. [Google Scholar] [CrossRef]
- Grard, G.; Fair, J.N.; Lee, D.; Slikas, E.; Steffen, I.; Muyembe, J.J.; Sittler, T.; Veeraraghavan, N.; Ruby, J.G.; Wang, C.; et al. A Novel Rhabdovirus Associated with Acute Hemorrhagic Fever in Central Africa. PLoS Pathog. 2012, 8. [Google Scholar] [CrossRef]
- Wiwanitkit, S.; Wiwanitkit, V. Acute viral hemorrhage disease: A summary on new viruses. J. Acute Dis. 2015. [Google Scholar] [CrossRef]
- Thissen, J.B.; Isshiki, M.; Jaing, C.; Nagao, Y.; Aldea, D.L.; Allen, J.E.; Izui, M.; Slezak, T.R.; Ishida, T.; Sano, T. A novel variant of torque teno virus 7 identified in patients with Kawasaki disease. PLoS ONE 2018, 13. [Google Scholar] [CrossRef]
- Zanella, M.C.; Lenggenhager, L.; Schrenzel, J.; Cordey, S.; Kaiser, L. High-throughput sequencing for the aetiologic identification of viral encephalitis, meningoencephalitis, and meningitis. A narrative review and clinical appraisal. Clin. Microbiol. Infect. 2019, 25, 422–430. [Google Scholar] [CrossRef]
- Moore, M.D.; Jaykus, L.A. Virus–bacteria interactions: Implications and potential for the applied and agricultural sciences. Viruses 2018, 10, 61. [Google Scholar] [CrossRef]
- Pfeiffer, J.K.; Virgin, H.W. Viral immunity: Transkingdom control of viral infection and immunity in the mammalian intestine. Science 2016, 351. [Google Scholar] [CrossRef]
- Breitbart, M.; Rohwer, F. Here a virus, there a virus, everywhere the same virus? Trends Microbiol. 2005, 13, 278–284. [Google Scholar] [CrossRef]
- Lim, E.S.; Wang, D.; Holtz, L.R. The Bacterial Microbiome and Virome Milestones of Infant Development. Trends Microbiol. 2016, 24, 801–810. [Google Scholar] [CrossRef]
- Minot, S.; Sinha, R.; Chen, J.; Li, H.; Keilbaugh, S.A.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. The human gut virome: Inter-individual variation and dynamic response to diet. Genome Res. 2011, 21, 1616–1625. [Google Scholar] [CrossRef]
- Park, E.J.; Kim, K.H.; Abell, G.C.J.; Kim, M.S.; Roh, S.W.; Bae, J.W. Metagenomic analysis of the viral communities in fermented foods. Appl. Environ. Microbiol. 2011, 77, 1284–1291. [Google Scholar] [CrossRef]
- Hannigan, G.D.; Meisel, J.S.; Tyldsley, A.S.; Zheng, Q.; Hodkinson, B.P.; SanMiguel, A.J.; Minot, S.; Bushman, F.D.; Grice, E.A. The Human Skin Double-Stranded DNA Virome: Topographical and Temporal Diversity, Genetic Enrichment, and Dynamic Associations with the Host Microbiome. MBio 2015, 6. [Google Scholar] [CrossRef]
- Abeles, S.R.; Robles-Sikisaka, R.; Ly, M.; Lum, A.G.; Salzman, J.; Boehm, T.K.; Pride, D.T. Human oral viruses are personal, persistent and gender-consistent. ISME J. 2014, 8, 1753–1767. [Google Scholar] [CrossRef]
- Clokie, M.R.J.; Millard, A.D.; Letarov, A.V.; Heaphy, S. Phages in nature. Bacteriophage 2011, 1, 31–45. [Google Scholar] [CrossRef]
- Thurber, R.V. Current insights into phage biodiversity and biogeography. Curr. Opin. Microbiol. 2009, 12, 582–587. [Google Scholar] [CrossRef]
- Barylski, J.; Enault, F.; Dutilh, B.E.; Schuller, M.B.; Edwards, R.A.; Gillis, A.; Klumpp, J.; Knezevic, P.; Krupovic, M.; Kuhn, J.H.; et al. Analysis of spounaviruses as a case study for the overdue reclassification of tailed bacteriophages. bioRxiv 2018. [Google Scholar] [CrossRef]
- Brüssow, H.; Hendrix, R.W. Phage genomics: Small is beautiful. Cell 2002, 108, 13–16. [Google Scholar] [CrossRef]
- Ly, M.; Abeles, S.R.; Boehm, T.K.; Robles-Sikisaka, R.; Naidu, M.; Santiago-Rodriguez, T.; Pride, D.T. Altered oral viral ecology in association with periodontal disease. MBio 2014. [Google Scholar] [CrossRef]
- Zuo, T.; Lu, X.J.; Zhang, Y.; Cheung, C.P.; Lam, S.; Zhang, F.; Tang, W.; Ching, J.Y.L.; Zhao, R.; Chan, P.K.S.; et al. Gut mucosal virome alterations in ulcerative colitis. Gut 2019. [Google Scholar] [CrossRef]
- Jiang, S.C.; Paul, J.H. Occurrence of lysogenic bacteria in marine microbial communities as determined by prophage induction. Mar. Ecol. Prog. Ser. 1996, 142, 27–38. [Google Scholar] [CrossRef]
- Weinbauer, M.G.; Suttle, C.A. Potential significance of lysogeny to bacteriophage production and bacterial mortality in coastal waters of the gulf of Mexico. Appl. Environ. Microbiol. 1996, 62, 4374–4380. [Google Scholar]
- Allué-Guardia, A.; Jofre, J.; Muniesa, M. Stability and infectivity of cytolethal distending toxin type V gene-carrying bacteriophages in a water mesocosm and under different inactivation conditions. Appl. Environ. Microbiol. 2012, 78, 5818–5823. [Google Scholar] [CrossRef]
- Chibani-Chennoufi, S.; Bruttin, A.; Dillmann, M.L.; Brüssow, H. Phage-host interaction: An ecological perspective. J. Bacteriol. 2004, 186, 3677–3686. [Google Scholar] [CrossRef]
- Stern, A.; Mick, E.; Tirosh, I.; Sagy, O.; Sorek, R. CRISPR targeting reveals a reservoir of common phages associated with the human gut microbiome. Genome Res. 2012, 22, 1985–1994. [Google Scholar] [CrossRef]
- Azam, A.H.; Tanji, Y. Bacteriophage-host arm race: An update on the mechanism of phage resistance in bacteria and revenge of the phage with the perspective for phage therapy. Appl. Microbiol. Biotechnol. 2019, 103, 2121–2131. [Google Scholar] [CrossRef]
- Bondy-Denomy, J.; Qian, J.; Westra, E.R.; Buckling, A.; Guttman, D.S.; Davidson, A.R.; Maxwell, K.L. Prophages mediate defense against phage infection through diverse mechanisms. ISME J. 2016, 10, 2854–2866. [Google Scholar] [CrossRef]
- Donaldson, G.P.; Lee, S.M.; Mazmanian, S.K. Gut biogeography of the bacterial microbiota. Nat. Rev. Microbiol. 2015, 14, 20–32. [Google Scholar] [CrossRef]
- Pereira, F.C.; Berry, D. Microbial nutrient niches in the gut. Environ. Microbiol. 2017, 19, 1366–1378. [Google Scholar] [CrossRef]
- Silveira, C.B.; Rohwer, F.L. Piggyback-The-Winner in host-Associated microbial Communities. npj Biofilms Microbiomes 2016, 2, 16010. [Google Scholar] [CrossRef]
- Barr, J.J.; Youle, M.; Rohwer, F. Innate and acquired bacteriophage-mediated immunity. Bacteriophage 2013, 3, e25857. [Google Scholar] [CrossRef]
- Howard-Varona, C.; Hargreaves, K.R.; Abedon, S.T.; Sullivan, M.B. Lysogeny in nature: Mechanisms, impact and ecology of temperate phages. ISME J. 2017, 11, 1511–1520. [Google Scholar] [CrossRef]
- Vaca Pacheco, S.; García González, O.; Paniagua Contreras, G.L. The lom gene of bacteriophage λ is involved in Escherichia coli K12 adhesion to human buccal epithelial cells. FEMS Microbiol. Lett. 1997, 156, 129–132. [Google Scholar] [CrossRef]
- Vaca-Pacheco, S.; Paniagua-Contreras, G.L.; García-González, O.; de La Garza, M. The clinically isolated FIZ15 bacteriophage causes lysogenic conversion in Pseudomonas aeruginosa PAO1. Curr. Microbiol. 1999, 38, 239–243. [Google Scholar] [CrossRef]
- Bensing, B.A.; Rubens, C.E.; Sullam, P.M. Genetic loci of Streptococcus mitis that mediate binding to human platelets. Infect. Immun. 2001, 69, 1373–1380. [Google Scholar] [CrossRef]
- Karaolis, D.K.R.; Somara, S.; Maneval, D.R.; Johnson, J.A.; Kaper, J.B. A bacteriophage encoding a pathogenicity island, a type-IV pilus and a phage receptor in cholera bacteria. Nature 1999, 399, 375–379. [Google Scholar] [CrossRef]
- Stanley, T.L.; Ellermeier, C.D.; Slauch, J.M. Tissue-specific gene expression identifies a gene in the lysogenic phage Gifsy-1 that affects Salmonella enterica serovar typhimurium survival in Peyer’s patches. J. Bacteriol. 2000, 182, 4406–4413. [Google Scholar] [CrossRef]
- Hynes, W.L.; Ferretti, J.J. Sequence analysis and expression in Escherichia coli of the hyaluronidase gene of Streptococcus pyogenes bacteriophage H4489A. Infect. Immun. 1989, 57, 533–539. [Google Scholar]
- Sako, T.; Sawaki, S.; Sakurai, T.; Ito, S.; Yoshizawa, Y.; Kondo, I. Cloning and expression of the staphylokinase gene of Staphylococcus aureus in Escherichia coli. MGG Mol. Gen. Genet. 1983, 190, 271–277. [Google Scholar] [CrossRef]
- Barondess, J.J.; Beckwfth, J. A bacterial virulence determinant encoded by lysogenic coliphage λ. Nature 1990, 346, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Holloway, B.W.; Cooper, G.N. Lysogenic conversion in Pseudomonas aeruginosa. J. Bacteriol. 1962, 84, 1321–1324. [Google Scholar] [PubMed]
- Figueroa-Bossi, N.; Bossi, L. Inducible prophages contribute to Salmonella virulence in mice. Mol. Microbiol. 1999, 33, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Mavris, M.; Manning, P.A.; Morona, R. Mechanism of bacteriophage SfII-mediated serotype conversion in Shigella flexneri. Mol. Microbiol. 1997, 26, 939–950. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, J.; Muramoto, K.; Kamio, Y. Gene of LukF-PV-like Component of Panton-Valentine Leukocidin in Staphylococcus aureus P83 is Linked with lukM. Biosci. Biotechnol. Biochem. 2009, 61, 541–544. [Google Scholar] [CrossRef] [PubMed]
- Spanier, J.G. Bacteriophage control of antiphagocytic determinants in group A streptococci. J. Exp. Med. 2004, 152, 1393–1406. [Google Scholar] [CrossRef]
- Fujii, N.; Oguma, K.; Yokosawa, N.; Kimura, K.; Tsuzuki, K. Characterization of bacteriophage nucleic acids obtained from Clostridium botulinum types C and D. Appl. Environ. Microbiol. 1988, 54, 69–73. [Google Scholar]
- Uchida, T.; Michael Gill, D.; Pappenheimer, A.M. Mutation in the structural gene for diphtheria toxin carried by temperate phage β. Nat. New Biol. 1971, 233, 8–11. [Google Scholar] [CrossRef]
- Willshaw, G.A.; Smith, H.R.; Scotland, S.M.; Rowe, B. Cloning of Genes Determining the Production of Vero Cytotoxin by Escherichia coli. Microbiology 2009, 131, 3047–3053. [Google Scholar] [CrossRef]
- Hayashi, T.; Baba, T.; Matsumoto, H.; Terawaki, Y. Phage—conversion of cytotoxin production in Pseudomonas aeruginosa. Mol. Microbiol. 1990, 4, 1703–1709. [Google Scholar] [CrossRef]
- McDonough, M.A.; Butterton, J.R. Spontaneous tandem amplification and deletion of the Shiga toxin operon in Shigella dysenteriae 1. Mol. Microbiol. 1999, 34, 1058–1069. [Google Scholar] [CrossRef]
- Betley, M.J.; Mekalanos, J.J. Staphylococcal enterotoxin a is encoded by phage. Science 1985, 229, 185–187. [Google Scholar] [CrossRef]
- Goshorn, S.C.; Schlievert, P.M. Bacteriophage association of streptococcal pyrogenic exotoxin type C. J. Bacteriol. 1989, 171, 3068–3073. [Google Scholar] [CrossRef] [PubMed]
- Waldor, M.K.; Mekalanos, J.J. Lysogenic conversion by a filamentous phage encoding cholera toxin. Science 1996, 272, 1910–1914. [Google Scholar] [CrossRef]
- Skurray, R.A.; Firth, N. Genetics: Accessory Elements and Genetic Exchange. In Gram-Positive Pathogens, 2nd ed.; Fischetti, V., Novick, R., Ferretti, J., Portnoy, D., Rood, J., Eds.; ASM Press: Washington, DC, USA, 2006; pp. 413–426. [Google Scholar]
- Caparon, M. Genetics of group A streptococci. In Gram-positive pathogens; ASM Press: Washington, DC, USA, 2000. [Google Scholar]
- Wagner, P.L.; Waldor, M.K. Bacteriophage control of bacterial virulence. Infect. Immun. 2002, 70, 3985–3993. [Google Scholar] [CrossRef]
- Balcazar, J.L. Bacteriophages as Vehicles for Antibiotic Resistance Genes in the Environment. PLoS Pathog. 2014, 10, e1004219. [Google Scholar] [CrossRef]
- Muniesa, M.; Lucena, F.; Jofre, J. Comparative survival of free Shiga toxin 2-encoding phages and Escherichia coli strains outside the gut. Appl. Environ. Microbiol. 1999, 65, 5615–5618. [Google Scholar]
- Yoo, B.B.; Liu, Y.; Juneja, V.; Huang, L.; Hwang, C.A. Effect of environmental stresses on the survival and cytotoxicity of Shiga toxin-producing Escherichia coli. Food Qual. Saf. 2017, 1, 139–146. [Google Scholar] [CrossRef]
- Lainhart, W.; Stolfa, G.; Koudelka, G.B. Shiga toxin as a bacterial defense against a eukaryotic predator, Tetrahymena thermophila. J. Bacteriol. 2009, 191, 5116–5122. [Google Scholar] [CrossRef]
- Lengeling, A.; Mahajan, A.; Gally, D.L. Bacteriophages as Pathogens and Immune Modulators? MBio 2013, 4. [Google Scholar] [CrossRef]
- Karmali, M.A. Emerging public health challenges of Shiga toxin-producing Escherichia coli related to changes in the pathogen, the population, and the environment. Clin. Infect. Dis. 2017, 64, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, S.; Baker, K.; Padman, B.S.; Patwa, R.; Dunstan, R.A.; Weston, T.A.; Schlosser, K.; Bailey, B.; Lithgow, T.; Lazarou, M.; et al. Bacteriophage Transcytosis Provides a Mechanism to Cross Epithelial Cell Layers. MBio 2017, 8. [Google Scholar] [CrossRef]
- Tetz, G.V.; Ruggles, K.V.; Zhou, H.; Heguy, A.; Tsirigos, A.; Tetz, V. Bacteriophages as potential new mammalian pathogens. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Morahan, G. Insights into type 1 diabetes provided by genetic analyses. Curr. Opin. Endocrinol. Diabetes Obes. 2012, 19, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Needell, J.C.; Zipris, D. The Role of the Intestinal Microbiome in Type 1 Diabetes Pathogenesis. Curr. Diab. Rep. 2016, 16, 89. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.T.; Davis-Richardson, A.G.; Giongo, A.; Gano, K.A.; Crabb, D.B.; Mukherjee, N.; Casella, G.; Drew, J.C.; Ilonen, J.; Knip, M.; et al. Gut microbiome metagenomics analysis suggests a functional model for the development of autoimmunity for type 1 diabetes. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Filippi, C.M.; Herrath, M.G. von Viral Trigger for Type 1 Diabetes. Diabetes 2008, 57, 2863–2871. [Google Scholar] [CrossRef]
- Zhao, G.; Vatanen, T.; Droit, L.; Park, A.; Kostic, A.D.; Poon, T.W.; Vlamakis, H.; Siljander, H.; Härkönen, T.; Hämäläinen, A.-M.; et al. Intestinal virome changes precede autoimmunity in type I diabetes-susceptible children. Proc. Natl. Acad. Sci. USA 2017, 114, E6166–E6175. [Google Scholar] [CrossRef]
- Tetz, G.; Brown, S.M.; Hao, Y.; Tetz, V. Type 1 Diabetes: An Association Between Autoimmunity the Dynamics of Gut Amyloid-producing E. coli and Their Phages. BioRxiv 2018. [Google Scholar] [CrossRef]
- Larsen, N.; Vogensen, F.K.; van Den Berg, F.W.J.; Nielsen, D.S.; Andreasen, A.S.; Pedersen, B.K.; Al-Soud, W.A.; Sørensen, S.J.; Hansen, L.H.; Jakobsen, M. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS ONE 2010, 5. [Google Scholar] [CrossRef]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; You, X.; Mai, G.; Tokuyasu, T.; Liu, C. A human gut phage catalog correlates the gut phageome with type 2 diabetes. Microbiome 2018, 6. [Google Scholar] [CrossRef] [PubMed]
- Halfvarson, J.; Brislawn, C.J.; Lamendella, R.; Vázquez-Baeza, Y.; Walters, W.A.; Bramer, L.M.; D’Amato, M.; Bonfiglio, F.; McDonald, D.; Gonzalez, A.; et al. Dynamics of the human gut microbiome in inflammatory bowel disease. Nat. Microbiol. 2017, 2, 17004. [Google Scholar] [CrossRef] [PubMed]
- Celiberto, L.S.; Graef, F.A.; Healey, G.R.; Bosman, E.S.; Jacobson, K.; Sly, L.M.; Vallance, B.A. Inflammatory bowel disease and immunonutrition: Novel therapeutic approaches through modulation of diet and the gut microbiome. Immunology 2018, 155, 36–52. [Google Scholar] [CrossRef] [PubMed]
- Morgan, X.C.; Tickle, T.L.; Sokol, H.; Gevers, D.; Devaney, K.L.; Ward, D.V.; Reyes, J.A.; Shah, S.A.; LeLeiko, N.; Snapper, S.B.; et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012, 13, R79. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Brocal, V.; García-López, R.; Nos, P.; Beltrán, B.; Moret, I.; Moya, A. Metagenomic analysis of Crohn’s disease patients identifies changes in the virome and microbiome related to disease status and therapy, and detects potential interactions and biomarkers. Inflamm. Bowel Dis. 2015, 21, 2515–2532. [Google Scholar] [CrossRef] [PubMed]
- Norman, J.M.; Handley, S.A.; Baldridge, M.T.; Droit, L.; Liu, C.Y.; Keller, B.C.; Kambal, A.; Monaco, C.L.; Zhao, G.; Fleshner, P.; et al. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell 2015, 160, 447–460. [Google Scholar] [CrossRef]
- Fernandes, M.A.; Verstraete, S.G.; Phan, T.G.; Deng, X.; Stekol, E.; LaMere, B.; Lynch, S.V.; Heyman, M.B.; Delwart, E. Enteric Virome and Bacterial Microbiota in Children With Ulcerative Colitis and Crohn Disease. J. Pediatr. Gastroenterol. Nutr. 2019, 68, 30–36. [Google Scholar] [CrossRef]
- Duerkop, B.A.; Kleiner, M.; Paez-Espino, D.; Zhu, W.; Bushnell, B.; Hassell, B.; Winter, S.E.; Kyrpides, N.C.; Hooper, L.V. Murine colitis reveals a disease-associated bacteriophage community. Nat. Microbiol. 2018, 3, 11023–11031. [Google Scholar] [CrossRef]
- Grulich, A.E.; van Leeuwen, M.T.; Falster, M.O.; Vajdic, C.M. Incidence of cancers in people with HIV/AIDS compared with immunosuppressed transplant recipients: a meta-analysis. Lancet 2007, 370, 59–67. [Google Scholar] [CrossRef]
- Ciledag, A.; Karnak, D. AIDS and Opportunistic Infections. In Microbes, Viruses and Parasites in AIDS Process; IntechOpen Ltd.: London, UK, 2012. [Google Scholar]
- Monaco, C.L.; Gootenberg, D.B.; Zhao, G.; Handley, S.A.; Ghebremichael, M.S.; Lim, E.S.; Lankowski, A.; Baldridge, M.T.; Wilen, C.B.; Flagg, M.; et al. Altered Virome and Bacterial Microbiome in Human Immunodeficiency Virus-Associated Acquired Immunodeficiency Syndrome. Cell Host Microbe 2016, 19, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Siqueira, J.D.; Curty, G.; Xutao, D.; Hofer, C.B.; Machado, E.S.; Seuánez, H.N.; Soares, M.A.; Delwart, E.; Soares, E.A. Composite Analysis of the Virome and Bacteriome of HIV/HPV Co-Infected Women Reveals Proxies for Immunodeficiency. Viruses 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Williams, V.; Filippova, M.; Filippov, V.; Duerksen-Hughes, P. Viral carcinogenesis: Factors inducing DNA damage and virus integration. Cancers (Basel). 2014, 6, 2155–2186. [Google Scholar] [CrossRef] [PubMed]
- Van Dyne, E.A.; Henley, S.J.; Saraiya, M.; Thomas, C.C.; Markowitz, L.E.; Benard, V.B. Trends in Human Papillomavirus–Associated Cancers—United States, 1999–2015. MMWR. Morb. Mortal. Wkly. Rep. 2018, 67, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Prado, J.; Monezi, T.; Amorim, A.; Lino, V.; Paladino, A.; Boccardo, E. Human polyomaviruses and cancer: An overview. Clinics 2018, 73. [Google Scholar] [CrossRef] [PubMed]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Lam, K.C.; Vyshenska, D.; Hu, J.; Rodrigues, R.R.; Nilsen, A.; Zielke, R.A.; Brown, N.S.; Aarnes, E.-K.; Sikora, A.E.; Shulzhenko, N.; et al. Transkingdom network reveals bacterial players associated with cervical cancer gene expression program. PeerJ 2018, 6, e5590. [Google Scholar] [CrossRef]
- Chua, L.L.; Rajasuriar, R.; Azanan, M.S.; Abdullah, N.K.; Tang, M.S.; Lee, S.C.; Woo, Y.L.; Lim, Y.A.L.; Ariffin, H.; Loke, P. Reduced microbial diversity in adult survivors of childhood acute lymphoblastic leukemia and microbial associations with increased immune activation. Microbiome 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.H.; Zhao, L.; Zhang, X.; Nakatsu, G.; Han, J.; Xu, W.; Xiao, X.; Kwong, T.N.Y.; Tsoi, H.; Wu, W.K.K.; et al. Gavage of Fecal Samples From Patients With Colorectal Cancer Promotes Intestinal Carcinogenesis in Germ-Free and Conventional Mice. Gastroenterology 2017, 153, 1621–1633. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Adolph, T.E.; Gerner, R.R.; Moschen, A.R. The Intestinal Microbiota in Colorectal Cancer. Cancer Cell 2018, 33, 954–964. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, M.R.; Wang, X.; Liu, W.; Hao, Y.; Cai, G.; Han, Y.W. Fusobacterium nucleatum Promotes Colorectal Carcinogenesis by Modulating E-Cadherin/β-Catenin Signaling via its FadA Adhesin. Cell Host Microbe 2013, 14, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Maisonneuve, C.; Irrazabal, T.; Martin, A.; Girardin, S.E.; Philpott, D.J. The Impact of the Gut Microbiome on Colorectal Cancer. Annu. Rev. Cancer Biol. 2017, 2, 229–249. [Google Scholar] [CrossRef]
- Veziant, J.; Gagnière, J.; Jouberton, E.; Bonnin, V.; Sauvanet, P.; Pezet, D.; Barnich, N.; Miot-Noirault, E.; Bonnet, M. Association of colorectal cancer with pathogenic Escherichia coli: Focus on mechanisms using optical imaging. World J. Clin. Oncol. 2016, 7, 293. [Google Scholar] [CrossRef] [PubMed]
- Hannigan, G.D.; Duhaime, M.B.; Ruffin, M.T.; Koumpouras, C.C.; Schloss, P.D. Diagnostic Potential and Interactive Dynamics of the Colorectal Cancer Virome. MBio 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Nakatsu, G.; Zhou, H.; Wu, W.K.K.; Wong, S.H.; Coker, O.O.; Dai, Z.; Li, X.; Szeto, C.H.; Sugimura, N.; Lam, T.Y.T.; et al. Alterations in Enteric Virome Are Associated With Colorectal Cancer and Survival Outcomes. Gastroenterology 2018, 155, 529–541.e5. [Google Scholar] [CrossRef] [PubMed]
- Kane, M.; Case, L.K.; Kopaskie, K.; Kozlova, A.; MacDearmid, C.; Chervonsky, A.V.; Golovkina, T.V. Successful transmission of a retrovirus depends on the commensal microbiota. Science 2011, 334, 245–249. [Google Scholar] [CrossRef]
- Wilks, J.; Lien, E.; Jacobson, A.N.; Fischbach, M.A.; Qureshi, N.; Chervonsky, A.V.; Golovkina, T.V. Mammalian Lipopolysaccharide Receptors Incorporated into the Retroviral Envelope Augment Virus Transmission. Cell Host Microbe 2015, 18, 456–462. [Google Scholar] [CrossRef]
- Robinson, C.M.; Jesudhasan, P.R.; Pfeiffer, J.K. Bacterial lipopolysaccharide binding enhances virion stability and promotes environmental fitness of an enteric virus. Cell Host Microbe 2014, 15, 36–46. [Google Scholar] [CrossRef]
- Osborne, L.C.; Monticelli, L.A.; Nice, T.J.; Sutherland, T.E.; Siracusa, M.C.; Hepworth, M.R.; Tomov, V.T.; Kobuley, D.; Tran, S.V.; Bittinger, K.; et al. Virus-helminthcoinfection reveals a microbiota-independent mechanism of immunomodulation. Science 2014, 345, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Cadwell, K.; Patel, K.K.; Maloney, N.S.; Liu, T.C.; Ng, A.C.Y.; Storer, C.E.; Head, R.D.; Xavier, R.; Stappenbeck, T.S.; Virgin, H.W. Virus-Plus-Susceptibility Gene Interaction Determines Crohn’s Disease Gene Atg16L1 Phenotypes in Intestine. Cell 2010, 141, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Sweere, J.M.; Van Belleghem, J.D.; Ishak, H.; Bach, M.S.; Popescu, M.; Sunkari, V.; Kaber, G.; Manasherob, R.; Suh, G.A.; Cao, X.; et al. Bacteriophage trigger antiviral immunity and prevent clearance of bacterial infection. Science 2019, 363, eaat9691. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, S.R.; Wang, D. Origins and challenges of viral dark matter. Virus Res. 2017, 239, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, M.; Hooper, L.V.; Duerkop, B.A. Evaluation of methods to purify virus-like particles for metagenomic sequencing of intestinal viromes. BMC Genomics 2015, 16, 7. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Murthy, S.; Kapoor, A. Evolution of selective-sequencing approaches for virus discovery and virome analysis. Virus Res. 2017, 239, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Daly, G.M.; Leggett, R.M.; Rowe, W.; Stubbs, S.; Wilkinson, M.; Ramirez-Gonzalez, R.H.; Caccamo, M.; Bernal, W.; Heeney, J.L. Host subtraction, filtering and assembly validations for novel viral discovery using next generation sequencing data. PLoS ONE 2015, 10, e0129059. [Google Scholar] [CrossRef] [PubMed]
- Eric Wommack, K.; Bhavsar, J.; Polson, S.W.; Chen, J.; Dumas, M.; Srinivasiah, S.; Furman, M.; Jamindar, S.; Nasko, D.J. VIROME: A standard operating procedure for analysis of viral metagenome sequences. Stand. Genomic Sci. 2012, 6, 427–439. [Google Scholar] [CrossRef]
- Zhao, G.; Wu, G.; Lim, E.S.; Droit, L.; Krishnamurthy, S.; Barouch, D.H.; Virgin, H.W.; Wang, D. VirusSeeker, a computational pipeline for virus discovery and virome composition analysis. Virology 2017, 503, 21–30. [Google Scholar] [CrossRef]
- Ho, T.; Tzanetakis, I.E. Development of a virus detection and discovery pipeline using next generation sequencing. Virology 2014, 471–473, 54–60. [Google Scholar] [CrossRef]
- Tithi, S.S.; Aylward, F.O.; Jensen, R.V.; Zhang, L. FastViromeExplorer: A pipeline for virus and phage identification and abundance profiling in metagenomics data. PeerJ 2018, 6, e4227. [Google Scholar] [CrossRef] [PubMed]
- Ajami, N.J.; Wong, M.C.; Ross, M.C.; Lloyd, R.E.; Petrosino, J.F. Maximal viral information recovery from sequence data using VirMAP. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Czeczko, P.; Greenway, S.C.; de Koning, A.P.J. EzMap: A simple pipeline for reproducible analysis of the human virome. Bioinformatics 2017, 33, 2573–2574. [Google Scholar] [CrossRef]
- Roux, S.; Tournayre, J.; Mahul, A.; Debroas, D.; Enault, F. Metavir 2: New tools for viral metagenome comparison and assembled virome analysis. BMC Bioinformatics 2014, 15. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Kramna, L.; Autio, R.; Hyöty, H.; Nykter, M.; Cinek, O. Vipie: Web pipeline for parallel characterization of viral populations from multiple NGS samples. BMC Genomics 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Rampelli, S.; Soverini, M.; Turroni, S.; Quercia, S.; Biagi, E.; Brigidi, P.; Candela, M. ViromeScan: A new tool for metagenomic viral community profiling. BMC Genomics 2016, 17. [Google Scholar] [CrossRef]
- Jarrett, R.F.; Gallagher, A.; Gatherer, D. Molecular methods of virus detection in lymphoma. Methods Mol. Biol. 2013, 971, 277–293. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Enault, F.; Hurwitz, B.L.; Sullivan, M.B. VirSorter: Mining viral signal from microbial genomic data. PeerJ 2015, 3, e985. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Gao, S.; Padmanabhan, C.; Li, R.; Galvez, M.; Gutierrez, D.; Fuentes, S.; Ling, K.S.; Kreuze, J.; Fei, Z. VirusDetect: An automated pipeline for efficient virus discovery using deep sequencing of small RNAs. Virology 2017, 500, 130–138. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, H.; Nie, K.; Zhang, C.; Zhang, Y.; Wang, J.; Niu, P.; Ma, X. VIP: An integrated pipeline for metagenomics of virus identification and discovery. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Ren, J.; Ahlgren, N.A.; Lu, Y.Y.; Fuhrman, J.A.; Sun, F. VirFinder: A novel k-mer based tool for identifying viral sequences from assembled metagenomic data. Microbiome 2017, 5, 69. [Google Scholar] [CrossRef]
- Sutton, T.D.S.; Clooney, A.G.; Ryan, F.J.; Ross, R.P.; Hill, C. Choice of assembly software has a critical impact on virome characterisation. Microbiome 2019, 7. [Google Scholar] [CrossRef]
- Highlander, S. Mock Community Analysis. In Encyclopedia of Metagenomics; Nelson, K., Ed.; Springer Nature: New York, NY, USA, 2015; pp. 497–503. [Google Scholar]
- Singer, E.; Andreopoulos, B.; Bowers, R.M.; Lee, J.; Deshpande, S.; Chiniquy, J.; Ciobanu, D.; Klenk, H.P.; Zane, M.; Daum, C.; et al. Next generation sequencing data of a defined microbial mock community. Sci. Data 2016. [Google Scholar] [CrossRef]
- Parada, A.E.; Needham, D.M.; Fuhrman, J.A. Every base matters: Assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 2016. [Google Scholar] [CrossRef]
- Venkataraman, A.; Parlov, M.; Hu, P.; Schnell, D.; Wei, X.; Tiesman, J.P. Spike-in genomic DNA for validating performance of metagenomics workflows. Biotechniques 2018, 65, 315–321. [Google Scholar] [CrossRef]
- Parras-Moltó, M.; Rodríguez-Galet, A.; Suárez-Rodríguez, P.; López-Bueno, A. Evaluation of bias induced by viral enrichment and random amplification protocols in metagenomic surveys of saliva DNA viruses. Microbiome 2018, 6. [Google Scholar] [CrossRef]
- Kutter, E.; de Vos, D.; Gvasalia, G.; Alavidze, Z.; Gogokhia, L.; Kuhl, S.; Abedon, S.T. Phage therapy in clinical practice: Treatment of human infections. Curr. Pharm. Biotechnol. 2010, 11, 69–86. [Google Scholar] [CrossRef]
- Laxminarayan, R.; Duse, A.; Wattal, C.; Zaidi, A.K.M.; Wertheim, H.F.L.; Sumpradit, N.; Vlieghe, E.; Hara, G.L.; Gould, I.M.; Goossens, H.; et al. Antibiotic resistance-the need for global solutions. Lancet Infect. Dis. 2013, 13, 1057–1098. [Google Scholar] [CrossRef]
- Jault, P.; Leclerc, T.; Jennes, S.; Pirnay, J.P.; Que, Y.A.; Resch, G.; Rousseau, A.F.; Ravat, F.; Carsin, H.; Le Floch, R.; et al. Efficacy and tolerability of a cocktail of bacteriophages to treat burn wounds infected by Pseudomonas aeruginosa (PhagoBurn): A randomised, controlled, double-blind phase 1/2 trial. Lancet Infect. Dis. 2019, 19, 35–45. [Google Scholar] [CrossRef]
- Wright, A.; Hawkins, C.H.; Änggård, E.E.; Harper, D.R. A controlled clinical trial of a therapeutic bacteriophage preparation in chronic otitis due to antibiotic-resistant Pseudomonas aeruginosa; A preliminary report of efficacy. Clin. Otolaryngol. 2009, 34, 349–357. [Google Scholar] [CrossRef]
- LaVergne, S.; Hamilton, T.; Biswas, B.; Kumaraswamy, M.; Schooley, R.T.; Wooten, D. Phage Therapy for a Multidrug-Resistant Acinetobacter baumannii Craniectomy Site Infection. Open Forum Infect. Dis. 2018, 5. [Google Scholar] [CrossRef]
- Haddad Kashani, H.; Schmelcher, M.; Sabzalipoor, H.; Seyed Hosseini, E.; Moniri, R. Recombinant endolysins as potential therapeutics against antibiotic-resistant Staphylococcus aureus: Current status of research and novel delivery strategies. Clin. Microbiol. Rev. 2018, 31. [Google Scholar] [CrossRef]
- Gordillo Altamirano, F.L.; Barr, J.J. Phage Therapy in the Postantibiotic Era. Clin. Microbiol. Rev. 2019, 32. [Google Scholar] [CrossRef]
- Lu, T.K.; Collins, J.J. Engineered bacteriophage targeting gene networks as adjuvants for antibiotic therapy. Proc. Natl. Acad. Sci. USA 2009, 106, 4629–4634. [Google Scholar] [CrossRef]
- Edgar, R.; Friedman, N.; Shahar, M.M.; Qimron, U. Reversing bacterial resistance to antibiotics by phage-mediated delivery of dominant sensitive genes. Appl. Environ. Microbiol. 2012, 78, 744–751. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Evolutionary Advantage through Lysogenic Conversion | Bacterial Host | Reference |
|---|---|---|
| Cell colonization and adhesion | Escherichia coli | [69] |
| Pseudomonas aeruginosa | [70] | |
| Streptococcus mitis | [71] | |
| Vibrio cholerae | [72] | |
| Promotion of cell invasion | Salmonella enterica | [73] |
| Streptococcus pyogenes | [74] | |
| Staphylococcus aureus | [75] | |
| Resistance to serum and phagocytes | Escherichia coli | [76] |
| Pseudomonas aeruginosa | [77] | |
| Salmonella enterica | [78] | |
| Shigella dysenteriae | [79] | |
| Staphyloccoccus aureus | [80] | |
| Streptococcus pyogenes | [81] | |
| Exotoxin production | Clostridium botulinum | [82] |
| Corynebacterium diphtheriae | [83] | |
| Escherichia coli | [84] | |
| Pseudomonas aeruginosa | [85] | |
| Shigella dysenteriae | [86] | |
| Staphylococcus aureus | [87] | |
| Streptococcus pyogenes | [88] | |
| Vibrio cholerae | [89] | |
| Antibiotic susceptibility | Staphylcoccus aureus | [90] |
| Streptococcus pyogenes | [91] |
| Bioinformatic Tool | Description | Source | Reference |
|---|---|---|---|
| Alignment-based | |||
| VIROME | Web-application interface for the classification of Open-Reading Frames (ORF) or assembled data, which receive one classification. | http://virome.dbi.udel.edu | [146] |
| VirusSeeker | Linux-based for the classification of sequences at the nucleotide and amino acid level. It is used for virus characterization and discovery; the latter requires assembled reads. | http://pathology.wustl.edu/virusseeker/index.htm | [147] |
| VirFind | Web-based tool that maps the reads to reference genomes and also performs de novo assembly to get longer contigs to identify known viruses and discover new ones. It performs Blastn and Blastx. | http://virfind.org/j/ | [148] |
| FastViromeExplorer | Pseudo-alignment tool that maps reads to a reference virus database, filters the alignment results based on minimal coverage criteria and reports virus types and abundances along with taxonomic annotation. | https://bench.cs.vt.edu/FastViromeExplorer/ | [149] |
| VirMap | Suitable for low coverage and highly divergent viruses in metagenomic datasets. Uses a mapping assembly algorithm with both nucleotide and amino-acid alignments to build virus-like super-scaffolds. It possesses a taxonomic classification algorithm based on bits-per-base scoring system. | https://github.com/cmmr/VirMAP | [150] |
| EZ-Map | Python-based to filter, align, and analyze viromes from cell-free DNA samples. | https://github.com/dekoning-lab/ezmap | [151] |
| MetaVir2 | Web-application interface that works with assembled reads to perform taxonomy assignments based on available sequences in RefSeq. | http://metavir-meb.univ-bpclermont.fr | [152] |
| Vipie | Web-application capable of analyzing datasets from different studies by performing de-novo assembly, followed by taxonomic classification. | https://binf.uta.fi/vipie/ | [153] |
| ViromeScan | Linux-based application that performs taxonomy classification from raw data. The tool uses hierarchical databases for eukaryotic viruses to assign reads to viral species. | http://sourceforge.net/projects/viromescan/ | [154] |
| Vanator | Perl-based pipeline designed for metagenomics and virus discovery projects using Illumina FASTQ-formatted deep sequencing reads as the input. | https://sourceforge.net/projects/vanator-cvr/ | [155] |
| VirSorter | Predicts prophage and viral sequences in a reference-dependent and -independent manner. Detects circular sequences, performs gene prediction, removes poor quality protein-predicted sequences and those remaining are compared to PFAM and RefSeqABVir or Viromes databases. | https://sourceforge.net/projects/viromescan/ | [156] |
| VirusDetect | Performs virus identification by aligning small RNA reads to a known virus reference database and also performs de novo assembly. | http://virusdetect.feilab.net/cgi-bin/virusdetect/index.cgi | [157] |
| PHASTER | The enhanced release of PHAST for the fast identification and annotation of prophage sequences from assembled metagenomic datasets. | http://phaster.ca | [158] |
| K-mer-based | |||
| MetaVir2 | Web-application interface that works with assembled reads to perform taxonomy assignments based on available sequences in RefSeq. Provides users the option to perform the analysis based on k-mers. | http://metavir-meb.univ-bpclermont.fr | [152] |
| VIP | Developed for identification of viral pathogens from metagenomic datasets. Removes background reads, classifies reads based on nucleotide and amino acid homology, and uses k-mer based de novo assembly for evolutionary studies. | https://github.com/keylabivdc/VIP/blob/master/README.md | [159] |
| VirFinder | K-mer-based tool to identify sequence signatures that distinguish viral sequences from host sequences. | https://github.com/jessieren/VirFinder | [160] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santiago-Rodriguez, T.M.; Hollister, E.B. Human Virome and Disease: High-Throughput Sequencing for Virus Discovery, Identification of Phage-Bacteria Dysbiosis and Development of Therapeutic Approaches with Emphasis on the Human Gut. Viruses 2019, 11, 656. https://doi.org/10.3390/v11070656
Santiago-Rodriguez TM, Hollister EB. Human Virome and Disease: High-Throughput Sequencing for Virus Discovery, Identification of Phage-Bacteria Dysbiosis and Development of Therapeutic Approaches with Emphasis on the Human Gut. Viruses. 2019; 11(7):656. https://doi.org/10.3390/v11070656
Chicago/Turabian StyleSantiago-Rodriguez, Tasha M., and Emily B. Hollister. 2019. "Human Virome and Disease: High-Throughput Sequencing for Virus Discovery, Identification of Phage-Bacteria Dysbiosis and Development of Therapeutic Approaches with Emphasis on the Human Gut" Viruses 11, no. 7: 656. https://doi.org/10.3390/v11070656
APA StyleSantiago-Rodriguez, T. M., & Hollister, E. B. (2019). Human Virome and Disease: High-Throughput Sequencing for Virus Discovery, Identification of Phage-Bacteria Dysbiosis and Development of Therapeutic Approaches with Emphasis on the Human Gut. Viruses, 11(7), 656. https://doi.org/10.3390/v11070656