IFITM3 Clusters on Virus Containing Endosomes and Lysosomes Early in the Influenza A Infection of Human Airway Epithelial Cells


Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cells, Reagents, Viruses and Plasmids
2.2. Immunofluorescence Staining
2.3. Immunoblotting (Western Blotting)
2.4. Microscopy and Data Analysis
3. Results
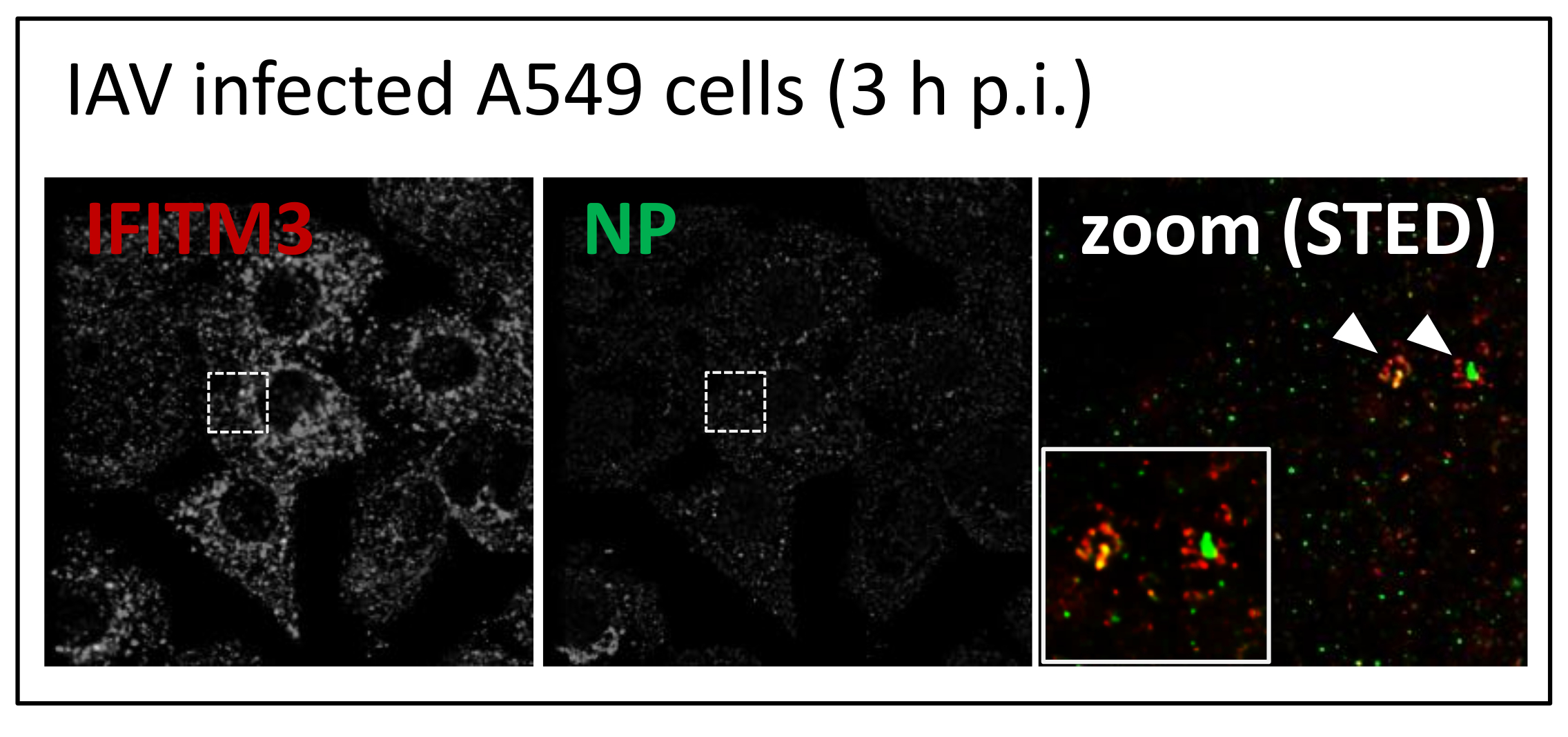
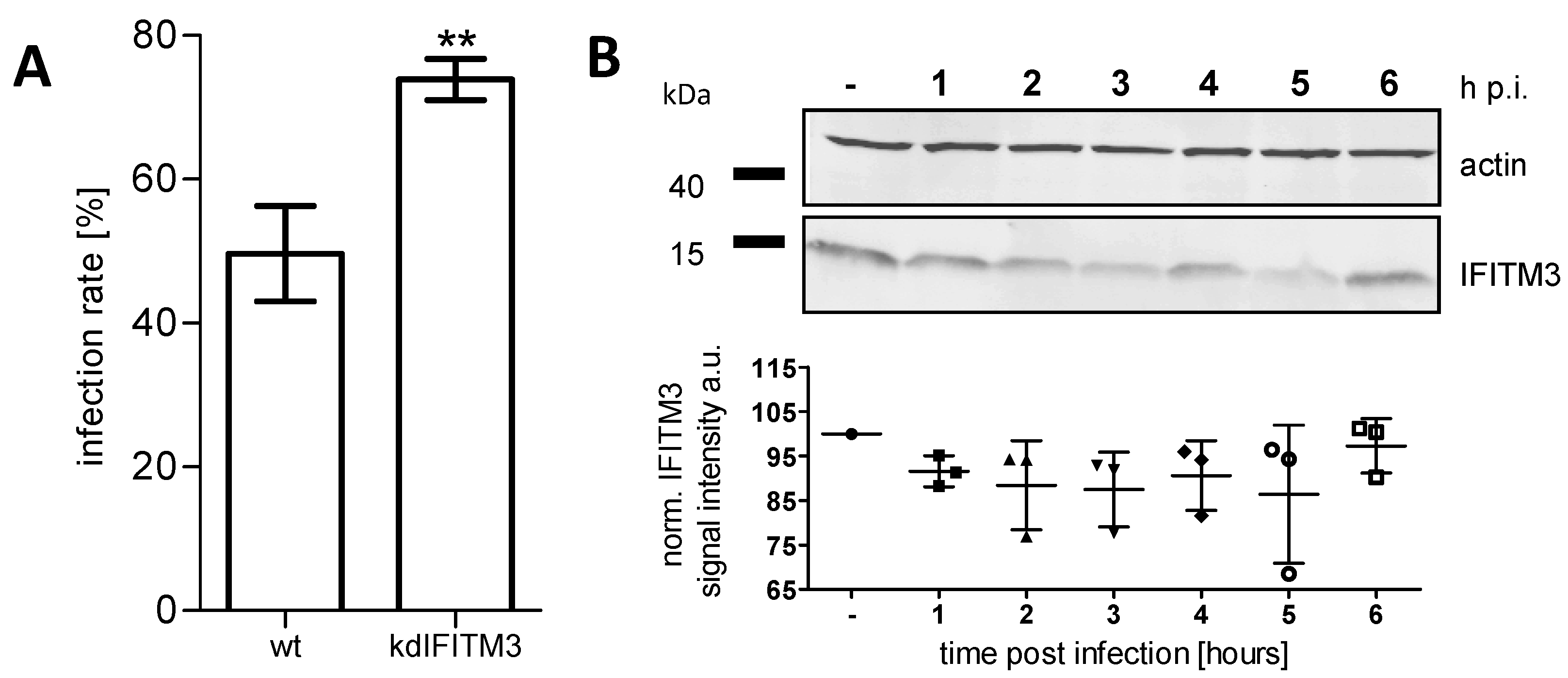
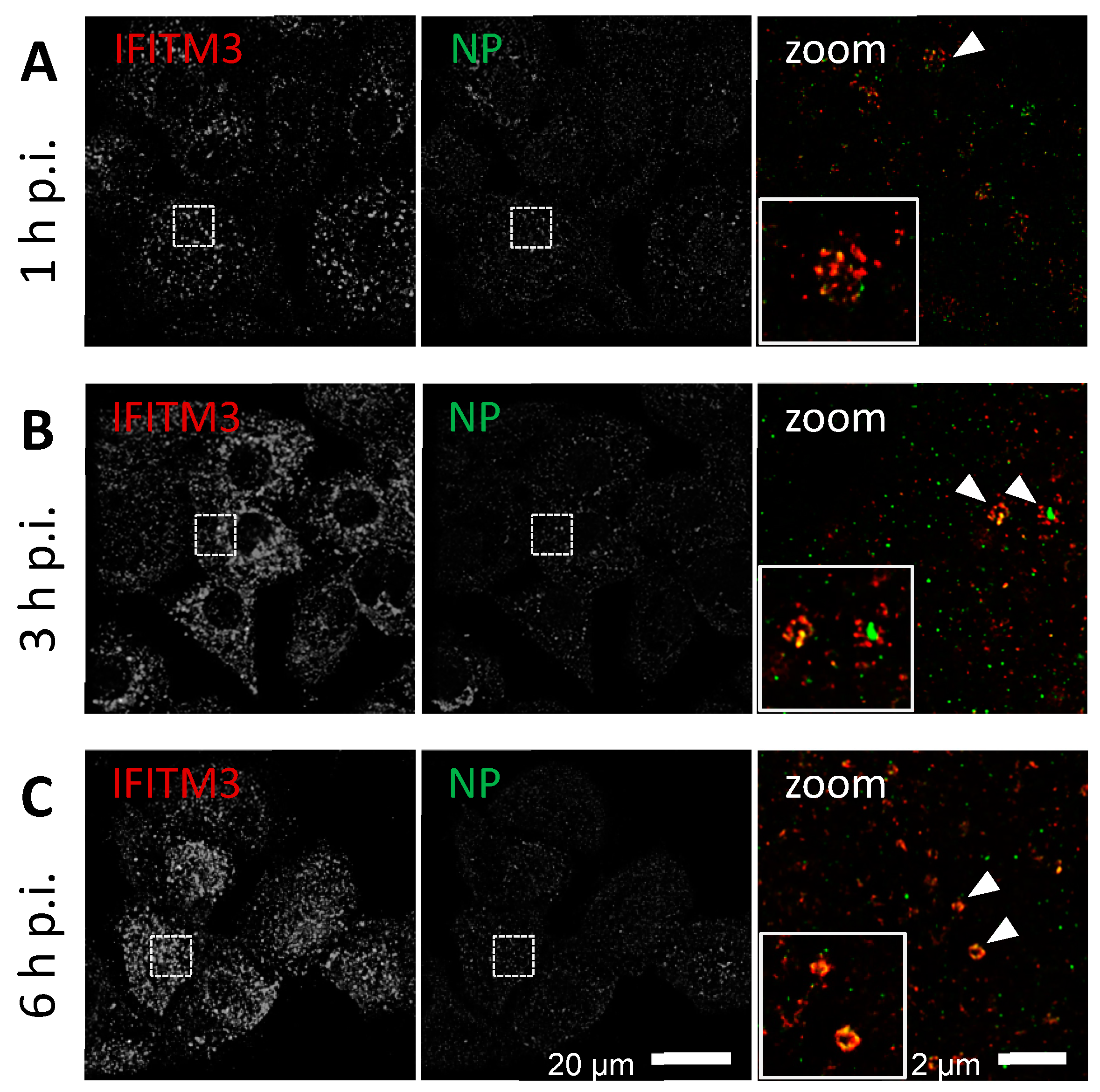
3.1. IFITM3 Is Re-Distributed upon IAV Infection
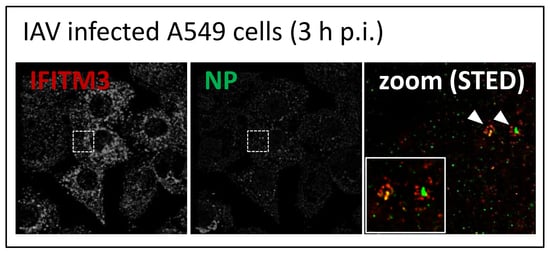
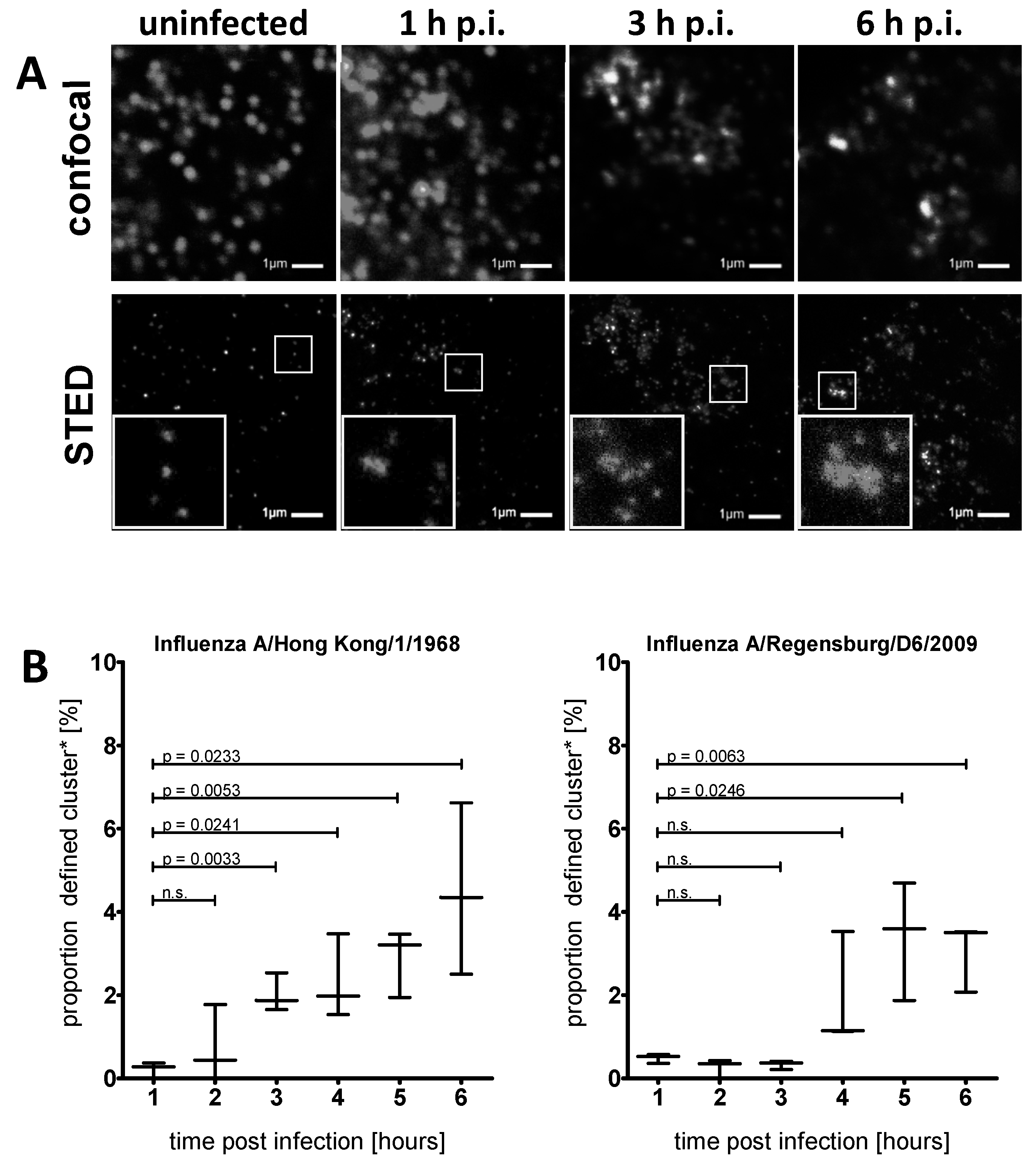
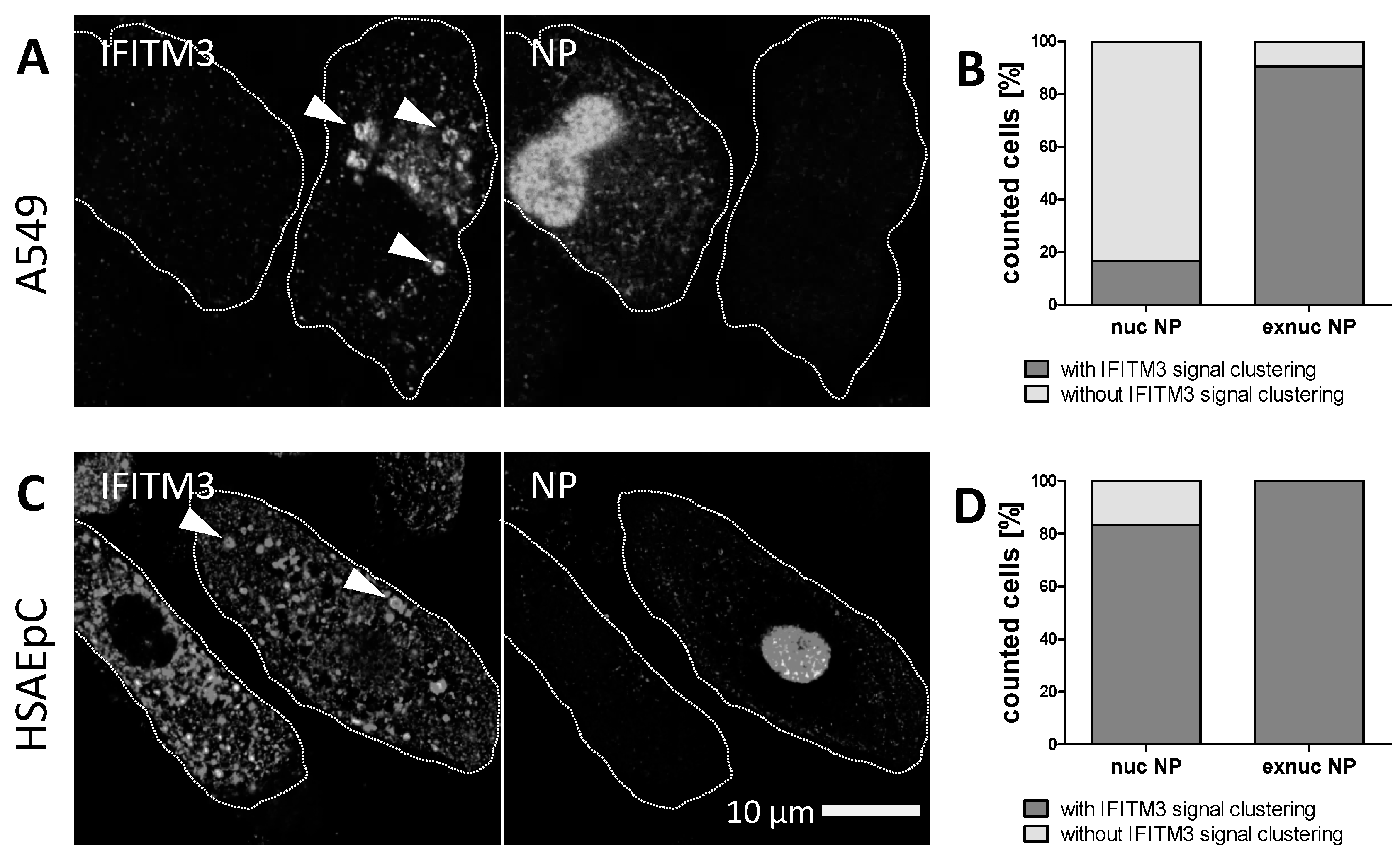
3.2. STED Analysis Reveals IFITM3 Clustering in IAV Infected A549 and Primary Human Respiratory Epithelial Cells
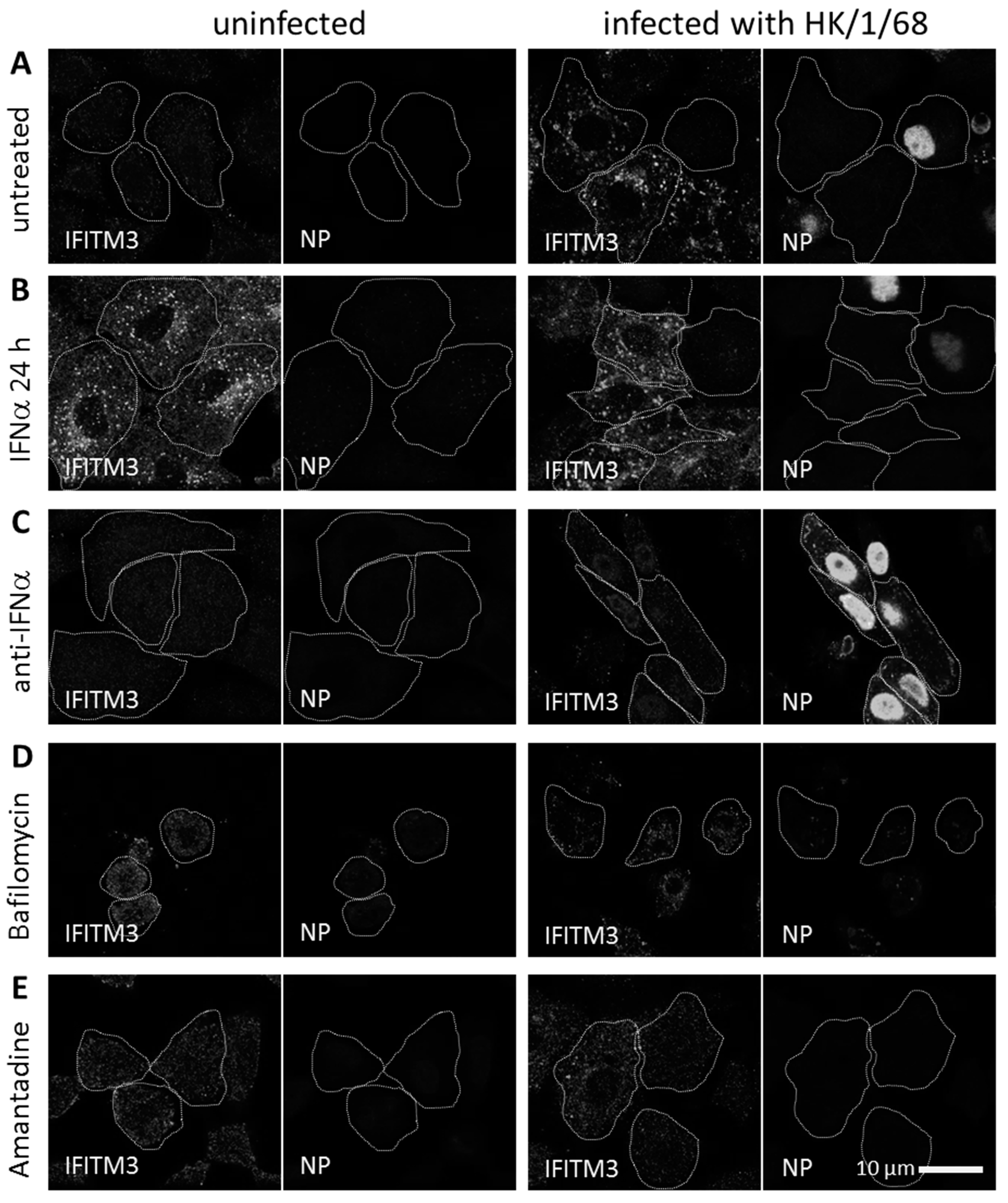
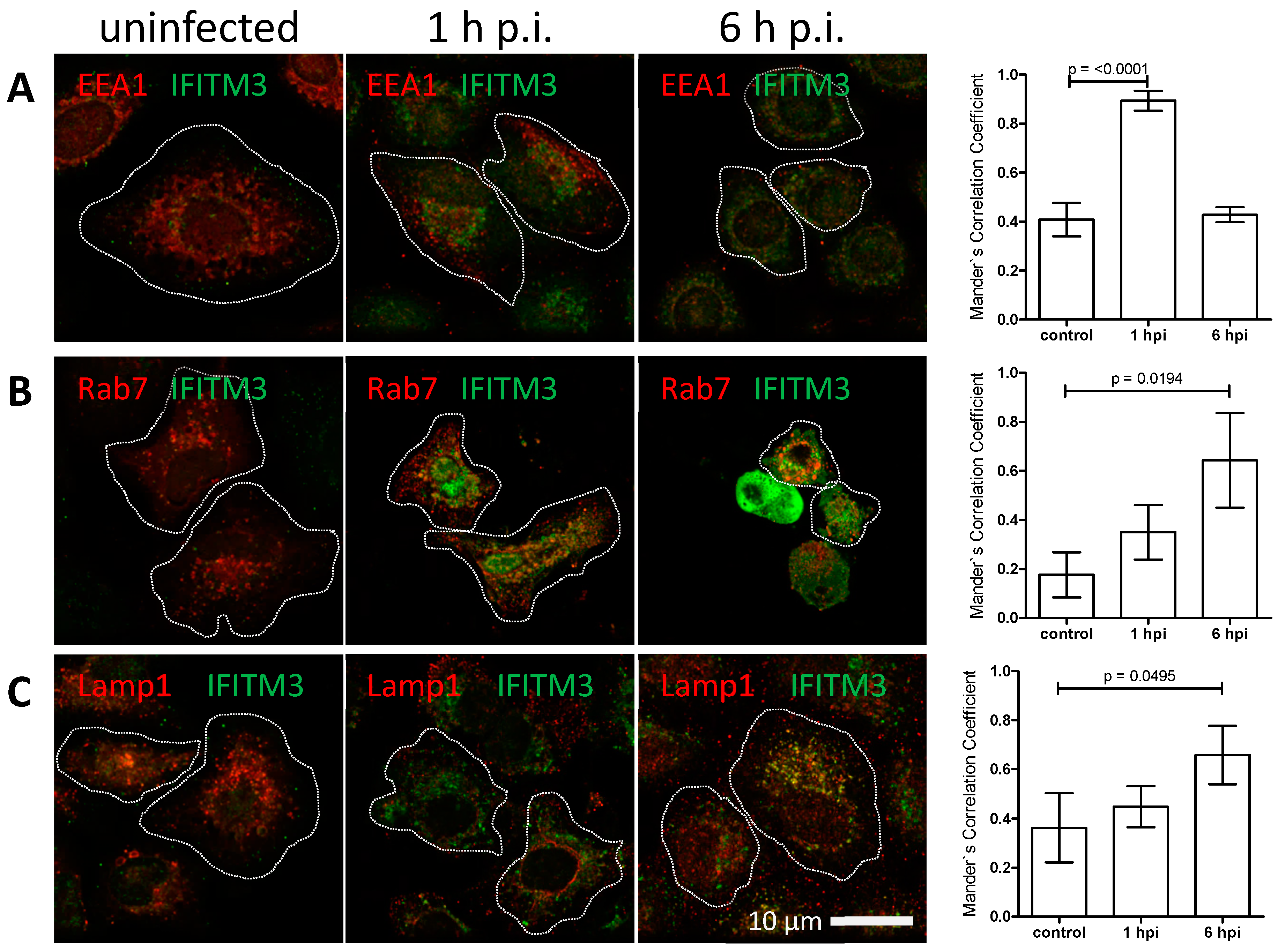
3.3. IFITM3 Is Recruited to Endosomes at Early Stages of IAV Infection
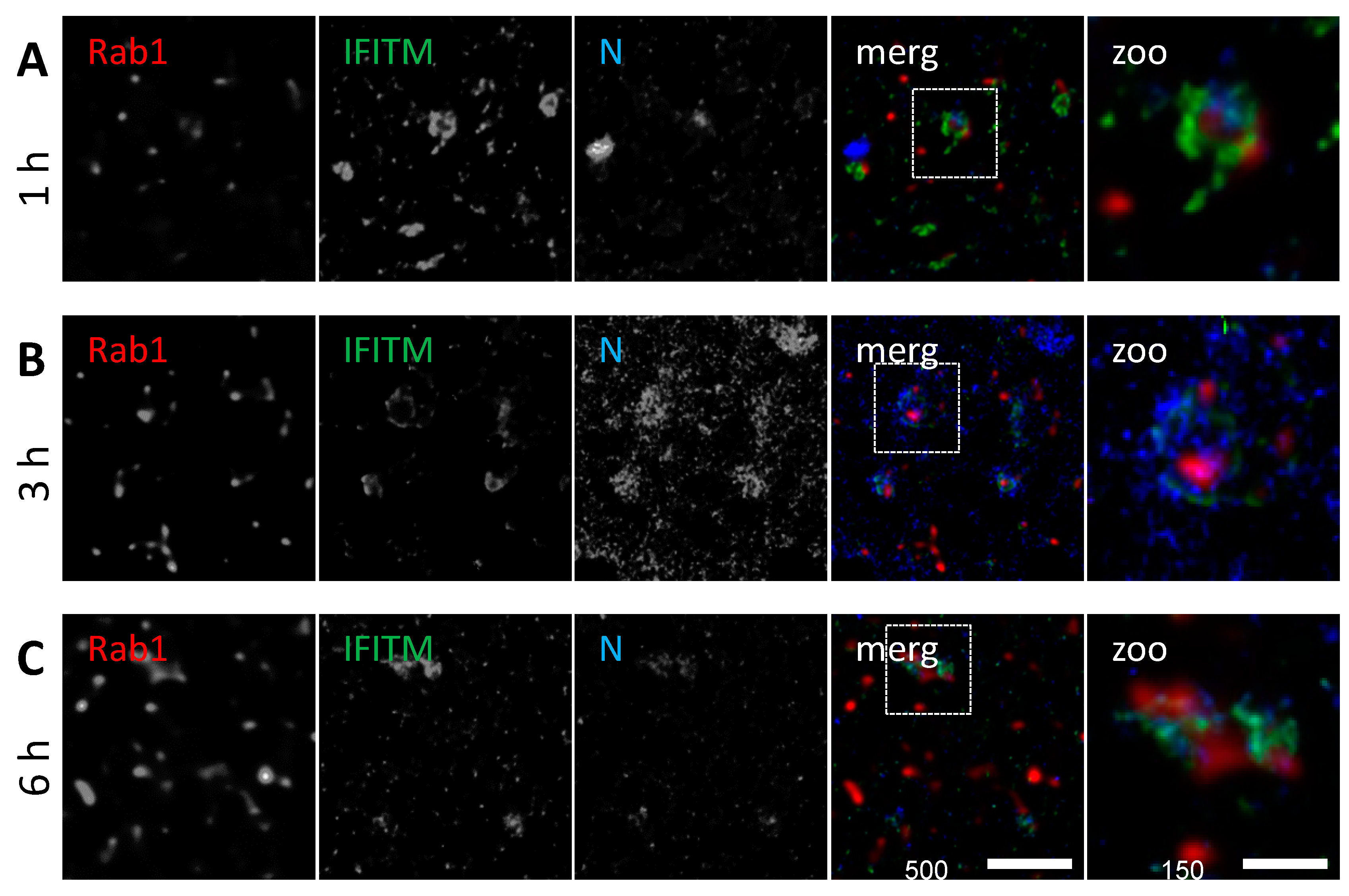
3.4. IFITM3 Is Present on IAV Containing Recycling Endosomes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Molinari, N.A.; Ortega-Sanchez, I.R.; Messonnier, M.L.; Thompson, W.W.; Wortley, P.M.; Weintraub, E.; Bridges, C.B. The annual impact of seasonal influenza in the US: Measuring disease burden and costs. Vaccine 2007, 25, 5086–5096. [Google Scholar] [CrossRef] [PubMed]
- Oxford, J.S. Influenza a pandemics of the 20th century with special reference to 1918: Virology, pathology and epidemiology. Rev. Med. Virol. 2000, 10, 119–133. [Google Scholar] [CrossRef]
- Potter, C.W. A history of influenza. J. Appl. Microbiol. 2001, 91, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Fouchier, R.A.; Munster, V.; Wallensten, A.; Bestebroer, T.M.; Herfst, S.; Smith, D.; Rimmelzwaan, G.F.; Olsen, B.; Osterhaus, A.D. Characterization of a novel influenza a virus hemagglutinin subtype (H16) obtained from black-headed gulls. J. Virol. 2005, 79, 2814–2822. [Google Scholar] [CrossRef] [PubMed]
- Gambotto, A.; Barratt-Boyes, S.M.; de Jong, M.D.; Neumann, G.; Kawaoka, Y. Human infection with highly pathogenic H5N1 influenza virus. Lancet 2008, 371, 1464–1475. [Google Scholar] [CrossRef]
- Ge, Y.; Cui, L.; Qi, X.; Shan, J.; Shan, Y.; Qi, Y.; Wu, B.; Wang, H.; Shi, Z. Detection of novel swine origin influenza A virus (H1N1) by real-time nucleic acid sequence-based amplification. J. Virol. Methods 2009, 163, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Salomon, R.; Webster, R.G. The influenza virus enigma. Cell 2009, 136, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Carroll, S.M.; Paulson, J.C. Differential infection of receptor-modified host cells by receptor-specific influenza viruses. Virus Res. 1985, 3, 165–179. [Google Scholar] [CrossRef]
- de Vries, E.; de Vries, R.P.; Wienholts, M.J.; Floris, C.E.; Jacobs, M.S.; van den Heuvel, A.; Rottier, P.J.; de Haan, C.A. Influenza a virus entry into cells lacking sialylated n-glycans. Proc. Nat. Acad. Sci. USA 2012, 109, 7457–7462. [Google Scholar] [CrossRef]
- Matlin, K.S.; Reggio, H.; Helenius, A.; Simons, K. Infectious entry pathway of influenza virus in a canine kidney cell line. J. Cell. Boil. 1981, 91, 601–613. [Google Scholar] [CrossRef]
- Edinger, T.O.; Pohl, M.O.; Stertz, S. Entry of influenza a virus: Host factors and antiviral targets. J. Gen. Virol. 2014, 95, 263–277. [Google Scholar] [CrossRef] [PubMed]
- de Vries, E.; Tscherne, D.M.; Wienholts, M.J.; Cobos-Jimenez, V.; Scholte, F.; Garcia-Sastre, A.; Rottier, P.J.; de Haan, C.A. Dissection of the influenza a virus endocytic routes reveals macropinocytosis as an alternative entry pathway. PLoS Pathog. 2011, 7, e1001329. [Google Scholar] [CrossRef] [PubMed]
- De Conto, F.; Covan, S.; Arcangeletti, M.C.; Orlandini, G.; Gatti, R.; Dettori, G.; Chezzi, C. Differential infectious entry of human influenza A/nws/33 virus (H1N1) in mammalian kidney cells. Virus Res. 2011, 155, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Sieczkarski, S.B.; Whittaker, G.R. Influenza virus can enter and infect cells in the absence of clathrin-mediated endocytosis. J. Virol. 2002, 76, 10455–10464. [Google Scholar] [CrossRef] [PubMed]
- Bouvier, N.M.; Palese, P. The biology of influenza viruses. Vaccine 2008, 26, D49–D53. [Google Scholar] [CrossRef] [PubMed]
- Skehel, J.J.; Bayley, P.M.; Brown, E.B.; Martin, S.R.; Waterfield, M.D.; White, J.M.; Wilson, I.A.; Wiley, D.C. Changes in the conformation of influenza virus hemagglutinin at the ph optimum of virus-mediated membrane fusion. Proc. Nat. Acad. Sci. USA 1982, 79, 968–972. [Google Scholar] [CrossRef]
- Steinman, R.M.; Mellman, I.S.; Muller, W.A.; Cohn, Z.A. Endocytosis and the recycling of plasma membrane. J. Cell. Boil. 1983, 96, 1–27. [Google Scholar] [CrossRef]
- Rust, M.J.; Lakadamyali, M.; Zhang, F.; Zhuang, X. Assembly of endocytic machinery around individual influenza viruses during viral entry. Nat. Struct. Mol. Boil. 2004, 11, 567–573. [Google Scholar] [CrossRef]
- Cocucci, E.; Aguet, F.; Boulant, S.; Kirchhausen, T. The first five seconds in the life of a clathrin-coated pit. Cell 2012, 150, 495–507. [Google Scholar] [CrossRef]
- Maeda, T.; Ohnishi, S. Activation of influenza virus by acidic media causes hemolysis and fusion of erythrocytes. FEBS Lett. 1980, 122, 283–287. [Google Scholar] [CrossRef]
- Daniels, R.S.; Downie, J.C.; Hay, A.J.; Knossow, M.; Skehel, J.J.; Wang, M.L.; Wiley, D.C. Fusion mutants of the influenza virus hemagglutinin glycoprotein. Cell 1985, 40, 431–439. [Google Scholar] [CrossRef]
- White, J.M.; Wilson, I.A. Anti-peptide antibodies detect steps in a protein conformational change: Low-ph activation of the influenza virus hemagglutinin. J. Cell. Boil. 1987, 105, 2887–2896. [Google Scholar] [CrossRef]
- Martin, K.; Helenius, A. Transport of incoming influenza virus nucleocapsids into the nucleus. J. Virol. 1991, 65, 232–244. [Google Scholar] [CrossRef]
- Lamb, R.A.; Krug , R.M. Orthomyxoviridae: The viruses and their replication. Fields Virol. 1996, 3, 1353–1445. [Google Scholar]
- Baudin, F.; Bach, C.; Cusack, S.; Ruigrok, R.W. Structure of influenza virus rnp. I. Influenza virus nucleoprotein melts secondary structure in panhandle RNA and exposes the bases to the solvent. EMBO J. 1994, 13, 3158–3165. [Google Scholar] [CrossRef] [PubMed]
- Jennings, P.A.; Finch, J.T.; Winter, G.; Robertson, J.S. Does the higher order structure of the influenza virus ribonucleoprotein guide sequence rearrangements in influenza viral RNA? Cell 1983, 34, 619–627. [Google Scholar] [CrossRef]
- Verhelst, J.; Parthoens, E.; Schepens, B.; Fiers, W.; Saelens, X. Interferon-inducible protein mx1 inhibits influenza virus by interfering with functional viral ribonucleoprotein complex assembly. J. Virol. 2012, 86, 13445–13455. [Google Scholar] [CrossRef]
- Karlas, A.; Machuy, N.; Shin, Y.; Pleissner, K.P.; Artarini, A.; Heuer, D.; Becker, D.; Khalil, H.; Ogilvie, L.A.; Hess, S.; et al. Genome-wide rnai screen identifies human host factors crucial for influenza virus replication. Nature 2010, 463, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Kummer, S.; Flottmann, M.; Schwanhausser, B.; Sieben, C.; Veit, M.; Selbach, M.; Klipp, E.; Herrmann, A. Alteration of protein levels during influenza virus H1N1 infection in host cells: A proteomic survey of host and virus reveals differential dynamics. PLoS ONE 2014, 9, e94257. [Google Scholar] [CrossRef]
- Coombs, K.M.; Berard, A.; Xu, W.; Krokhin, O.; Meng, X.; Cortens, J.P.; Kobasa, D.; Wilkins, J.; Brown, E.G. Quantitative proteomic analyses of influenza virus-infected cultured human lung cells. J. Virol. 2010, 84, 10888–10906. [Google Scholar] [CrossRef]
- Dove, B.K.; Surtees, R.; Bean, T.J.H.; Munday, D.; Wise, H.M.; Digard, P.; Carroll, M.W.; Ajuh, P.; Barr, J.N.; Hiscox, J.A. A quantitative proteomic analysis of lung epithelial (a549) cells infected with 2009 pandemic influenza A virus using stable isotope labelling with amino acids in cell culture. Proteomics 2012, 12, 1431–1436. [Google Scholar] [CrossRef] [PubMed]
- Brass, A.L.; Huang, I.C.; Benita, Y.; John, S.P.; Krishnan, M.N.; Feeley, E.M.; Ryan, B.J.; Weyer, J.L.; van der Weyden, L.; Fikrig, E.; et al. The ifitm proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and Dengue virus. Cell 2009, 139, 1243–1254. [Google Scholar] [CrossRef] [PubMed]
- Shapira, S.D.; Gat-Viks, I.; Shum, B.O.; Dricot, A.; de Grace, M.M.; Wu, L.; Gupta, P.B.; Hao, T.; Silver, S.J.; Root, D.E.; et al. A physical and regulatory map of host-influenza interactions reveals pathways in H1N1 infection. Cell 2009, 139, 1255–1267. [Google Scholar] [CrossRef] [PubMed]
- Brem, R.; Oraszlan-Szovik, K.; Foser, S.; Bohrmann, B.; Certa, U. Inhibition of proliferation by 1-8U in interferon-alpha-responsive and non-responsive cell lines. Cell. Mol. Life Sci 2003, 60, 1235–1248. [Google Scholar] [CrossRef] [PubMed]
- Zucchi, I.; Prinetti, A.; Scotti, M.; Valsecchi, V.; Valaperta, R.; Mento, E.; Reinbold, R.; Vezzoni, P.; Sonnino, S.; Albertini, A.; et al. Association of rat8 with fyn protein kinase via lipid rafts is required for rat mammary cell differentiation in vitro. Proc. Nat. Acad. Sci. USA 2004, 101, 1880–1885. [Google Scholar] [CrossRef]
- Huang, I.C.; Bailey, C.C.; Weyer, J.L.; Radoshitzky, S.R.; Becker, M.M.; Chiang, J.J.; Brass, A.L.; Ahmed, A.A.; Chi, X.; Dong, L.; et al. Distinct patterns of IFITM-mediated restriction of filoviruses, SARS coronavirus, and influenza A virus. PLoS Pathog. 2011, 7, e1001258. [Google Scholar] [CrossRef]
- Feeley, E.M.; Sims, J.S.; John, S.P.; Chin, C.R.; Pertel, T.; Chen, L.M.; Gaiha, G.D.; Ryan, B.J.; Donis, R.O.; Elledge, S.J.; et al. IFITM3 inhibits influenza A virus infection by preventing cytosolic entry. PLoS Pathog. 2011, 7, e1002337. [Google Scholar] [CrossRef]
- Amini-Bavil-Olyaee, S.; Choi, Y.J.; Lee, J.H.; Shi, M.; Huang, I.C.; Farzan, M.; Jung, J.U. The antiviral effector ifitm3 disrupts intracellular cholesterol homeostasis to block viral entry. Cell. Host Microbe 2013, 13, 452–464. [Google Scholar] [CrossRef]
- John, S.P.; Chin, C.R.; Perreira, J.M.; Feeley, E.M.; Aker, A.M.; Savidis, G.; Smith, S.E.; Elia, A.E.; Everitt, A.R.; Vora, M.; et al. The CD225 domain of IFITM3 is required for both ifitm protein association and inhibition of influenza A virus and dengue virus replication. J. Virol. 2013, 87, 7837–7852. [Google Scholar] [CrossRef]
- Mudhasani, R.; Tran, J.P.; Retterer, C.; Radoshitzky, S.R.; Kota, K.P.; Altamura, L.A.; Smith, J.M.; Packard, B.Z.; Kuhn, J.H.; Costantino, J.; et al. IFITM-2 and IFITM-3 but not IFITM-1 restrict rift valley fever virus. J. Virol. 2013, 87, 8451–8464. [Google Scholar] [CrossRef]
- Desai, T.M.; Marin, M.; Chin, C.R.; Savidis, G.; Brass, A.L.; Melikyan, G.B. IFITM3 restricts influenza A virus entry by blocking the formation of fusion pores following virus-endosome hemifusion. PLoS Pathog. 2014, 10, e1004048. [Google Scholar] [CrossRef] [PubMed]
- Everitt, A.R.; Clare, S.; Pertel, T.; John, S.P.; Wash, R.S.; Smith, S.E.; Chin, C.R.; Feeley, E.M.; Sims, J.S.; Adams, D.J.; et al. IFITM3 restricts the morbidity and mortality associated with influenza. Nature 2012, 484, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Randolph, A.G.; Yip, W.K.; Allen, E.K.; Rosenberger, C.M.; Agan, A.A.; Ash, S.A.; Zhang, Y.; Bhangale, T.R.; Finkelstein, D.; Cvijanovich, N.Z.; et al. Evaluation of IFITM3 RS12252 association with severe pediatric influenza infection. J. Infect. Dis. 2017, 216, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Mills, T.C.; Rautanen, A.; Elliott, K.S.; Parks, T.; Naranbhai, V.; Ieven, M.M.; Butler, C.C.; Little, P.; Verheij, T.; Garrard, C.S.; et al. IFITM3 and susceptibility to respiratory viral infections in the community. J. Infect. Dis. 2014, 209, 1028–1031. [Google Scholar] [CrossRef] [PubMed]
- Carter, T.C.; Hebbring, S.J.; Liu, J.; Mosley, J.D.; Shaffer, C.M.; Ivacic, L.C.; Kopitzke, S.; Stefanski, E.L.; Strenn, R.; Sundaram, M.E.; et al. Pilot screening study of targeted genetic polymorphisms for association with seasonal influenza hospital admission. J. Med. Virol. 2018, 90, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Rodriguez, M.; Herrera-Ramos, E.; Sole-Violan, J.; Ruiz-Hernandez, J.J.; Borderias, L.; Horcajada, J.P.; Lerma-Chippirraz, E.; Rajas, O.; Briones, M.; Perez-Gonzalez, M.C.; et al. Ifitm3 and severe influenza virus infection. No evidence of genetic association. Eur. J. Clin. Microbiol. Infect. Dis. 2016, 35, 1811–1817. [Google Scholar] [CrossRef]
- David, S.; Correia, V.; Antunes, L.; Faria, R.; Ferrao, J.; Faustino, P.; Nunes, B.; Maltez, F.; Lavinha, J.; Rebelo de Andrade, H. Population genetics of IFITM3 in portugal and Central Africa reveals a potential modifier of influenza severity. Immunogenetics 2018, 70, 169–177. [Google Scholar] [CrossRef]
- Xu-Yang, Z.; Pei-Yu, B.; Chuan-Tao, Y.; Wei, Y.; Hong-Wei, M.; Kang, T.; Chun-Mei, Z.; Ying-Feng, L.; Xin, W.; Ping-Zhong, W.; et al. Interferon-induced transmembrane protein 3 inhibits hantaan virus infection, and its single nucleotide polymorphism RS12252 influences the severity of hemorrhagic fever with renal syndrome. Front. Immunol. 2016, 7, 535. [Google Scholar] [CrossRef]
- Allen, E.K.; Randolph, A.G.; Bhangale, T.; Dogra, P.; Ohlson, M.; Oshansky, C.M.; Zamora, A.E.; Shannon, J.P.; Finkelstein, D.; Dressen, A.; et al. SNP-mediated disruption of CTCF binding at the IFITM3 promoter is associated with risk of severe influenza in humans. Nat. Med. 2017, 23, 975–983. [Google Scholar] [CrossRef]
- Yount, J.S.; Moltedo, B.; Yang, Y.Y.; Charron, G.; Moran, T.M.; Lopez, C.B.; Hang, H.C. Palmitoylome profiling reveals s-palmitoylation-dependent antiviral activity of IFITM3. Nat. Chem. Boil. 2010, 6, 610–614. [Google Scholar] [CrossRef]
- Bailey, C.C.; Zhong, G.; Huang, I.C.; Farzan, M. IFITM-family proteins: The cell’s first line of antiviral defense. Annu. Rev. Virol. 2014, 1, 261–283. [Google Scholar] [CrossRef] [PubMed]
- Perreira, J.M.; Chin, C.R.; Feeley, E.M.; Brass, A.L. IFITMS restrict the replication of multiple pathogenic viruses. J. Mol. Boil. 2013, 425, 4937–4955. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Markosyan, R.M.; Zheng, Y.M.; Golfetto, O.; Bungart, B.; Li, M.; Ding, S.; He, Y.; Liang, C.; Lee, J.C.; et al. IFITM proteins restrict viral membrane hemifusion. PLoS Pathog. 2013, 9, e1003124. [Google Scholar] [CrossRef] [PubMed]
- Bolard, J. Interaction of polyene antibiotics with membrane lipids: Physicochemical studies of the molecular basis of selectivity. Drugs Under Exp. Clin. Res. 1986, 12, 613–618. [Google Scholar]
- Rojek, J.M.; Sanchez, A.B.; Nguyen, N.T.; de la Torre, J.C.; Kunz, S. Different mechanisms of cell entry by human-pathogenic old world and new world arenaviruses. J. Virol. 2008, 82, 7677–7687. [Google Scholar] [CrossRef]
- Beer, C.; Andersen, D.S.; Rojek, A.; Pedersen, L. Caveola-dependent endocytic entry of amphotropic murine leukemia virus. J. Virol. 2005, 79, 10776–10787. [Google Scholar] [CrossRef]
- Kuhnl, A.; Musiol, A.; Heitzig, N.; Johnson, D.E.; Ehrhardt, C.; Grewal, T.; Gerke, V.; Ludwig, S.; Rescher, U. Late endosomal/lysosomal cholesterol accumulation is a host cell-protective mechanism inhibiting endosomal escape of influenza a virus. MBio 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Chesarino, N.M.; Compton, A.A.; McMichael, T.M.; Kenney, A.D.; Zhang, L.; Soewarna, V.; Davis, M.; Schwartz, O.; Yount, J.S. IFITM3 requires an amphipathic helix for antiviral activity. EMBO Rep. 2017, 18, 1740–1751. [Google Scholar] [CrossRef] [PubMed]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelson, T.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-scale crispr-cas9 knockout screening in human cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Tang, S.N.; Marsh, J.L.; Shankar, S.; Srivastava, R.K. Ellagic acid inhibits human pancreatic cancer growth in BALB C nude mice. Cancer Lett. 2013, 337, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Sobrido, L.; Garcia-Sastre, A. Generation of recombinant influenza virus from plasmid DNA. J. Vis. Exp. JoVE 2010. [Google Scholar] [CrossRef] [PubMed]
- Morrill, J.C.; Ikegami, T.; Yoshikawa-Iwata, N.; Lokugamage, N.; Won, S.; Terasaki, K.; Zamoto-Niikura, A.; Peters, C.J.; Makino, S. Rapid accumulation of virulent rift valley fever virus in mice from an attenuated virus carrying a single nucleotide substitution in the mRNA. PLoS ONE 2010, 5, e9986. [Google Scholar] [CrossRef]
- Dunn, K.W.; Kamocka, M.M.; McDonald, J.H. A practical guide to evaluating colocalization in biological microscopy. Am. J. Physiol. Cell Physiol. 2011, 300, C723–C742. [Google Scholar] [CrossRef] [PubMed]
- Gruenberg, J. The endocytic pathway: A mosaic of domains. Nat. Rev. Mol. Cell Biol. 2001, 2, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Khor, R.; McElroy, L.J.; Whittaker, G.R. The ubiquitin-vacuolar protein sorting system is selectively required during entry of influenza virus into host cells. Traffic 2003, 4, 857–868. [Google Scholar] [CrossRef]
- Takahashi, S.; Kubo, K.; Waguri, S.; Yabashi, A.; Shin, H.W.; Katoh, Y.; Nakayama, K. Rab11 regulates exocytosis of recycling vesicles at the plasma membrane. J. Cell. Sci. 2012, 125, 4049–4057. [Google Scholar] [CrossRef]
- Amorim, M.J.; Bruce, E.A.; Read, E.K.; Foeglein, A.; Mahen, R.; Stuart, A.D.; Digard, P. A RAB11- and microtubule-dependent mechanism for cytoplasmic transport of influenza a virus viral RNA. J. Virol. 2011, 85, 4143–4156. [Google Scholar] [CrossRef]
- Momose, F.; Sekimoto, T.; Ohkura, T.; Jo, S.; Kawaguchi, A.; Nagata, K.; Morikawa, Y. Apical transport of influenza A virus ribonucleoprotein requires RAB11-positive recycling endosome. PLoS ONE 2011, 6, e21123. [Google Scholar] [CrossRef]
- Vale-Costa, S.; Amorim, M.J. Clustering of rab11 vesicles in influenza a virus infected cells creates hotspots containing the 8 viral ribonucleoproteins. Small GTPases 2017, 8, 71–77. [Google Scholar] [CrossRef]
- Suddala, K.C.; Lee, C.C.; Meraner, P.; Marin, M.; Markosyan, R.M.; Desai, T.M.; Cohen, F.S.; Brass, A.L.; Melikyan, G.B. Interferon-induced transmembrane protein 3 blocks fusion of sensitive but not resistant viruses by partitioning into virus-carrying endosomes. PLoS Pathog. 2019, 15, e1007532. [Google Scholar] [CrossRef] [PubMed]
- Narayana, S.K.; Helbig, K.J.; McCartney, E.M.; Eyre, N.S.; Bull, R.A.; Eltahla, A.; Lloyd, A.R.; Beard, M.R. The interferon-induced transmembrane proteins, IFITM1, IFITM2, and IFITM3 inhibit hepatitis C virus entry. J. Boil. Chem. 2015, 290, 25946–25959. [Google Scholar] [CrossRef] [PubMed]
- Knossow, M.; Skehel, J.J. Variation and infectivity neutralization in influenza. Immunology 2006, 119, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Jia, R.; Xu, F.; Qian, J.; Yao, Y.; Miao, C.; Zheng, Y.M.; Liu, S.L.; Guo, F.; Geng, Y.; Qiao, W.; et al. Identification of an endocytic signal essential for the antiviral action of IFITM3. Cell. Microbiol 2014, 16, 1080–1093. [Google Scholar] [CrossRef] [PubMed]
- Chesarino, N.M.; McMichael, T.M.; Hach, J.C.; Yount, J.S. Phosphorylation of the antiviral protein interferon-inducible transmembrane protein 3 (IFITM3) dually regulates its endocytosis and ubiquitination. J. Boil. Chem. 2014, 289, 11986–11992. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.F.; Chen, L.; Luo, J.; He, H.X. KLF5 is involved in regulation of IFITM1, 2, and 3 genes during H5N1 virus infection in A549 cells. Cell. Mol. Boil. 2016, 62, 65–70. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Spence, J.S.; He, R.; Hoffmann, H.H.; Das, T.; Thinon, E.; Rice, C.M.; Peng, T.; Chandran, K.; Hang, H.C. IFITM3 directly engages and shuttles incoming virus particles to lysosomes. Nat. Chem. Boil. 2019, 15, 259–268. [Google Scholar] [CrossRef]
- Taguchi, T. Emerging roles of recycling endosomes. J. Biochem. 2013, 153, 505–510. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kummer, S.; Avinoam, O.; Kräusslich, H.-G. IFITM3 Clusters on Virus Containing Endosomes and Lysosomes Early in the Influenza A Infection of Human Airway Epithelial Cells. Viruses 2019, 11, 548. https://doi.org/10.3390/v11060548
Kummer S, Avinoam O, Kräusslich H-G. IFITM3 Clusters on Virus Containing Endosomes and Lysosomes Early in the Influenza A Infection of Human Airway Epithelial Cells. Viruses. 2019; 11(6):548. https://doi.org/10.3390/v11060548
Chicago/Turabian StyleKummer, Susann, Ori Avinoam, and Hans-Georg Kräusslich. 2019. "IFITM3 Clusters on Virus Containing Endosomes and Lysosomes Early in the Influenza A Infection of Human Airway Epithelial Cells" Viruses 11, no. 6: 548. https://doi.org/10.3390/v11060548
APA StyleKummer, S., Avinoam, O., & Kräusslich, H.-G. (2019). IFITM3 Clusters on Virus Containing Endosomes and Lysosomes Early in the Influenza A Infection of Human Airway Epithelial Cells. Viruses, 11(6), 548. https://doi.org/10.3390/v11060548