HIV-1-Based Virus-like Particles that Morphologically Resemble Mature, Infectious HIV-1 Virions



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells, Enzymes, Oligonucleotides and Chemicals
2.2. Monoclonal and Polyclonal Antibodies
2.3. Recombinant gp120 and p24
2.4. Plasmids
2.5. Production of Viral Particles
2.6. Rate Velocity Centrifugation
2.7. Transmission Electron Microscopy
2.8. Western Blotting
2.9. Enzyme-Linked Immunosorbent Assays (ELISAs)
2.9.1. p24 ELISA
2.9.2. VLP ELISA
2.10. Reverse Transcriptase Assay
2.11. Virus Fusion Assay
3. Results
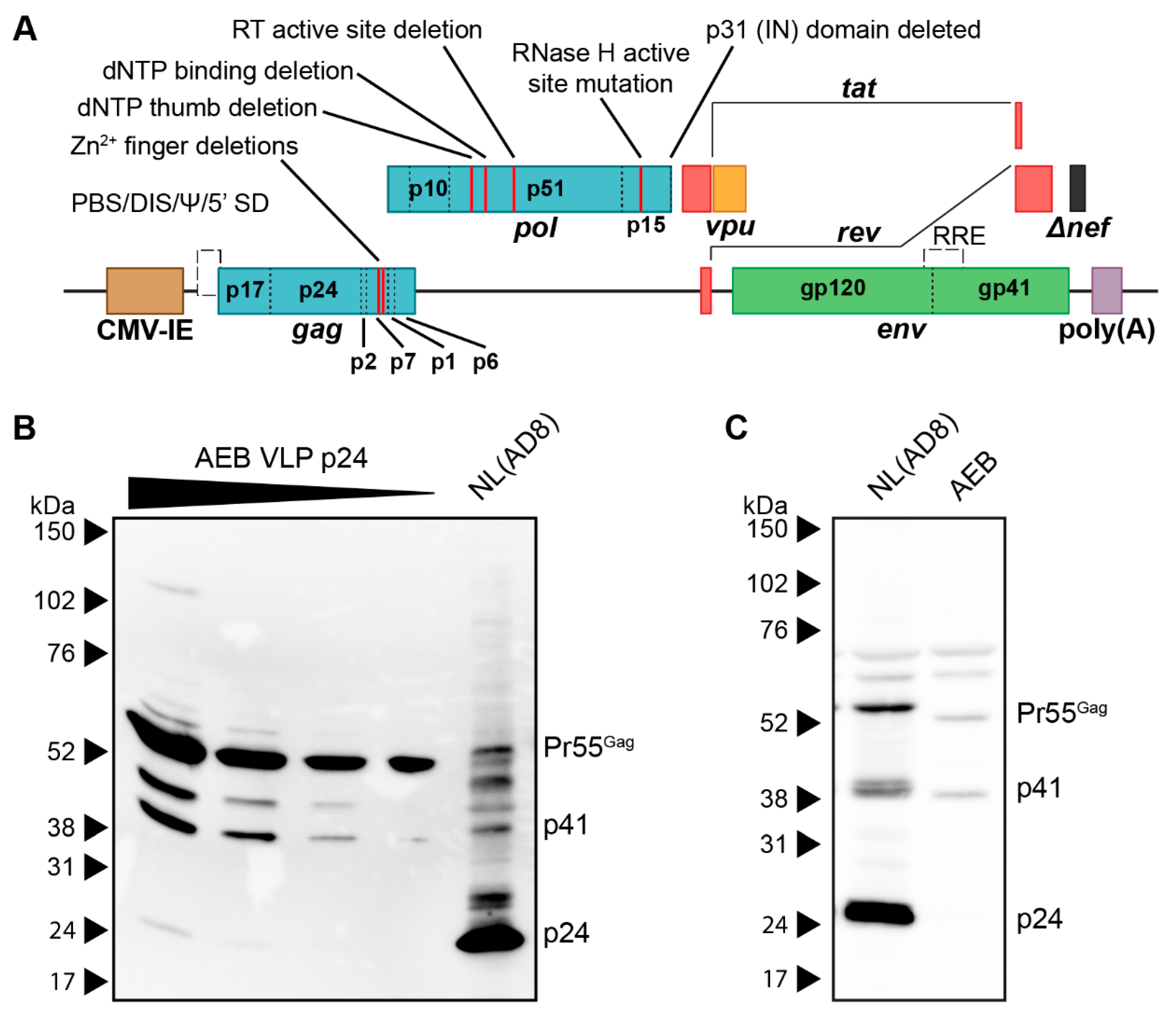
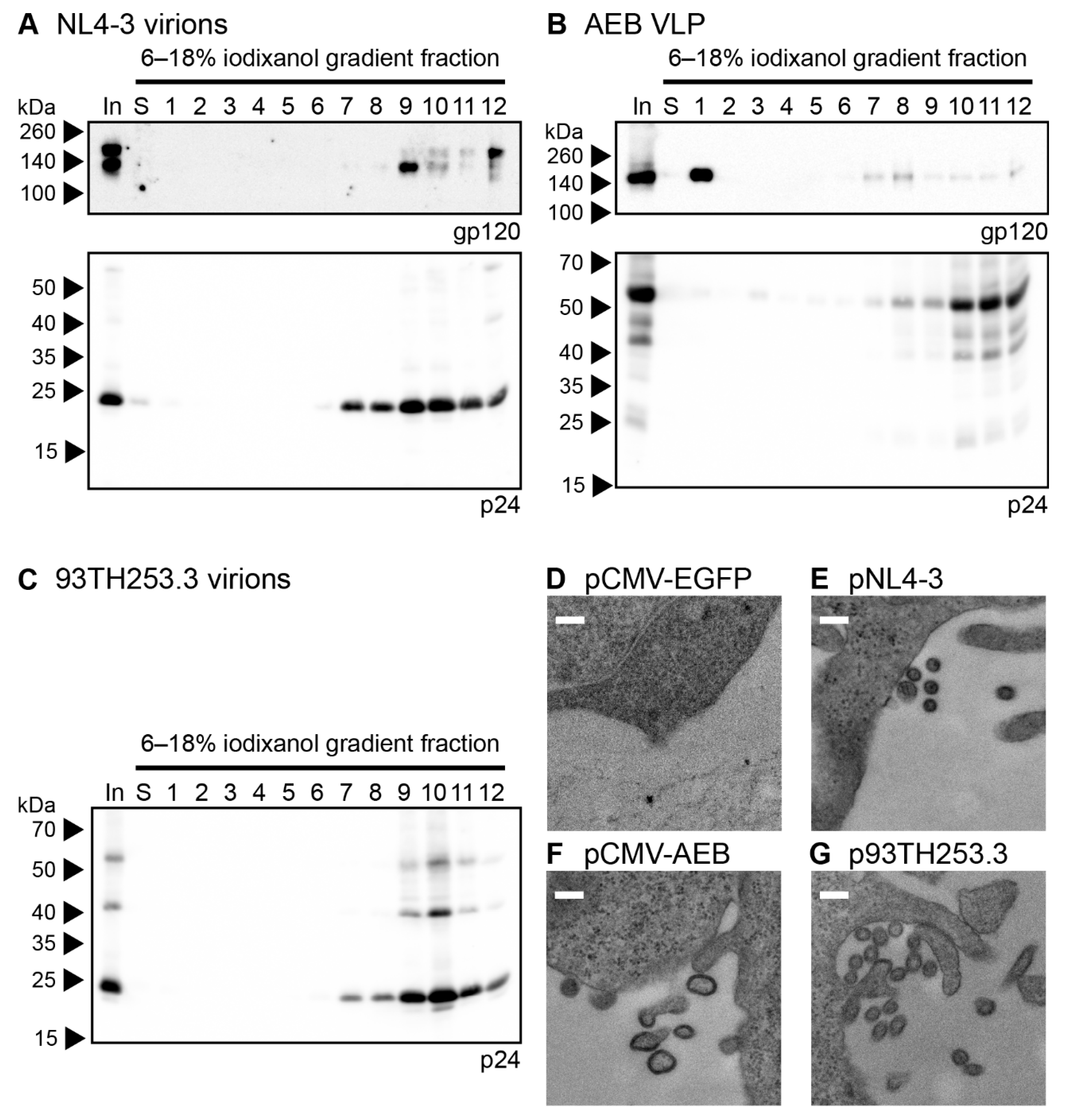
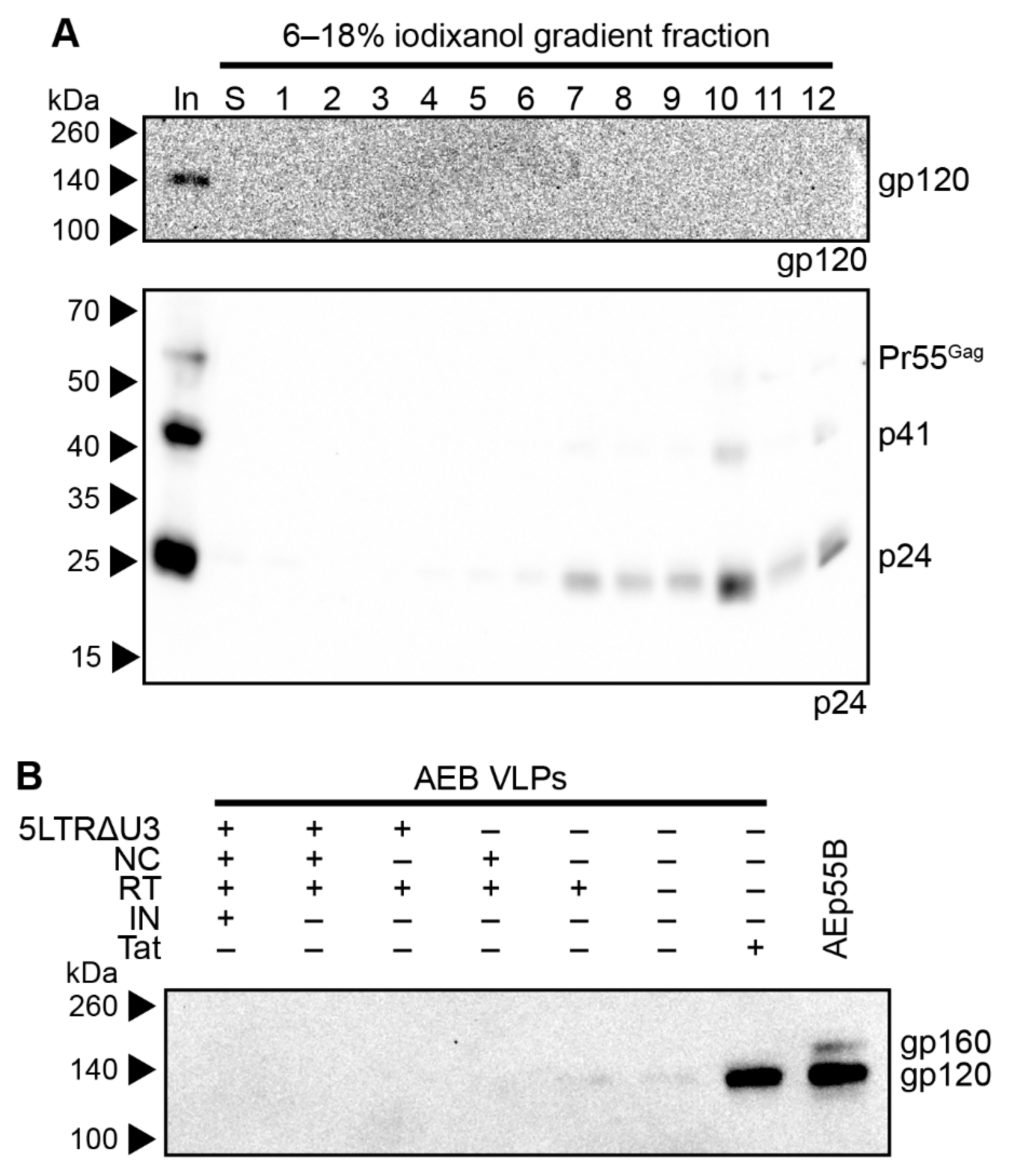
3.1. HIV-1 AEB VLPs Are Mostly Immature and Irregularly Sized
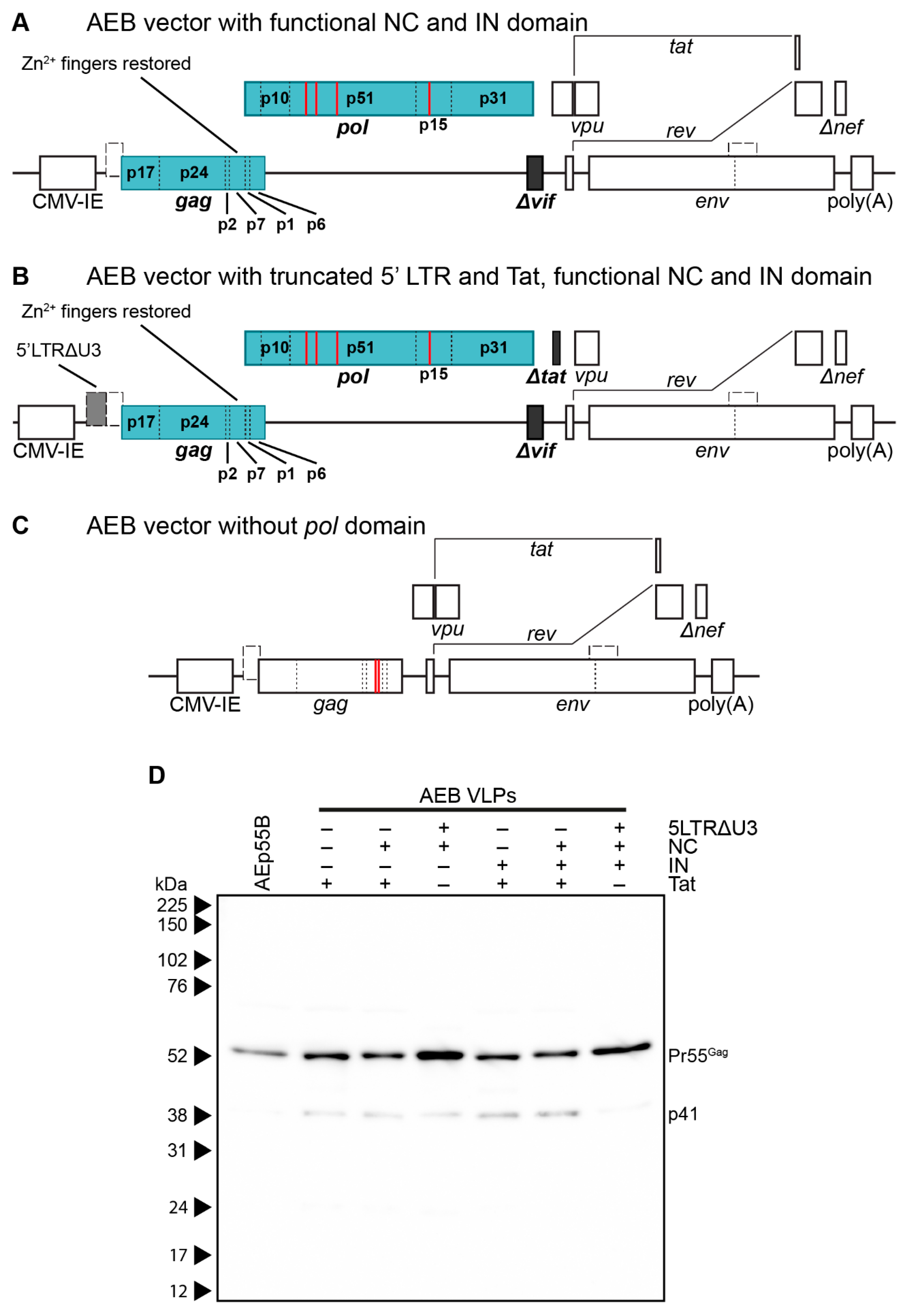
3.2. Restoration of RNA Stem Loop Motifs and RNA-Interacting Protein Domains Does Not Enhance Particle Assembly and Maturation
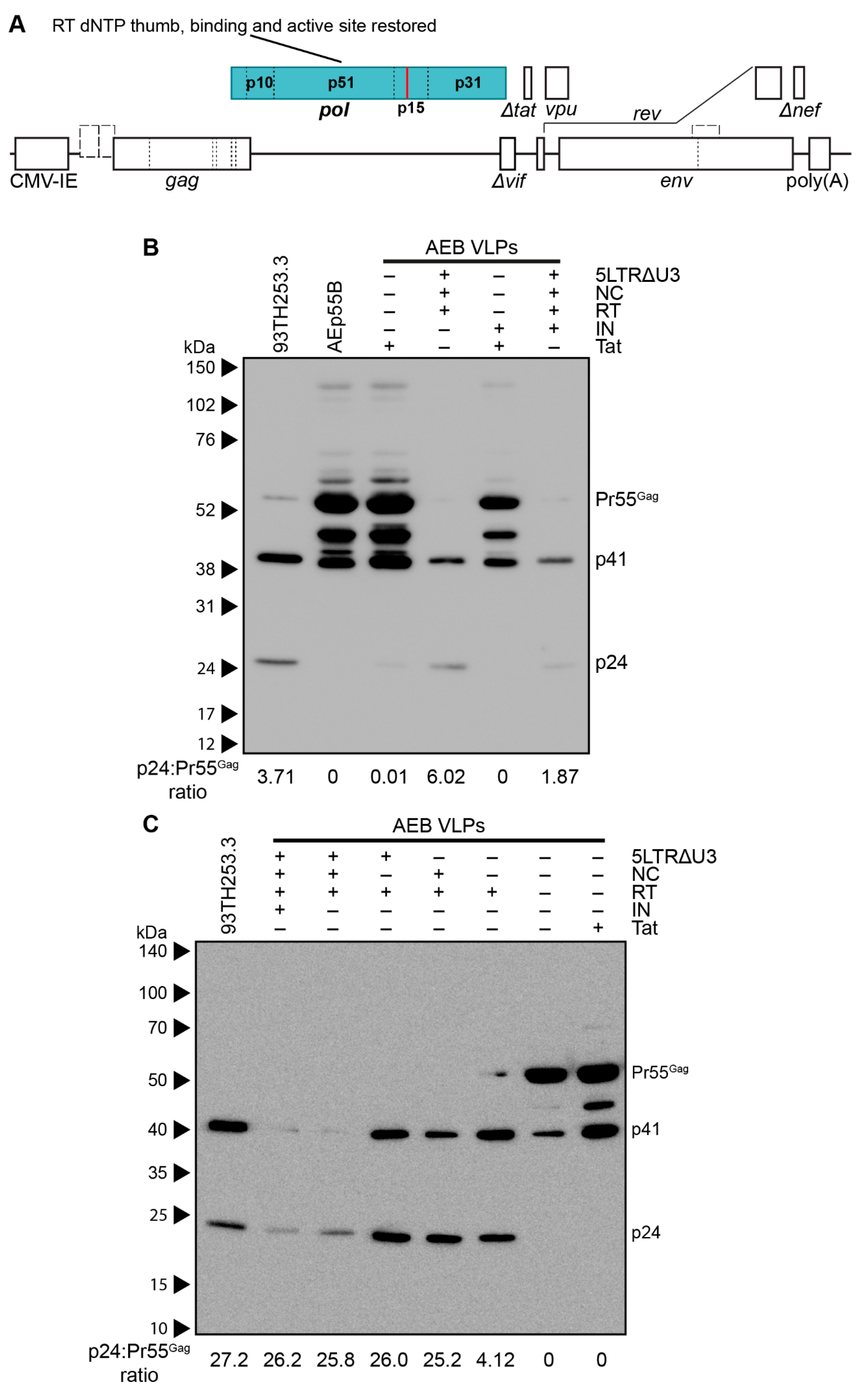
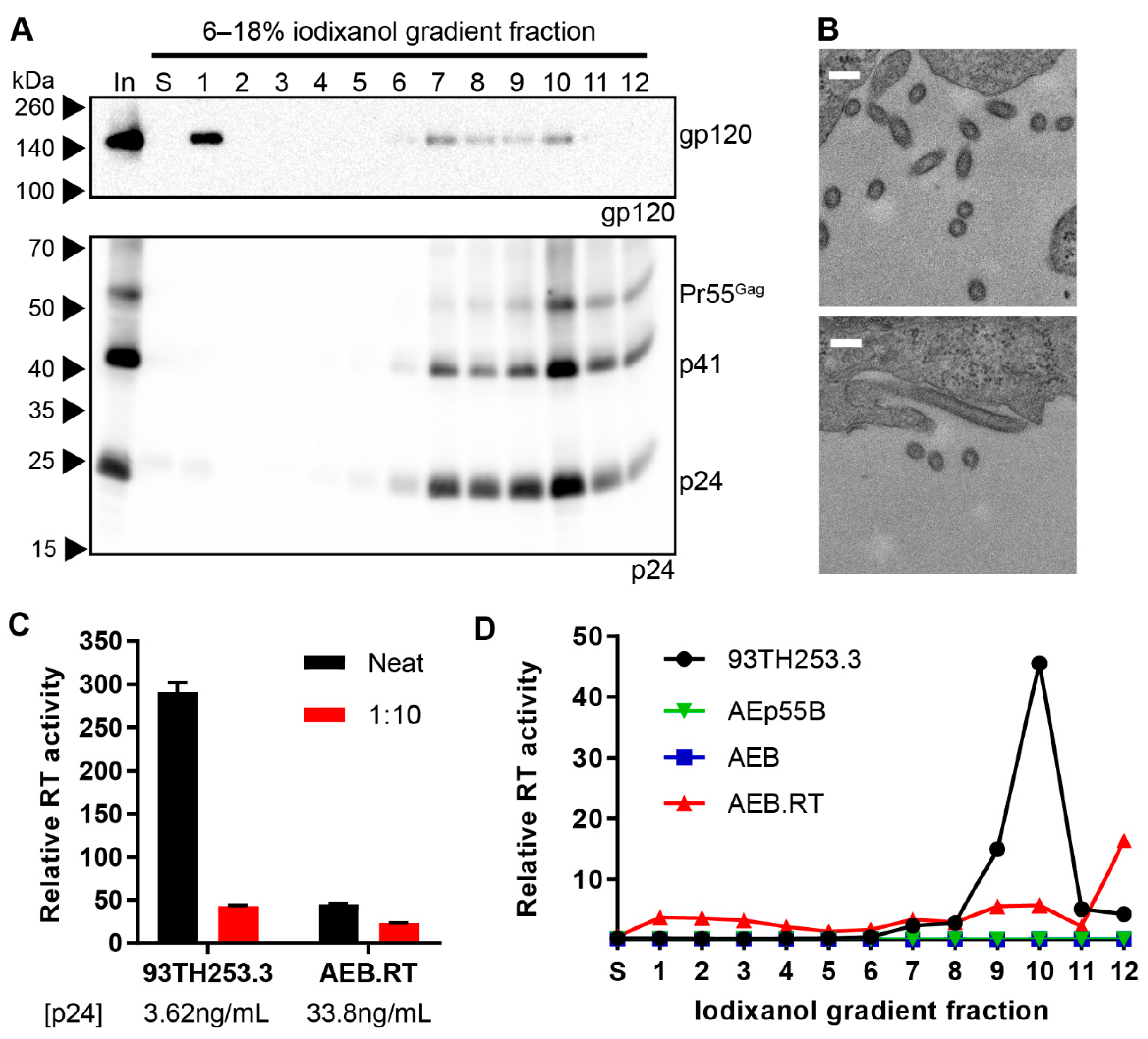
3.3. Amino Acid Deletions within the RT Domain Block Particle Assembly and Maturation
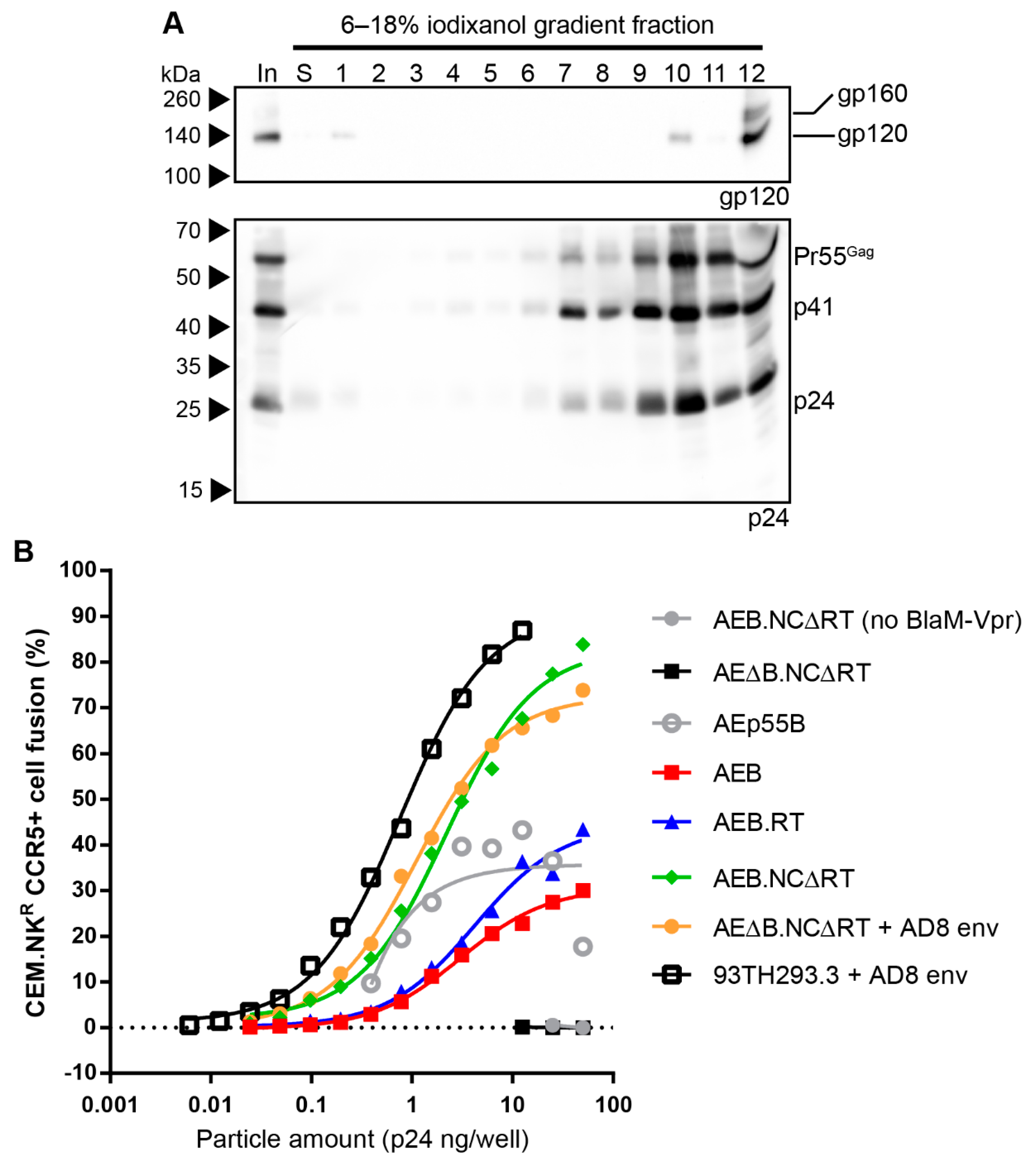
3.4. Removal of the Entire RT Domain Facilitates Expression of VLPs Resembling Mature Virions
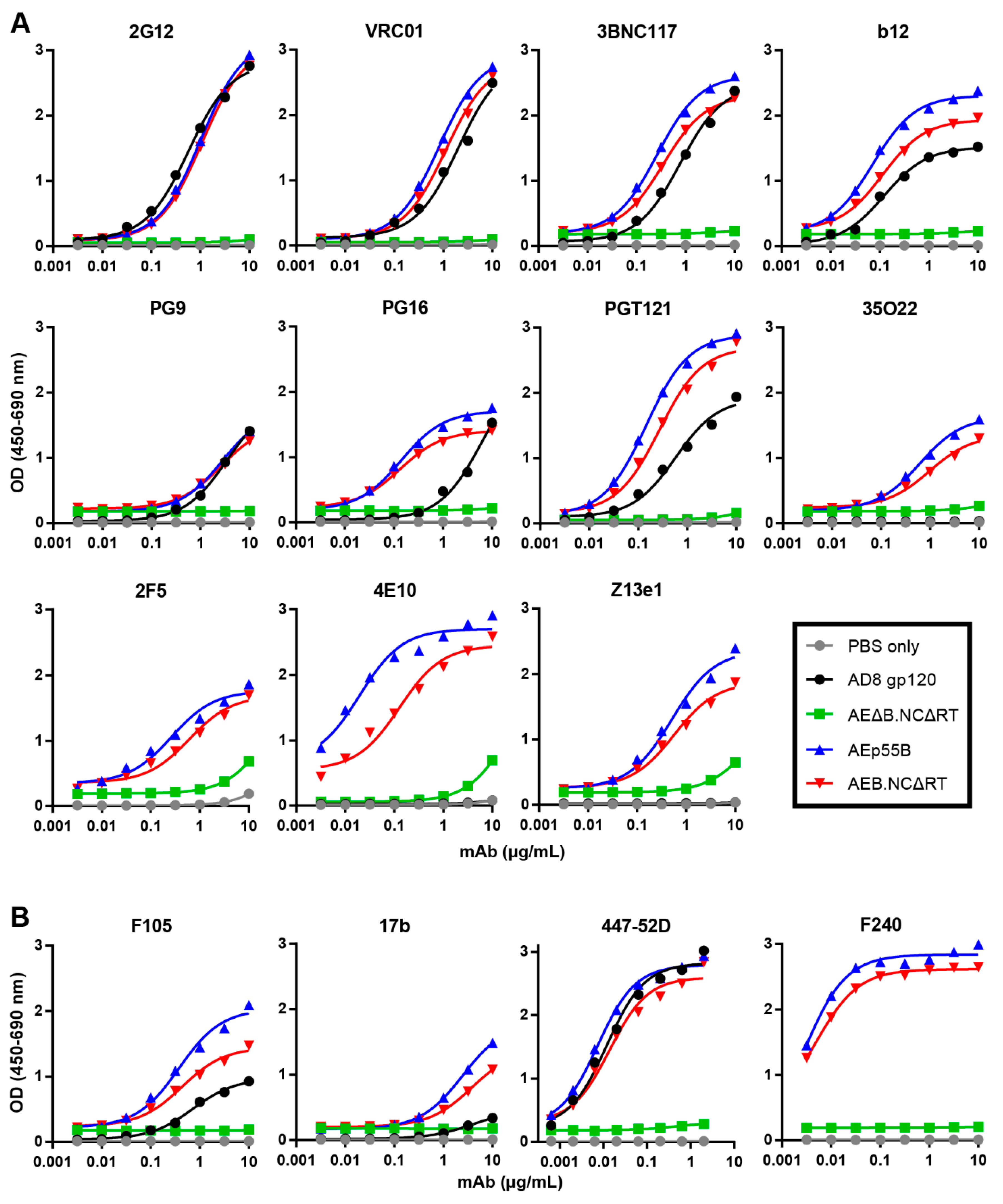
3.5. Mature-form VLPs Can Present Anti-Env Antibody Epitopes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Celum, C.; Baeten, J.M. Antiretroviral-based HIV-1 prevention: Antiretroviral treatment and pre-exposure prophylaxis. Antivir. Ther. 2012, 17, 1483–1493. [Google Scholar] [CrossRef] [PubMed]
- Lynch, R.M.; Shen, T.; Gnanakaran, S.; Derdeyn, C.A. Appreciating HIV type 1 diversity: Subtype differences in Env. AIDS Res. Hum. Retrovir. 2009, 25, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, M.; Potz, J.; Basiripour, L.; Dorfman, T.; Goh, W.C.; Terwilliger, E.; Dayton, A.; Rosen, C.; Haseltine, W.; Sodroski, J. Functional regions of the envelope glycoprotein of human immunodeficiency virus type 1. Science 1987, 237, 1351–1355. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Blacklow, S.C.; Kim, P.S. A trimeric structural domain of the HIV-1 transmembrane glycoprotein. Nat. Struct. Biol. 1995, 2, 1075–1082. [Google Scholar] [CrossRef] [PubMed]
- Flynn, N.M.; Forthal, D.N.; Harro, C.D.; Judson, F.N.; Mayer, K.H.; Para, M.F.; rgp120 HIV Vaccine Study Group. Placebo-controlled phase 3 trial of a recombinant glycoprotein 120 vaccine to prevent HIV-1 infection. J. Infect. Dis. 2005, 191, 654–665. [Google Scholar] [PubMed]
- Berman, P.W.; Murthy, K.K.; Wrin, T.; Vennari, J.C.; Cobb, E.K.; Eastman, D.J.; Champe, M.; Nakamura, G.R.; Davison, D.; Powell, M.F.; et al. Protection of MN-rgp120-immunized chimpanzees from heterologous infection with a primary isolate of human immunodeficiency virus type 1. J. Infect. Dis. 1996, 173, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Rerks-Ngarm, S.; Pitisuttithum, P.; Nitayaphan, S.; Kaewkungwal, J.; Chiu, J.; Paris, R.; Premsri, N.; Namwat, C.; de Souza, M.; Adams, E.; et al. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N. Engl. J. Med. 2009, 361, 2209–2220. [Google Scholar] [CrossRef]
- Earl, P.L.; Broder, C.C.; Long, D.; Lee, S.A.; Peterson, J.; Chakrabarti, S.; Doms, R.W.; Moss, B. Native oligomeric human immunodeficiency virus type 1 envelope glycoprotein elicits diverse monoclonal antibody reactivities. J. Virol. 1994, 68, 3015–3026. [Google Scholar]
- Center, R.J.; Lebowitz, J.; Leapman, R.D.; Moss, B. Promoting trimerization of soluble human immunodeficiency virus type 1 (HIV-1) Env through the use of HIV-1/simian immunodeficiency virus chimeras. J. Virol. 2004, 78, 2265–2276. [Google Scholar] [CrossRef]
- Binley, J.M.; Sanders, R.W.; Clas, B.; Schuelke, N.; Master, A.; Guo, Y.; Kajumo, F.; Anselma, D.J.; Maddon, P.J.; Olson, W.C.; et al. A recombinant human immunodeficiency virus type 1 envelope glycoprotein complex stabilized by an intermolecular disulfide bond between the gp120 and gp41 subunits is an antigenic mimic of the trimeric virion-associated structure. J. Virol. 2000, 74, 627–643. [Google Scholar] [CrossRef]
- Sanders, R.W.; Vesanen, M.; Schuelke, N.; Master, A.; Schiffner, L.; Kalyanaraman, R.; Paluch, M.; Berkhout, B.; Maddon, P.J.; Olson, W.C.; et al. Stabilization of the soluble, cleaved, trimeric form of the envelope glycoprotein complex of human immunodeficiency virus type 1. J. Virol. 2002, 76, 8875–8889. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, J.M.; Nkolola, J.P.; Peng, H.; Cheung, A.; Perry, J.; Miller, C.A.; Seaman, M.S.; Barouch, D.H.; Chen, B. HIV-1 envelope trimer elicits more potent neutralizing antibody responses than monomeric gp120. Proc. Natl. Acad. Sci. USA 2012, 109, 12111–12116. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Svehla, K.; Mathy, N.L.; Voss, G.; Mascola, J.R.; Wyatt, R. Characterization of antibody responses elicited by human immunodeficiency virus type 1 primary isolate trimeric and monomeric envelope glycoproteins in selected adjuvants. J. Virol. 2006, 80, 1414–1426. [Google Scholar] [CrossRef] [PubMed]
- Beddows, S.; Franti, M.; Dey, A.K.; Kirschner, M.; Iyer, S.P.; Fisch, D.C.; Ketas, T.; Yuste, E.; Desrosiers, R.C.; Klasse, P.J.; et al. A comparative immunogenicity study in rabbits of disulfide-stabilized, proteolytically cleaved, soluble trimeric human immunodeficiency virus type 1 gp140, trimeric cleavage-defective gp140 and monomeric gp120. Virology 2007, 360, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Sanders, R.W.; van Gils, M.J.; Derking, R.; Sok, D.; Ketas, T.J.; Burger, J.A.; Ozorowski, G.; Cupo, A.; Simonich, C.; Goo, L.; et al. HIV-1 neutralizing antibodies induced by native-like envelope trimers. Science 2015, 349, aac4223. [Google Scholar] [CrossRef] [PubMed]
- Tong, T.; Crooks, E.T.; Osawa, K.; Robinson, J.E.; Barnes, M.; Apetrei, C.; Binley, J.M. Multi-parameter exploration of HIV-1 virus-like particles as neutralizing antibody immunogens in guinea pigs, rabbits and macaques. Virology 2014, 456–457, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Buonaguro, L.; Tagliamonte, M.; Visciano, M.L.; Andersen, H.; Lewis, M.; Pal, R.; Tornesello, M.L.; Schroeder, U.; Hinkula, J.; Wahren, B.; et al. Immunogenicity of HIV virus-like particles in rhesus macaques by intranasal administration. Clin. Vaccine Immunol. 2012, 19, 970–973. [Google Scholar] [CrossRef]
- Nguyen, H.T.; Madani, N.; Ding, H.; Elder, E.; Princiotto, A.; Gu, C.; Darby, P.; Alin, J.; Herschhorn, A.; Kappes, J.C.; et al. Evaluation of the contribution of the transmembrane region to the ectodomain conformation of the human immunodeficiency virus (HIV-1) envelope glycoprotein. Virol. J. 2017, 14, 33. [Google Scholar] [CrossRef] [PubMed]
- Manolova, V.; Flace, A.; Bauer, M.; Schwarz, K.; Saudan, P.; Bachmann, M.F. Nanoparticles target distinct dendritic cell populations according to their size. Eur. J. Immunol. 2008, 38, 1404–1413. [Google Scholar] [CrossRef] [PubMed]
- Buonaguro, L.; Buonaguro, F.M.; Tornesello, M.L.; Mantas, D.; Beth-Giraldo, E.; Wagner, R.; Michelson, S.; Prevost, M.C.; Wolf, H.; Giraldo, G. High efficient production of Pr55(gag) virus-like particles expressing multiple HIV-1 epitopes, including a gp120 protein derived from an Ugandan HIV-1 isolate of subtype A. Antivir. Res. 2001, 49, 35–47. [Google Scholar] [CrossRef]
- Guo, L.; Lu, X.; Kang, S.M.; Chen, C.; Compans, R.W.; Yao, Q. Enhancement of mucosal immune responses by chimeric influenza HA/SHIV virus-like particles. Virology 2003, 313, 502–513. [Google Scholar] [CrossRef]
- Wang, B.Z.; Liu, W.; Kang, S.M.; Alam, M.; Huang, C.; Ye, L.; Sun, Y.; Li, Y.; Kothe, D.L.; Pushko, P.; et al. Incorporation of high levels of chimeric human immunodeficiency virus envelope glycoproteins into virus-like particles. J. Virol. 2007, 81, 10869–10878. [Google Scholar] [CrossRef] [PubMed]
- Valley-Omar, Z.; Meyers, A.E.; Shephard, E.G.; Williamson, A.L.; Rybicki, E.P. Abrogation of contaminating RNA activity in HIV-1 Gag VLPs. Virol. J. 2011, 8, 462. [Google Scholar] [CrossRef] [PubMed]
- Debouck, C.; Gorniak, J.G.; Strickler, J.E.; Meek, T.D.; Metcalf, B.W.; Rosenberg, M. Human immunodeficiency virus protease expressed in Escherichia coli exhibits autoprocessing and specific maturation of the gag precursor. Proc. Natl. Acad. Sci. USA 1987, 84, 8903–8906. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Ho, B.K.; Chang, T.W.; Chang, N.T. Role of human immunodeficiency virus type 1-specific protease in core protein maturation and viral infectivity. J. Virol. 1989, 63, 2550–2556. [Google Scholar] [PubMed]
- Chojnacki, J.; Staudt, T.; Glass, B.; Bingen, P.; Engelhardt, J.; Anders, M.; Schneider, J.; Muller, B.; Hell, S.W.; Krausslich, H.G. Maturation-dependent HIV-1 surface protein redistribution revealed by fluorescence nanoscopy. Science 2012, 338, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Chertova, E.; Bess, J., Jr.; Lifson, J.D.; Arthur, L.O.; Liu, J.; Taylor, K.A.; Roux, K.H. Electron tomography analysis of envelope glycoprotein trimers on HIV and simian immunodeficiency virus virions. Proc. Natl. Acad. Sci. USA 2003, 100, 15812–15817. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Fernandez, C.; Crespo Guardo, A.; Garcia-Perez, J.; Garcia, F.; Blanco, J.; Escriba-Garcia, L.; Gatell, J.M.; Alcami, J.; Plana, M.; Sanchez-Palomino, S. Generation and characterization of a defective HIV-1 Virus as an immunogen for a therapeutic vaccine. PLoS ONE 2012, 7, e48848. [Google Scholar] [CrossRef] [PubMed]
- Crooks, E.T.; Tong, T.; Chakrabarti, B.; Narayan, K.; Georgiev, I.S.; Menis, S.; Huang, X.; Kulp, D.; Osawa, K.; Muranaka, J.; et al. Vaccine-Elicited Tier 2 HIV-1 Neutralizing Antibodies Bind to Quaternary Epitopes Involving Glycan-Deficient Patches Proximal to the CD4 Binding Site. PLoS Pathog. 2015, 11, e1004932. [Google Scholar] [CrossRef]
- Rossio, J.L.; Esser, M.T.; Suryanarayana, K.; Schneider, D.K.; Bess, J.W., Jr.; Vasquez, G.M.; Wiltrout, T.A.; Chertova, E.; Grimes, M.K.; Sattentau, Q.; et al. Inactivation of human immunodeficiency virus type 1 infectivity with preservation of conformational and functional integrity of virion surface proteins. J. Virol. 1998, 72, 7992–8001. [Google Scholar]
- Bilska, M.; Tang, H.; Montefiori, D.C. Short Communication: Potential Risk of Replication-Competent Virus in HIV-1 Env-Pseudotyped Virus Preparations. AIDS Res. Hum. Retrovir. 2017, 33, 368–372. [Google Scholar] [CrossRef] [PubMed]
- Steppert, P.; Burgstaller, D.; Klausberger, M.; Kramberger, P.; Tover, A.; Berger, E.; Nobauer, K.; Razzazi-Fazeli, E.; Jungbauer, A. Separation of HIV-1 gag virus-like particles from vesicular particles impurities by hydroxyl-functionalized monoliths. J. Sep. Sci. 2017, 40, 979–990. [Google Scholar] [CrossRef] [PubMed]
- Steppert, P.; Burgstaller, D.; Klausberger, M.; Berger, E.; Aguilar, P.P.; Schneider, T.A.; Kramberger, P.; Tover, A.; Nobauer, K.; Razzazi-Fazeli, E.; et al. Purification of HIV-1 gag virus-like particles and separation of other extracellular particles. J. Chromatogr. A 2016, 1455, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Effio, C.L.; Hubbuch, J. Next generation vaccines and vectors: Designing downstream processes for recombinant protein-based virus-like particles. Biotechnol. J. 2015, 10, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Esser, M.T.; Graham, D.R.; Coren, L.V.; Trubey, C.M.; Bess, J.W., Jr.; Arthur, L.O.; Ott, D.E.; Lifson, J.D. Differential incorporation of CD45, CD80 (B7-1), CD86 (B7-2), and major histocompatibility complex class I and II molecules into human immunodeficiency virus type 1 virions and microvesicles: Implications for viral pathogenesis and immune regulation. J. Virol. 2001, 75, 6173–6182. [Google Scholar] [CrossRef]
- Linde, M.E.; Colquhoun, D.R.; Ubaida Mohien, C.; Kole, T.; Aquino, V.; Cotter, R.; Edwards, N.; Hildreth, J.E.; Graham, D.R. The conserved set of host proteins incorporated into HIV-1 virions suggests a common egress pathway in multiple cell types. J. Proteome Res. 2013, 12, 2045–2054. [Google Scholar] [CrossRef] [PubMed]
- Center, R.J.; Wheatley, A.K.; Campbell, S.M.; Gaeguta, A.J.; Peut, V.; Alcantara, S.; Siebentritt, C.; Kent, S.J.; Purcell, D.F. Induction of HIV-1 subtype B and AE-specific neutralizing antibodies in mice and macaques with DNA prime and recombinant gp140 protein boost regimens. Vaccine 2009, 27, 6605–6612. [Google Scholar] [CrossRef]
- Hemachandra, A.; Puls, R.L.; Sirivichayakul, S.; Kerr, S.; Thantiworasit, P.; Ubolyam, S.; Cooper, D.A.; Emery, S.; Phanuphak, P.; Kelleher, A.; et al. An HIV-1 clade A/E DNA prime, recombinant fowlpox virus boost vaccine is safe, but non-immunogenic in a randomized phase I/IIa trial in Thai volunteers at low risk of HIV infection. Hum. Vaccine 2010, 6, 835–840. [Google Scholar] [CrossRef]
- Kelleher, A.D.; Puls, R.L.; Bebbington, M.; Boyle, D.; Ffrench, R.; Kent, S.J.; Kippax, S.; Purcell, D.F.; Thomson, S.; Wand, H.; et al. A randomized, placebo-controlled phase I trial of DNA prime, recombinant fowlpox virus boost prophylactic vaccine for HIV-1. AIDS 2006, 20, 294–297. [Google Scholar] [CrossRef]
- Simon, J.H.; Fouchier, R.A.; Southerling, T.E.; Guerra, C.B.; Grant, C.K.; Malim, M.H. The Vif and Gag proteins of human immunodeficiency virus type 1 colocalize in infected human T cells. J. Virol. 1997, 71, 5259–5267. [Google Scholar]
- Hatch, W.C.; Tanaka, K.E.; Calvelli, T.; Rashbaum, W.K.; Kress, Y.; Lyman, W.D. Persistent productive HIV-1 infection of a CD4- human fetal thymocyte line. J. Immunol. 1992, 148, 3055–3061. [Google Scholar] [PubMed]
- Thali, M.; Moore, J.P.; Furman, C.; Charles, M.; Ho, D.D.; Robinson, J.; Sodroski, J. Characterization of conserved human immunodeficiency virus type 1 gp120 neutralization epitopes exposed upon gp120-CD4 binding. J. Virol. 1993, 67, 3978–3988. [Google Scholar] [PubMed]
- Huang, J.; Kang, B.H.; Pancera, M.; Lee, J.H.; Tong, T.; Feng, Y.; Imamichi, H.; Georgiev, I.S.; Chuang, G.Y.; Druz, A.; et al. Broad and potent HIV-1 neutralization by a human antibody that binds the gp41-gp120 interface. Nature 2014, 515, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Gorny, M.K.; Conley, A.J.; Karwowska, S.; Buchbinder, A.; Xu, J.Y.; Emini, E.A.; Koenig, S.; Zolla-Pazner, S. Neutralization of diverse human immunodeficiency virus type 1 variants by an anti-V3 human monoclonal antibody. J. Virol. 1992, 66, 7538–7542. [Google Scholar] [PubMed]
- Posner, M.R.; Cavacini, L.A.; Emes, C.L.; Power, J.; Byrn, R. Neutralization of HIV-1 by F105, a human monoclonal antibody to the CD4 binding site of gp120. J. Acquir. Immune Defic. Syndr. 1993, 6, 7–14. [Google Scholar] [PubMed]
- Cavacini, L.A.; Emes, C.L.; Wisnewski, A.V.; Power, J.; Lewis, G.; Montefiori, D.; Posner, M.R. Functional and molecular characterization of human monoclonal antibody reactive with the immunodominant region of HIV type 1 glycoprotein 41. AIDS Res. Hum. Retrovir. 1998, 14, 1271–1280. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.M.; Huber, M.; Doores, K.J.; Falkowska, E.; Pejchal, R.; Julien, J.P.; Wang, S.K.; Ramos, A.; Chan-Hui, P.Y.; Moyle, M.; et al. Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature 2011, 477, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Yang, Z.Y.; Li, Y.; Hogerkorp, C.M.; Schief, W.R.; Seaman, M.S.; Zhou, T.; Schmidt, S.D.; Wu, L.; Xu, L.; et al. Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science 2010, 329, 856–861. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.D.; Brunel, F.M.; Jensen, R.; Crooks, E.T.; Cardoso, R.M.; Wang, M.; Hessell, A.; Wilson, I.A.; Binley, J.M.; Dawson, P.E.; et al. An affinity-enhanced neutralizing antibody against the membrane-proximal external region of human immunodeficiency virus type 1 gp41 recognizes an epitope between those of 2F5 and 4E10. J. Virol. 2007, 81, 4033–4043. [Google Scholar] [CrossRef]
- Freed, E.O.; Englund, G.; Martin, M.A. Role of the basic domain of human immunodeficiency virus type 1 matrix in macrophage infection. J. Virol. 1995, 69, 3949–3954. [Google Scholar]
- Center, R.J.; Earl, P.L.; Lebowitz, J.; Schuck, P.; Moss, B. The human immunodeficiency virus type 1 gp120 V2 domain mediates gp41-independent intersubunit contacts. J. Virol. 2000, 74, 4448–4455. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Robertson, D.L.; Morrison, S.G.; Hui, H.; Craig, S.; Decker, J.; Fultz, P.N.; Girard, M.; Shaw, G.M.; Hahn, B.H.; et al. The heterosexual human immunodeficiency virus type 1 epidemic in Thailand is caused by an intersubtype (A/E) recombinant of African origin. J. Virol. 1996, 70, 7013–7029. [Google Scholar] [PubMed]
- Adachi, A.; Gendelman, H.E.; Koenig, S.; Folks, T.; Willey, R.; Rabson, A.; Martin, M.A. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 1986, 59, 284–291. [Google Scholar] [PubMed]
- Kramski, M.; Center, R.J.; Wheatley, A.K.; Jacobson, J.C.; Alexander, M.R.; Rawlin, G.; Purcell, D.F. Hyperimmune bovine colostrum as a low-cost, large-scale source of antibodies with broad neutralizing activity for HIV-1 envelope with potential use in microbicides. Antimicrob. Agents Chemother. 2012, 56, 4310–4319. [Google Scholar] [CrossRef] [PubMed]
- Dale, C.J.; De Rose, R.; Wilson, K.M.; Croom, H.A.; Thomson, S.; Coupar, B.E.; Ramsay, A.; Purcell, D.F.; Ffrench, R.; Law, M.; et al. Evaluation in macaques of HIV-1 DNA vaccines containing primate CpG motifs and fowlpoxvirus vaccines co-expressing IFNgamma or IL-12. Vaccine 2004, 23, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Cavrois, M.; De Noronha, C.; Greene, W.C. A sensitive and specific enzyme-based assay detecting HIV-1 virion fusion in primary T lymphocytes. Nat. Biotechnol. 2002, 20, 1151–1154. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Huang, L.M.; Joshi, A.; Willey, R.; Orenstein, J.; Jeang, K.T. Human immunodeficiency viruses regulated by alternative trans-activators: Genetic evidence for a novel non-transcriptional function of Tat in virion infectivity. EMBO J. 1994, 13, 2886–2896. [Google Scholar] [CrossRef]
- Christensen, H.S.; Daher, A.; Soye, K.J.; Frankel, L.B.; Alexander, M.R.; Laine, S.; Bannwarth, S.; Ong, C.L.; Chung, S.W.; Campbell, S.M.; et al. Small interfering RNAs against the TAR RNA binding protein, TRBP, a Dicer cofactor, inhibit human immunodeficiency virus type 1 long terminal repeat expression and viral production. J. Virol. 2007, 81, 5121–5131. [Google Scholar] [CrossRef]
- Dettenhofer, M.; Yu, X.F. Highly purified human immunodeficiency virus type 1 reveals a virtual absence of Vif in virions. J. Virol. 1999, 73, 1460–1467. [Google Scholar]
- Cantin, R.; Diou, J.; Belanger, D.; Tremblay, A.M.; Gilbert, C. Discrimination between exosomes and HIV-1: Purification of both vesicles from cell-free supernatants. J. Immunol. Methods 2008, 338, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.K.; Hart, T.K.; Jonak, Z.L.; Bugelski, P.J. Physicochemical dissociation of CD4-mediated syncytium formation and shedding of human immunodeficiency virus type 1 gp120. J. Virol. 1993, 67, 3818–3825. [Google Scholar] [PubMed]
- Quillent, C.; Borman, A.M.; Paulous, S.; Dauguet, C.; Clavel, F. Extensive regions of pol are required for efficient human immunodeficiency virus polyprotein processing and particle maturation. Virology 1996, 219, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Larson, D.R.; Ma, Y.M.; Vogt, V.M.; Webb, W.W. Direct measurement of Gag-Gag interaction during retrovirus assembly with FRET and fluorescence correlation spectroscopy. J. Cell Biol. 2003, 162, 1233–1244. [Google Scholar] [CrossRef] [PubMed]
- Mattei, S.; Flemming, A.; Anders-Osswein, M.; Krausslich, H.G.; Briggs, J.A.; Muller, B. RNA and Nucleocapsid Are Dispensable for Mature HIV-1 Capsid Assembly. J. Virol. 2015, 89, 9739–9747. [Google Scholar] [CrossRef]
- El Meshri, S.E.; Dujardin, D.; Godet, J.; Richert, L.; Boudier, C.; Darlix, J.L.; Didier, P.; Mely, Y.; de Rocquigny, H. Role of the nucleocapsid domain in HIV-1 Gag oligomerization and trafficking to the plasma membrane: A fluorescence lifetime imaging microscopy investigation. J. Mol. Biol. 2015, 427, 1480–1494. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Sturgeon, T.; Weisz, O.A.; Mothes, W.; Montelaro, R.C. HIV-1 matrix dependent membrane targeting is regulated by Gag mRNA trafficking. PLoS ONE 2009, 4, e6551. [Google Scholar] [CrossRef]
- Nikolaitchik, O.A.; Dilley, K.A.; Fu, W.; Gorelick, R.J.; Tai, S.H.; Soheilian, F.; Ptak, R.G.; Nagashima, K.; Pathak, V.K.; Hu, W.S. Dimeric RNA recognition regulates HIV-1 genome packaging. PLoS Pathog. 2013, 9, e1003249. [Google Scholar] [CrossRef]
- Chen, J.; Rahman, S.A.; Nikolaitchik, O.A.; Grunwald, D.; Sardo, L.; Burdick, R.C.; Plisov, S.; Liang, E.; Tai, S.; Pathak, V.K.; et al. HIV-1 RNA genome dimerizes on the plasma membrane in the presence of Gag protein. Proc. Natl. Acad. Sci. USA 2016, 113, E201–E208. [Google Scholar] [CrossRef]
- Huthoff, H.; Berkhout, B. Two alternating structures of the HIV-1 leader RNA. RNA 2001, 7, 143–157. [Google Scholar] [CrossRef]
- Lu, K.; Heng, X.; Garyu, L.; Monti, S.; Garcia, E.L.; Kharytonchyk, S.; Dorjsuren, B.; Kulandaivel, G.; Jones, S.; Hiremath, A.; et al. NMR detection of structures in the HIV-1 5′-leader RNA that regulate genome packaging. Science 2011, 334, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Ott, M.; Geyer, M.; Zhou, Q. The control of HIV transcription: Keeping RNA polymerase II on track. Cell Host Microbe 2011, 10, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Ottmann, M.; Pechoux, C.; Le Grice, S.; Darlix, J.L. Mutations in the primer grip of human immunodeficiency virus type 1 reverse transcriptase impair proviral DNA synthesis and virion maturation. J. Virol. 1998, 72, 7676–7680. [Google Scholar] [PubMed]
- Pang, H.B.; Hevroni, L.; Kol, N.; Eckert, D.M.; Tsvitov, M.; Kay, M.S.; Rousso, I. Virion stiffness regulates immature HIV-1 entry. Retrovirology 2013, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Sanders, R.W.; Venturi, M.; Schiffner, L.; Kalyanaraman, R.; Katinger, H.; Lloyd, K.O.; Kwong, P.D.; Moore, J.P. The mannose-dependent epitope for neutralizing antibody 2G12 on human immunodeficiency virus type 1 glycoprotein gp120. J. Virol. 2002, 76, 7293–7305. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Gao, F.; Mascola, J.R.; Stamatatos, L.; Polonis, V.R.; Koutsoukos, M.; Voss, G.; Goepfert, P.; Gilbert, P.; Greene, K.M.; et al. Human immunodeficiency virus type 1 env clones from acute and early subtype B infections for standardized assessments of vaccine-elicited neutralizing antibodies. J. Virol. 2005, 79, 10108–10125. [Google Scholar] [CrossRef]
- Crooks, E.T.; Moore, P.L.; Franti, M.; Cayanan, C.S.; Zhu, P.; Jiang, P.; de Vries, R.P.; Wiley, C.; Zharkikh, I.; Schulke, N.; et al. A comparative immunogenicity study of HIV-1 virus-like particles bearing various forms of envelope proteins, particles bearing no envelope and soluble monomeric gp120. Virology 2007, 366, 245–262. [Google Scholar] [CrossRef] [PubMed]
- Sedegah, M.; Charoenvit, Y.; Minh, L.; Belmonte, M.; Majam, V.F.; Abot, S.; Ganeshan, H.; Kumar, S.; Bacon, D.J.; Stowers, A.; et al. Reduced immunogenicity of DNA vaccine plasmids in mixtures. Gene Ther. 2004, 11, 448–456. [Google Scholar] [CrossRef]
- Haffar, O.; Garrigues, J.; Travis, B.; Moran, P.; Zarling, J.; Hu, S.L. Human immunodeficiency virus-like, nonreplicating, gag-env particles assemble in a recombinant vaccinia virus expression system. J. Virol. 1990, 64, 2653–2659. [Google Scholar]
- Gheysen, D.; Jacobs, E.; de Foresta, F.; Thiriart, C.; Francotte, M.; Thines, D.; De Wilde, M. Assembly and release of HIV-1 precursor Pr55gag virus-like particles from recombinant baculovirus-infected insect cells. Cell 1989, 59, 103–112. [Google Scholar] [CrossRef]
- Smith, A.J.; Cho, M.I.; Hammarskjöld, M.L.; Rekosh, D. Human immunodeficiency virus type 1 Pr55gag and Pr160gag-pol expressed from a simian virus 40 late replacement vector are efficiently processed and assembled into viruslike particles. J. Virol. 1990, 64, 2743–2750. [Google Scholar] [PubMed]
- Kohl, N.E.; Emini, E.A.; Schleif, W.A.; Davis, L.J.; Heimbach, J.C.; Dixon, R.A.; Scolnick, E.M.; Sigal, I.S. Active human immunodeficiency virus protease is required for viral infectivity. Proc. Natl. Acad. Sci. USA 1988, 85, 4686–4690. [Google Scholar] [CrossRef] [PubMed]
- Ross, E.K.; Fuerst, T.R.; Orenstein, J.M.; O’Neill, T.; Martin, M.A.; Venkatesan, S. Maturation of human immunodeficiency virus particles assembled from the gag precursor protein requires in situ processing by gag-pol protease. AIDS Res. Hum. Retrovir. 1991, 7, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Briggs, J.A.G.; Wilk, T.; Welker, R.; Kräusslich, H.-G.; Fuller, S.D. Structural organization of authentic, mature HIV-1 virions and cores. EMBO J. 2003, 22, 1707–1715. [Google Scholar] [CrossRef] [PubMed]
- Schiller, J.; Chackerian, B. Why HIV virions have low numbers of envelope spikes: Implications for vaccine development. PLoS Pathog. 2014, 10, e1004254. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Dintzis, R.Z.; Dintzis, H.M. Specific cellular stimulation in the primary immune response: A quantized model. Proc. Natl. Acad. Sci. USA 1982, 79, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Hat, B.; Kazmierczak, B.; Lipniacki, T. B cell activation triggered by the formation of the small receptor cluster: A computational study. PLoS Comput. Biol. 2011, 7, e1002197. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, M.F.; Jennings, G.T. Vaccine delivery: A matter of size, geometry, kinetics and molecular patterns. Nat. Rev. Immunol. 2010, 10, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Louis, J.M.; Clore, G.M.; Gronenborn, A.M. Autoprocessing of HIV-1 protease is tightly coupled to protein folding. Nat. Struct. Biol. 1999, 6, 868–875. [Google Scholar] [PubMed]
- Chatterjee, A.; Mridula, P.; Mishra, R.K.; Mittal, R.; Hosur, R.V. Folding regulates autoprocessing of HIV-1 protease precursor. J. Biol. Chem. 2005, 280, 11369–11378. [Google Scholar] [CrossRef] [PubMed]
- Wapling, J.; Moore, K.L.; Sonza, S.; Mak, J.; Tachedjian, G. Mutations that abrogate human immunodeficiency virus type 1 reverse transcriptase dimerization affect maturation of the reverse transcriptase heterodimer. J. Virol. 2005, 79, 10247–10257. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Wakefield, J.K.; Liu, H.; Xiao, H.; Kralovics, R.; Prchal, J.T.; Kappes, J.C. Development of a novel trans-lentiviral vector that affords predictable safety. Mol. Ther. 2000, 2, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Shehu-Xhilaga, M.; Hill, M.; Marshall, J.A.; Kappes, J.; Crowe, S.M.; Mak, J. The conformation of the mature dimeric human immunodeficiency virus type 1 RNA genome requires packaging of pol protein. J. Virol. 2002, 76, 4331–4340. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Accola, M.A.; Ohagen, A.; Gottlinger, H.G. Isolation of human immunodeficiency virus type 1 cores: Retention of Vpr in the absence of p6(gag). J. Virol. 2000, 74, 6198–6202. [Google Scholar] [CrossRef] [PubMed]
- Müller, B.; Anders, M.; Akiyama, H.; Welsch, S.; Glass, B.; Nikovics, K.; Clavel, F.; Tervo, H.-M.; Keppler, O.T.; Kräusslich, H.-G. HIV-1 Gag Processing Intermediates Trans-dominantly Interfere with HIV-1 Infectivity. J. Biol. Chem. 2009, 284, 29692–29703. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.L.; Johnson, A.T.; Howard, J.L.; Purcell, D.F. Both linear and discontinuous ribosome scanning are used for translation initiation from bicistronic human immunodeficiency virus type 1 env mRNAs. J. Virol. 2007, 81, 4664–4676. [Google Scholar] [CrossRef]
- Moore, P.L.; Crooks, E.T.; Porter, L.; Zhu, P.; Cayanan, C.S.; Grise, H.; Corcoran, P.; Zwick, M.B.; Franti, M.; Morris, L.; et al. Nature of nonfunctional envelope proteins on the surface of human immunodeficiency virus type 1. J. Virol. 2006, 80, 2515–2528. [Google Scholar] [CrossRef]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonelli, C.A.; Khoury, G.; Center, R.J.; Purcell, D.F.J. HIV-1-Based Virus-like Particles that Morphologically Resemble Mature, Infectious HIV-1 Virions. Viruses 2019, 11, 507. https://doi.org/10.3390/v11060507
Gonelli CA, Khoury G, Center RJ, Purcell DFJ. HIV-1-Based Virus-like Particles that Morphologically Resemble Mature, Infectious HIV-1 Virions. Viruses. 2019; 11(6):507. https://doi.org/10.3390/v11060507
Chicago/Turabian StyleGonelli, Christopher A., Georges Khoury, Rob J. Center, and Damian F.J. Purcell. 2019. "HIV-1-Based Virus-like Particles that Morphologically Resemble Mature, Infectious HIV-1 Virions" Viruses 11, no. 6: 507. https://doi.org/10.3390/v11060507
APA StyleGonelli, C. A., Khoury, G., Center, R. J., & Purcell, D. F. J. (2019). HIV-1-Based Virus-like Particles that Morphologically Resemble Mature, Infectious HIV-1 Virions. Viruses, 11(6), 507. https://doi.org/10.3390/v11060507