Enteroviruses: A Gut-Wrenching Game of Entry, Detection, and Evasion


Abstract

1. Introduction
1.1. Enteroviruses
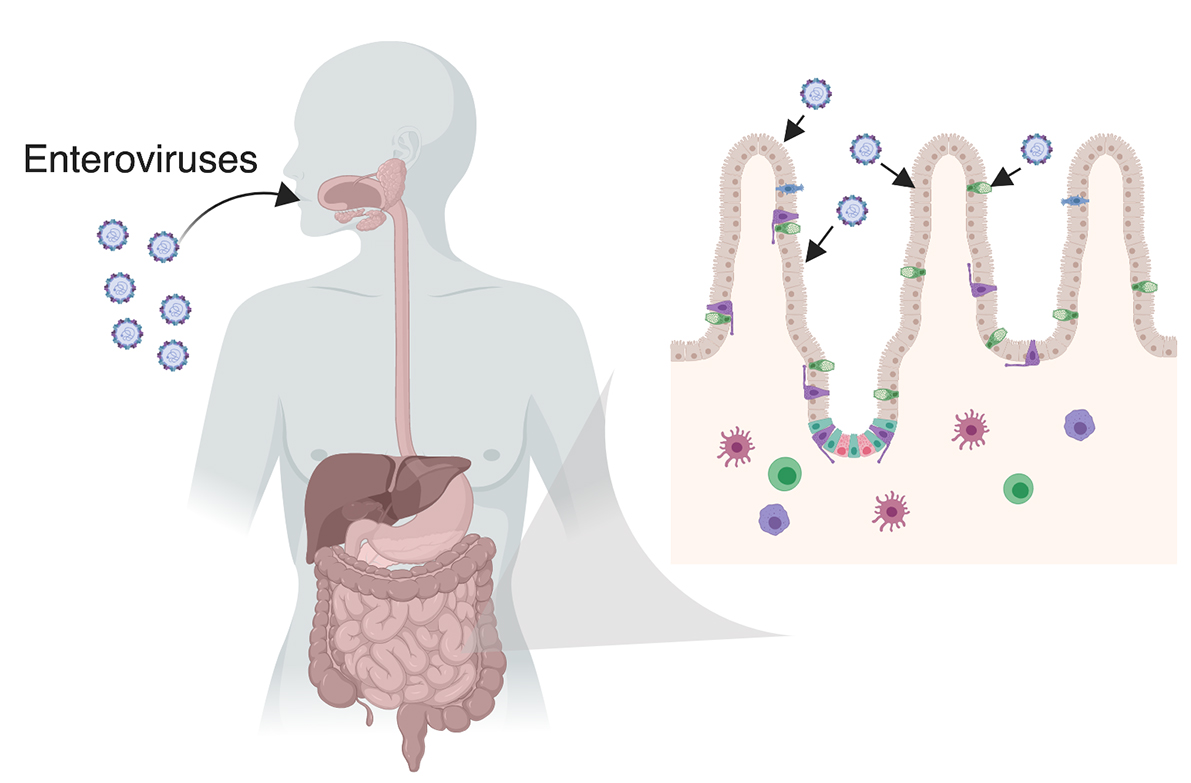
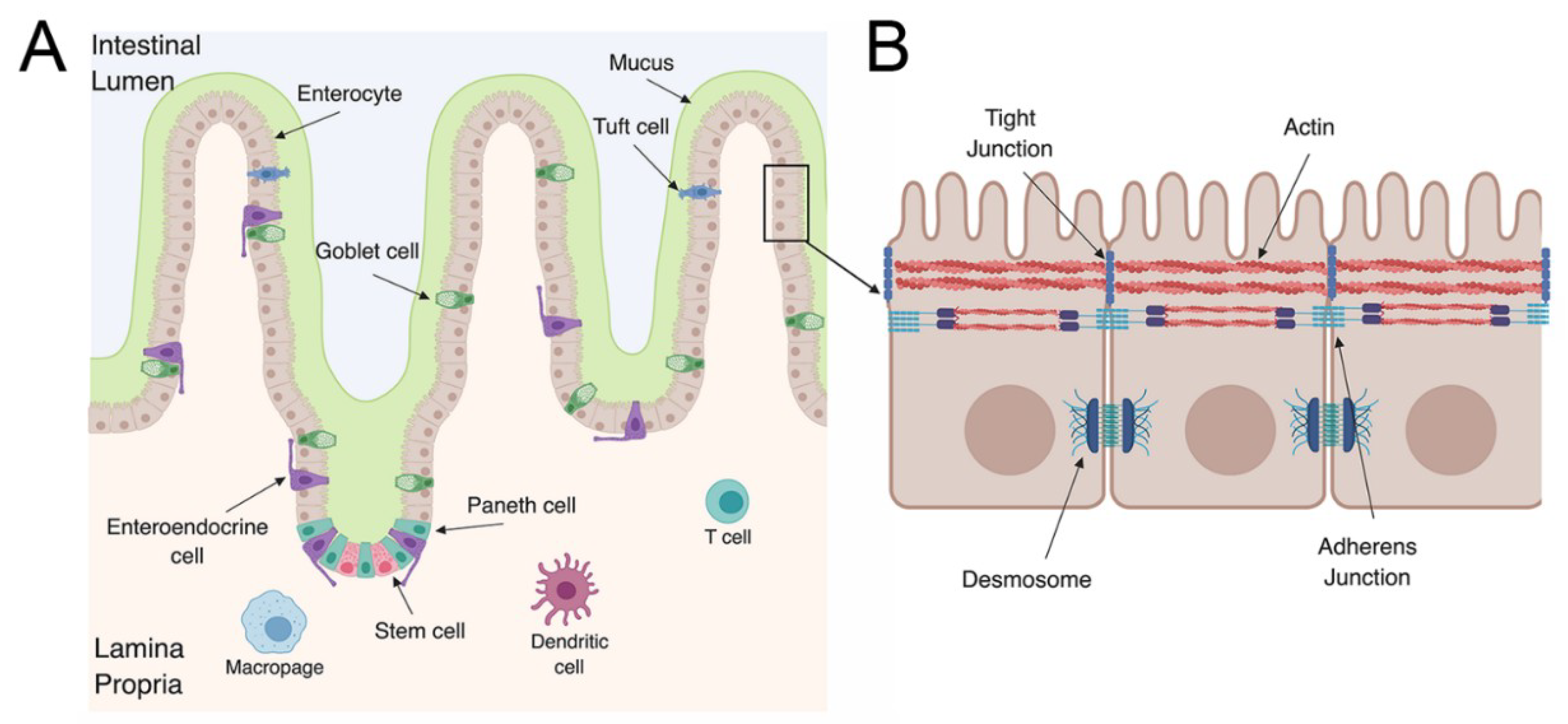
1.2. The Gastrointestinal Tract
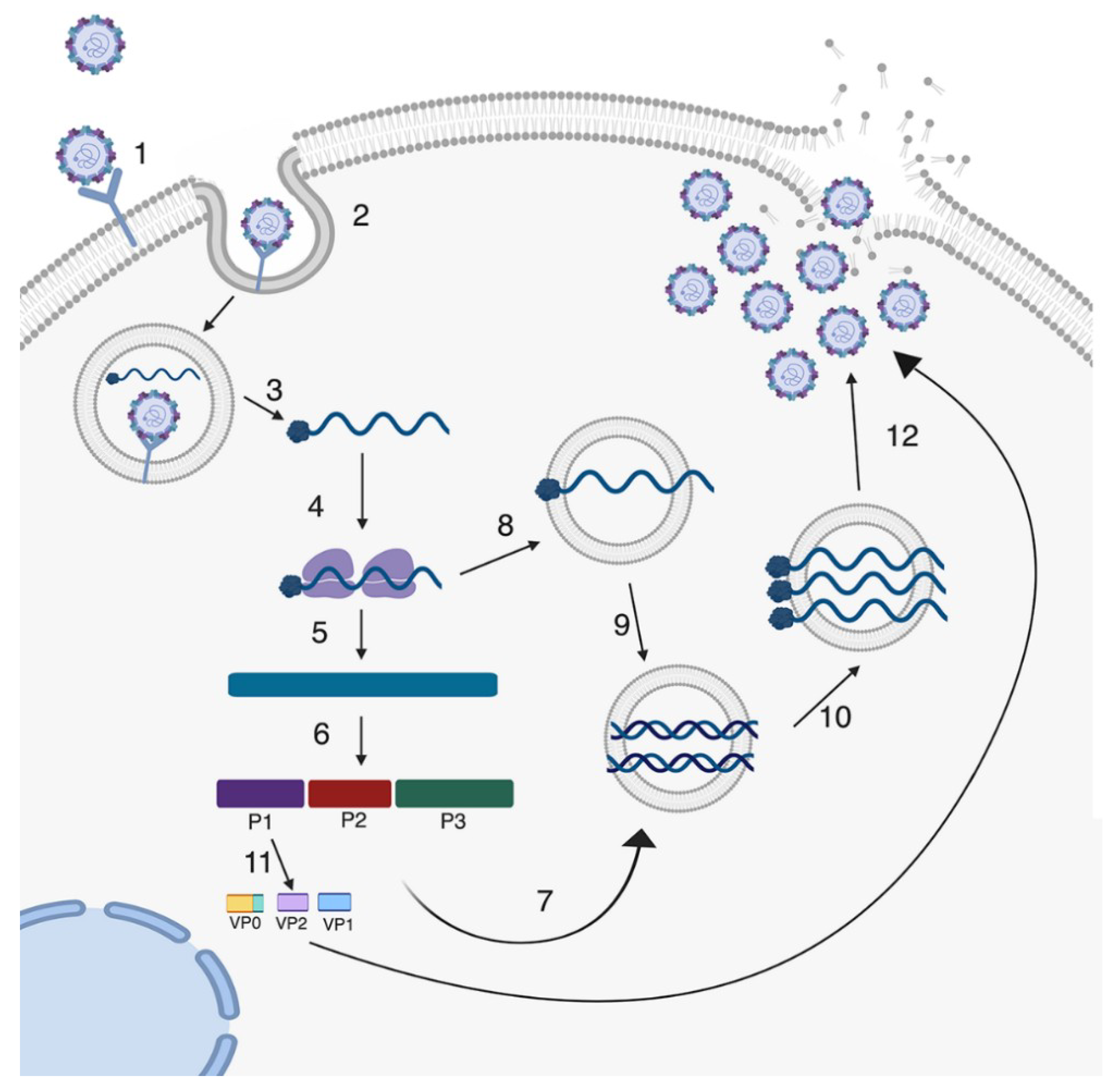
2. Enterovirus Infections in the GI Tract
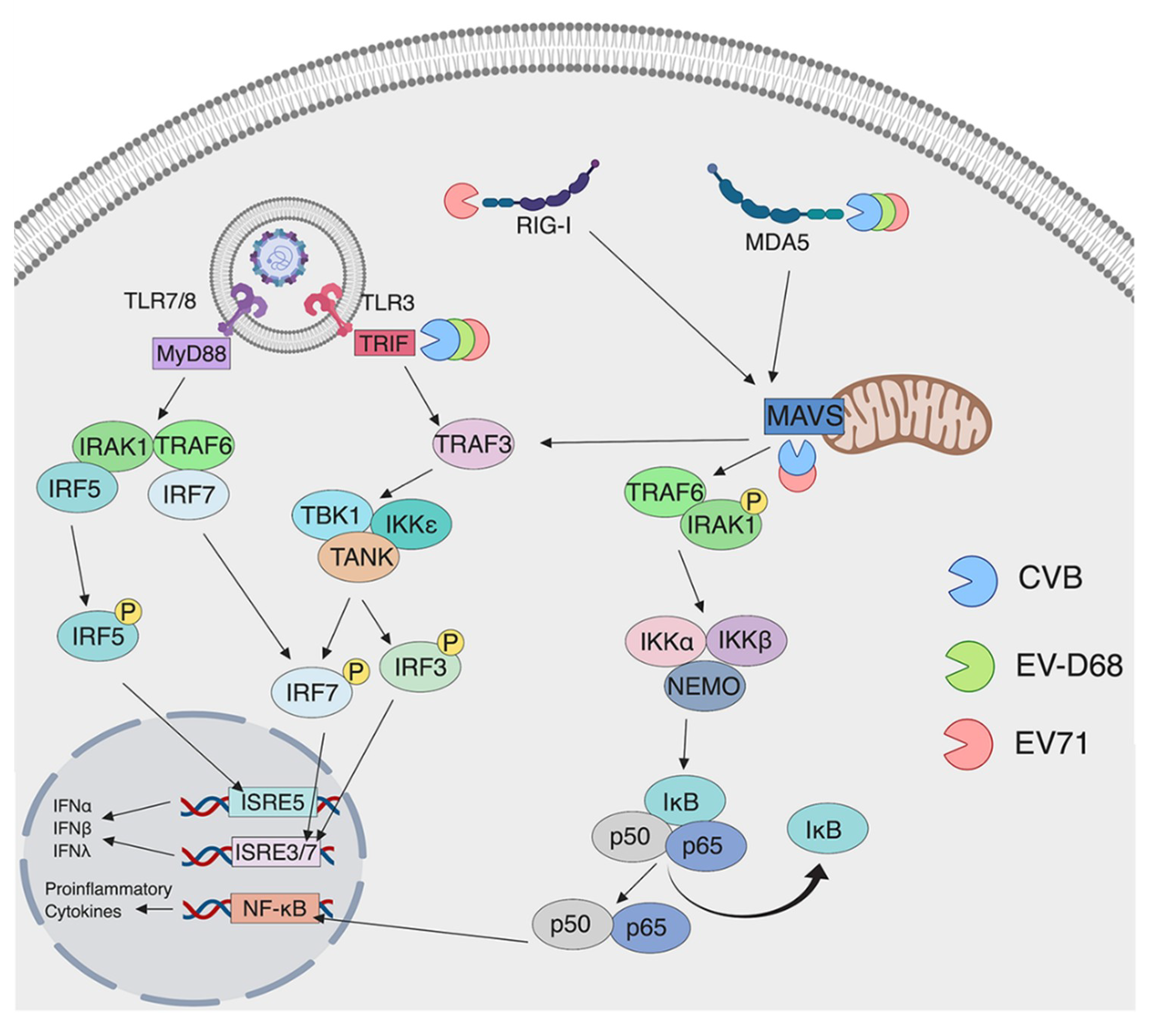
3. Detection of Enteroviruses by Pattern Recognition Receptors
3.1. Detection by TLRs
3.2. Detection by RLRs
4. Evasion of Innate Immunity by Enteroviruses
4.1. Evasion of TLRs by Enteroviruses
4.2. Evasion of RLRs by Enteroviruses
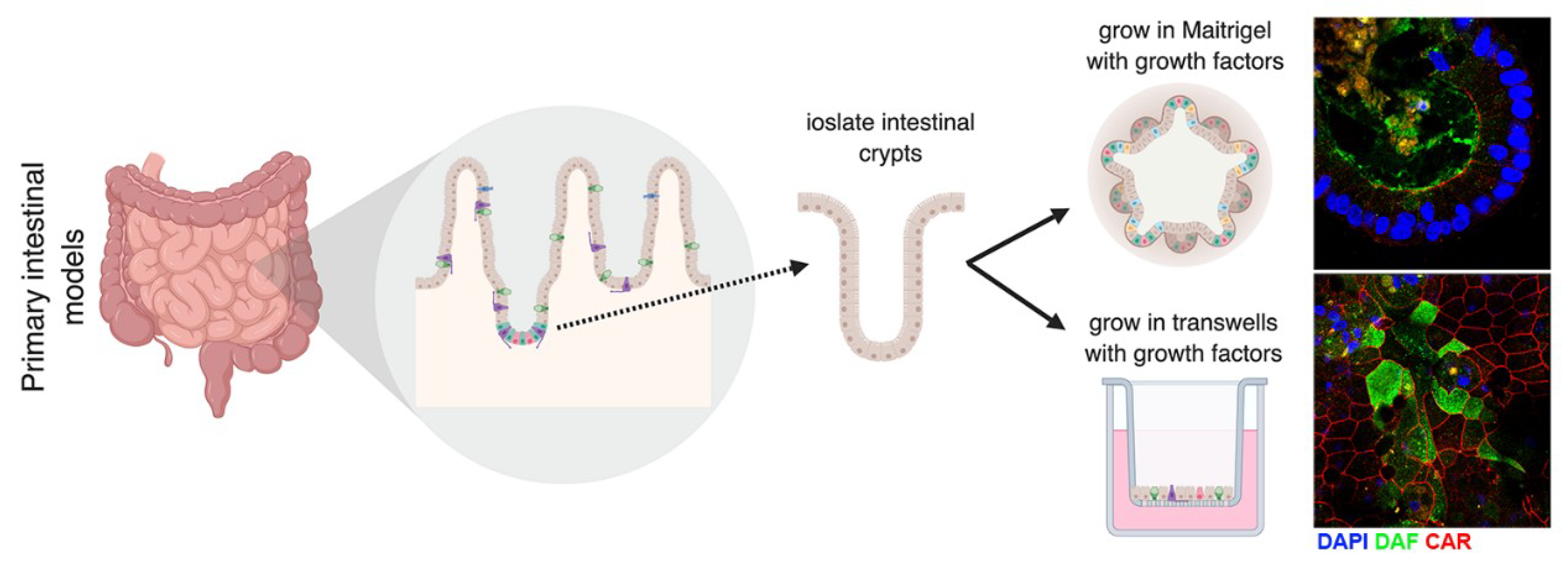
5. Models to Study Enteroviruses in the Gut
5.1. In Vitro and Ex Vivo Models to Study Enterovirus Infection
5.2. In Vivo Models to Study Enterovirus Infection
6. Concluding Remarks
Funding
Conflicts of Interest
References
- Division of Viral Diseases CDC Non-Polio Enterovirus Infection. Available online: https://www.cdc.gov/non-polio-enterovirus/index.html (accessed on 28 December 2018).
- Pons-Salort, M.; Parker, E.P.K.; Grassly, N.C. The epidemiology of non-polio enteroviruses: Recent advances and outstanding questions. Curr. Opin. Infect. Dis. 2015, 28, 479–487. [Google Scholar] [CrossRef]
- Lie, S.-L.; Pan, H.; Liu, P.; Amer, S.; Chan, T.-C.; Zhan, J.; Huo, X.; Liu, Y.; Teng, Z.; Wang, L.; Zhuang, H. Comparative epidemiology and virology of fatal and nonfatal cases of hand, foot and mouth disease in mainland China from 2008 to 2014. Rev. Med. Virol. 2015, 25, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Atchison, R.W.; Walpusk, J.; Jaffe, R. Echovirus hepatic failure in infancy: Report of four cases with speculation on the pathogenesis. Pediatr. Dev. Pathol. 2001, 4, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Huber, S.; Ramsingh, A.I. Coxsackievirus-Induced Pancreatitis. Viral Immunol. 2004, 17, 358–369. [Google Scholar] [CrossRef]
- Ph Syriopoulou, V.; Hadjichristodoulou, C.; Daikos, G.L.; Pirounakiy, M.; Chatzicou, V.; Pavlopoulou, I.; Anagnostakou, M.; Theodoridou, M.; Dellagrammaticas, H. Clinical and epidemiological aspects of an enterovirus outbreak in a neonatal unit. J. Hosp. Infect. 2002, 51, 275–280. [Google Scholar] [CrossRef]
- Haston, J.C.; Dixon, T.C. Nonpolio Enterovirus Infections in Neonates. Pediatr. Ann. 2015, 44, e103–e107. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, D.; Dobson, S.R.; Wilkinson, A.R.; Hope, P.L.; Eglin, R.; Moxon, E.R. Conservative management of an echovirus 11 outbreak in a neonatal unit. Lancet 1989, 1, 543–545. [Google Scholar] [CrossRef]
- Civardi, E.; Tzialla, C.; Baldanti, F.; Strocchio, L.; Manzoni, P.; Stronati, M. Viral outbreaks in neonatal intensive care units: What we do not know. Am. J. Infect. Control 2013, 41, 854–856. [Google Scholar] [CrossRef] [PubMed]
- Naing, Z.; Rayner, B.; Killikulangara, A.; Vunnam, K.; Leach, S.; McIver, C.J.; Scott, G.M.; Craig, M.E.; Lui, K.; Rawlinson, W.D. Prevalence of viruses in stool of premature neonates at a neonatal intensive care unit. J. Paediatr. Child Health 2013, 49. [Google Scholar] [CrossRef]
- Xing, W.; Liao, Q.; Viboud, C.; Zhang, J.; Sun, J.; Wu, J.T.; Chang, Z.; Liu, F.; Fang, V.J.; Zheng, Y.; et al. Hand, foot, and mouth disease in China, 2008-12: An epidemiological study. Lancet Infect. Dis. 2014, 14, 308–318. [Google Scholar] [CrossRef]
- Cassidy, H.; Poelman, R.; Knoester, M.; Van Leer-Buter, C.C.; Niesters, H.G.M. Enterovirus D68 -the new polio? Front. Microbiol. 2018, 9, 1–11. [Google Scholar] [CrossRef]
- Centers for Disease Control & Prevention Acute Flaccid Myelitis. Available online: https://www.cdc.gov/acute-flaccid-myelitis/ (accessed on 23 March 2019).
- Sejvar, J.J.; Lopez, A.S.; Cortese, M.M.; Leshem, E.; Pastula, D.M.; Miller, L.; Glaser, C.; Kambhampati, A.; Shioda, K.; Aliabadi, N.; et al. Acute Flaccid Myelitis in the United States, August-December 2014: Results of Nationwide Surveillance. Clin. Infect. Dis. 2016, 63, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Zhu, R.; Cheng, T.; Yin, Z.; Liu, D.; Xu, L.; Li, Y.; Wang, W.; Liu, J.; Que, Y.; Ye, X.; et al. Serological survey of neutralizing antibodies to eight major enteroviruses among healthy population. Emerg. Microbes Infect. 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Wallace, G.S.; Curns, A.T.; Weldon, W.C.; Oberste, M.S. Seroprevalence of Poliovirus Antibodies in the United States Population, 2009-2010. BMC Public Health 2016, 16, 1–8. [Google Scholar] [CrossRef]
- Sabin, A.B. Oral Poliovirus Vaccine: History of Its Development and Use and Current Challenge to Eliminate Poliomyelitis from the World. J. Infect. Dis. 1985, 151, 420–436. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Molodecky, N.A.; Verma, H.; Sharma, P.; Yang, J.S.; Saletti, G.; Ahmad, M.; Bahl, S.K.; Wierzba, T.F.; Nandy, R.K.; et al. Human circulating antibody-producing B cell as a predictive measure of mucosal immunity to poliovirus. PLoS ONE 2016, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Bearden, D.; Collett, M.; Quan, P.L.; Costa-Carvalho, B.T.; Sullivan, K.E. Enteroviruses in X-Linked Agammaglobulinemia: Update on Epidemiology and Therapy. J. Allergy Clin. Immunol. Pract. 2016, 4, 1059–1065. [Google Scholar] [CrossRef]
- Luk, A.D.W.; Ni, K.; Wu, Y.; Lam, K.T.; Chan, K.W.; Lee, P.P.; Tu, W.; Mao, H.; Lau, Y.L. Type I and III interferon productions are impaired in X-linked agammaglobulinemia patients toward poliovirus but not influenza virus. Front. Immunol. 2018, 9, 1–16. [Google Scholar] [CrossRef]
- Mena, I.; Perry, C.M.; Harkins, S.; Rodriguez, F.; Gebhard, J.; Whitton, J.L. The role of B lymphocytes in coxsackievirus B3 infection. Am. J. Pathol. 1999, 155, 1205–1215. [Google Scholar] [CrossRef]
- Williams, J.M.; Duckworth, C.A.; Burkitt, M.D.; Watson, A.J.M.; Campbell, B.J.; Pritchard, D.M. Epithelial Cell Shedding and Barrier Function: A Matter of Life and Death at the Small Intestinal Villus Tip. Vet. Pathol. 2015, 52, 445–455. [Google Scholar] [CrossRef]
- Makala, L.H.C.; Suzuki, N.; Nagasawa, H. Peyer’s patches: Organized lymphoid structures for the induction of mucosal immune responses in the intestine. Pathobiology 2002, 70, 55–68. [Google Scholar] [CrossRef]
- Heel, A.K.; McCauley, R.; Papadimitriou, J.M.; Hall, J.C. Review: Peyer’s patches. J. Gastroenterol. 1997, 12, 122–136. [Google Scholar] [CrossRef]
- Finke, D.; Kraehenbuhl, J.P. Formation of Peyer’s patches. Curr. Opin. Genet. Dev. 2001, 11, 561–567. [Google Scholar] [CrossRef]
- Farquhar, M.G.; Palade, G.E. Junctional complexes in various epithelia. J. Cell Biol. 1963, 17, 375–412. [Google Scholar] [CrossRef]
- Green, K.J.; Getsios, S.; Troyanovsky, S.; Godsel, L.M. Intercellular junction assembly, dynamics, and homeostasis. Cold Spring Harb. Perspect. Biol. 2010, 2, 1–24. [Google Scholar] [CrossRef]
- Delorme-Axford, E.; Coyne, C.B. The Actin Cytoskeleton as a Barrier to Virus Infection of Polarized Epithelial Cells. Viruses 2011, 3, 2462–2477. [Google Scholar] [CrossRef]
- Wells, A.I.; Coyne, C.B. Type III Interferons in Antiviral Defenses at Barrier Surfaces. Trends Immunol. 2018, 39, 848–858. [Google Scholar] [CrossRef]
- Zanoni, I.; Arduini, A.; Baldridge, M.T.; Hornef, M.W.; Selvakumar, T.A.; Bhushal, S.; Kalinke, U.; Wirth, D.; Hauser, H.; Köster, M. Identification of a Predominantly interferon-λ-induced Transcriptional Profile in Murine intestinal epithelial cells. Front. Immunol 2017, 8, 1302. [Google Scholar] [CrossRef]
- Kotredes, K.P.; Thomas, B.; Gamero, A.M. The Protective Role of Type I interferons in the Gastrointestinal Tract. Front. Immunol 2017, 8, 1–10. [Google Scholar] [CrossRef]
- Henning, S.J.; Von Furstenberg, R.J. GI stem cells—new insights into roles in physiology and pathophysiology. J Physiol 2016, 594, 4769–4779. [Google Scholar] [CrossRef]
- Barker, N.; van Es, J.H.; Kuipers, J.; Kujala, P.; van den Born, M.; Cozijnsen, M.; Haegebarth, A.; Korving, J.; Begthel, H.; Peters, P.J.; et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007, 449, 1003–1007. [Google Scholar] [CrossRef]
- Nephrol, W.J. Paneth cells in intestinal physiology and pathophysiology. World J. Gastrointest. Pathophysiol. 2017, 8. [Google Scholar]
- Sato, T.; Van Es, J.H.; Snippert, H.J.; Stange, D.E.; Vries, R.G.; Van Den Born, M.; Barker, N.; Shroyer, N.F.; Van De Wetering, M.; Clevers, H. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 2011, 469, 415–418. [Google Scholar] [CrossRef]
- Karpus, O.N.; Westendorp, B.F.; Vermeulen, J.L.M.; Meisner, S.; Koster, J.; Muncan, V.; Wildenberg, M.E.; van den Brink, G.R. Colonic CD90+ Crypt Fibroblasts Secrete Semaphorins to Support Epithelial Growth. Cell Rep. 2019, 26, 3698–3708. [Google Scholar] [CrossRef]
- Neutra, M.R. Current Concepts in Mucosal Immunity V. Role of M cells in transepithelial transport of antigens and pathogens to the mucosal immune system. Am J Physiol Gastrointest Liver Physiol 1998, 274, 785–791. [Google Scholar] [CrossRef]
- Cheng, H.; Leblond, C.P. Origin, differentiation and renewal of the four main epithelial cell types in the mouse small intestine. Am. J. Anat. 1974, 141, 537–561. [Google Scholar] [CrossRef]
- Cornick, S.; Tawiah, A.; Chadee, K. Roles and regulation of the mucus barrier in the gut. Tissue Barriers 2015, 3. [Google Scholar] [CrossRef]
- Cherry, J.; Krogstad, P. Enteroviruses, Parechoviruses, and Saffold Viruses. In Feigin and Cherry’s Textbook of Pediatric Infectious Diseases, 8th ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2019. [Google Scholar]
- Pallansch, M.A.; Oberste, M.S.; Whitton, J.L. Field’s Virology, 6th ed.; Knipe, D., Howley, P., Cohen, J., Lamb, R., Martin, M., Racaniello, V., Roizman, B., Eds.; Wolters Kluwer/Lippincott Williams & Wilkins Health: Philadelpha, PA, USA, 2013. [Google Scholar]
- Liu, M.Y.; Liu, J.; Lai, W.; Luo, J.; Liu, Y.; Vu, G.-P.; Yang, Z.; Trang, P.; Li, H.; Wu, J. Characterization of enterovirus 71 infection and associated outbreak of Hand, Foot, and Mouth Disease in Shawo of China in 2012. Nat. Publ. Gr. 2016. [Google Scholar] [CrossRef]
- Rapmund, G.; Gauld, J.R.; Rogers, N.G.; Holmes, G.E. Neonatal Myocarditis and Meningoencephalitis Due to Coxsackie Virus Group B, Type 4. N. Engl. J. Med. 1959, 260, 819–821. [Google Scholar] [CrossRef]
- Vanek, J.; Lukes, J.; Potuznik, V.; Polednikova, I.; Vilim, V. Myocarditis and encephalitis in newborn infants, caused by Coxsackie B virus. J. Hyg. Epidemiol. Microbiol. Immunol. 1959, 3, 283–291. [Google Scholar]
- Centers for Disease Control & Prevention Enterovirus surveillance-United States 2002–2004. MMWR Morb. Mortal. Wkly. Rep. 2006, 55, 153–156.
- Yamayoshi, S.; Yamashita, Y.; Li, J.; Hanagata, N.; Minowa, T.; Takemura, T.; Koike, S. Scavenger receptor B2 is a cellular receptor for enterovirus 71. Nat. Med. 2009, 15, 798–802. [Google Scholar] [CrossRef]
- Nishimura, Y.; Shimojima, M.; Tano, Y.; Miyamura, T.; Wakita, T.; Shimizu, H. Human P-selectin glycoprotein ligand-1 is a functional receptor for enterovirus 71. Nat. Microbiol. 2009, 15, 794–798. [Google Scholar] [CrossRef]
- Yamayoshi, S.; Ohka, S.; Fujii, K.; Koike, S. Functional Comparison of SCARB2 and PSGL1 as Receptors for Enterovirus 71. J. Virol. 2013, 87, 3335–3347. [Google Scholar] [CrossRef]
- Bergelson, J.M.; Droguett, G.; Kurt-jones, E.A.; Krithivas, A.; Hong, J.S.; Horwitz, M.S.; Crowell, R.L.; Finberg, R.W. Isolation of a Common Receptor for Coxsackie B Viruses and Adenoviruses 2 and 5. Science 1997, 275, 1320–1324. [Google Scholar] [CrossRef]
- Carson, S.D.; Chapman, N.N.; Tracy, S.M. Purification of the putative coxsackievirus B receptor from HeLa cells. Biochem. Biophys. Res. Commun. 1997, 233, 325–328. [Google Scholar] [CrossRef]
- Tomko, R.P.; Xu, R.; Philipson, L. HCAR and MCAR: The human and mouse cellular receptors for subgroup C adenoviruses and group B coxsackieviruses. Proc. Natl. Acad. Sci. USA 1997, 94, 3352–3356. [Google Scholar] [CrossRef]
- Mendelsohn, C.L.; Wimmer, E.; Racaniello, V.R. Cellular receptor for poliovirus: Molecular cloning, nucleotide sequence, and expression of a new member of the immunoglobulin superfamily. Cell 1989, 56, 855–865. [Google Scholar] [CrossRef]
- Morosky, S.; Wells, A.I.; Lemon, K.; Evans, A.S.; Schamus, S.; Bakkenist, C.J.; Coyne, C.B. The neonatal Fc receptor is a pan-echovirus receptor. Proc. Natl. Acad. Sci. USA 2019, 116. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, G.; Liu, S.; Chen, X.; Peng, R.; Dai, L.; Qu, X.; Li, S.; Song, H.; Gao, Z.; et al. Human Neonatal Fc Receptor is the Cellular Uncoating Receptor for Enterovirus B. Cell 2019, 177, 1–13. [Google Scholar] [CrossRef]
- Bergelson, J.M.; Chan, M.; Solomon, K.R.; John, N.F.S.T.; Lin, H.; Finberg, R.W. Decay-accelerating factor (CD55), a glycosylphosphatidylinositol- anchored complement regulatory protein, is a receptor for several. Proc. Natl. Acad. Sci. USA 1994, 91, 6245–6248. [Google Scholar] [CrossRef] [PubMed]
- Bergelson, J.M.; Mohanty, J.G.; Crowell, R.L.; St John, N.F.; Lublin, D.M.; Finberg, R.W. Coxsackievirus B3 adapted to growth in RD cells binds to decay-accelerating factor (CD55). J. Virol. 1995, 69, 1903–1906. [Google Scholar]
- Coyne, C.B.; Bergelson, J.M. Virus-induced Abl and Fyn kinase signals permit coxsackievirus entry through epithelial tight junctions. Cell 2006, 124, 119–131. [Google Scholar] [CrossRef]
- Rubbia-Brandt, L.; Brown, T.D.K.; Stuart, A.D.; McKee, T.A.; Sobo, K. Decay-Accelerating Factor Binding Determines the Entry Route of Echovirus 11 in Polarized Epithelial Cells. J. Virol. 2011, 85, 12376–12386. [Google Scholar] [CrossRef]
- Nishimura, Y.; Shimizu, H. Cellular receptors for human enterovirus species A. Front. Microbiol. 2012, 3, 1–5. [Google Scholar] [CrossRef]
- Staring, J.; van den Hengel, L.G.; Raaben, M.; Blomen, V.A.; Carette, J.E.; Brummelkamp, T.R. KREMEN1 Is a Host Entry Receptor for a Major Group of Enteroviruses. Cell Host Microbe 2018, 23, 636–643. [Google Scholar] [CrossRef] [PubMed]
- Su, P.-Y.; Liu, Y.-T.; Chang, H.-Y.; Huang, S.-W.; Wang, Y.-F.; Yu, C.-K.; Wang, J.-R.; Chang, C.-F. Cell surface sialylation affects binding of enterovirus 71 to rhabdomyosarcoma and neuroblastoma cells. BMC Microbiol. 2012, 12, 162. [Google Scholar] [CrossRef]
- Wei, W.; Guo, H.; Chang, J.; Yu, Y.; Liu, G.; Zhang, N.; Willard, S.H.; Zheng, S.; Yu, X.F. ICAM-5/Telencephalin Is a Functional Entry Receptor for Enterovirus D68. Cell Host Microbe 2016, 20, 631–641. [Google Scholar] [CrossRef]
- Bergelson, J.M.; Shepley, M.P.; Chan, B.M.C.; Hemler, M.E.; Bergelson, J.M.; Shepley, M.P. Identification of the Integrin VLA-2 as a Receptor for Echovirus 1. Science 1992, 255, 1718–1720. [Google Scholar] [CrossRef]
- Brandenburg, B.; Lee, L.Y.; Lakadamyali, M.; Rust, M.J.; Zhuang, X.; Hogle, J.M. Imaging poliovirus entry in live cells. PLoS Biol. 2007, 5, 1543–1555. [Google Scholar] [CrossRef]
- Crowther, D.; Melnick, J.L. The incorporation of neutral red and acridine orange into developing poliovirus particles making them photosensitive. Virology 1961, 14, 11–21. [Google Scholar] [CrossRef]
- Coyne, C.B.; Kim, K.S.; Bergelson, J.M. Poliovirus entry into human brain microvascular cells requires receptor-induced activation of SHP-2. EMBO J. 2007, 26, 4016–4028. [Google Scholar] [CrossRef]
- Lulla, V.; Dinan, A.M.; Hosmillo, M.; Chaudhry, Y.; Sherry, L.; Irigoyen, N.; Nayak, K.M.; Stonehouse, N.J.; Zilbauer, M.; Goodfellow, I.; Firth, A.E. An upstream protein-coding region in enteroviruses modulates virus infection in gut epithelial cells. Nat. Microbiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Van Der Schaar, H.M.; Dorobantu, C.M.; Albulescu, L.; Strating, J.R.P.M.; Van Kuppeveld, F.J.M. Fat(al) attraction: Picornaviruses Usurp Lipid Transfer at Membrane Contact Sites to Create Replication Organelles. Trends Microbiol. 2016. [Google Scholar] [CrossRef]
- Baggen, J.; Thibaut, H.; P M Strating, J.R.; van Kuppeveld, F.J. The life cycle of non-polio enteroviruses and how to target it. Nat. Rev. Microbiol. 2018, 16. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Hensley, L.; McKnight, K.L.; Hu, F.; Madden, V.; Ping, L.; Jeong, S.H.; Walker, C.; Lanford, R.E.; Lemon, S.M. A pathogenic picornavirus acquires an envelope by hijacking cellular membranes. Nature 2013, 496, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Du, W.; Hagemeijer, M.C.; Takvorian, P.M.; Pau, C.; Cali, A.; Brantner, C.A.; Stempinski, E.S.; Connelly, P.S.; Ma, H.C.; et al. Phosphatidylserine vesicles enable efficient en bloc transmission of enteroviruses. Cell 2015, 160, 619–630. [Google Scholar] [CrossRef]
- Bird, S.W.; Maynard, N.D.; Covert, M.W.; Kirkegaard, K. Nonlytic viral spread enhanced by autophagy components. Proc. Natl. Acad. Sci. USA 2014, 111, 13081–13086. [Google Scholar] [CrossRef]
- Chi, C.; Sun, Q.; Wang, S.; Zhang, Z.; Li, X.; Cardona, C.J.; Jin, Y.; Xing, Z. Robust antiviral responses to enterovirus 71 infection in human intestinal epithelial cells. Virus Res. 2013, 176, 53–60. [Google Scholar] [CrossRef]
- Drummond, C.G.; Bolock, A.M.; Ma, C.; Luke, C.J.; Good, M.; Coyne, C.B. Enteroviruses infect human enteroids and induce antiviral signaling in a cell lineage-specific manner. PNAS 2017, 114, 1672–1677. [Google Scholar] [CrossRef]
- Good, C.; Wells, A.I.; Coyne, C.B. Type III interferon signaling restricts Enterovirus 71 infection of goblet cells. Sci. Adv. 2019, 5, 1–11. [Google Scholar] [CrossRef]
- Jensen, S.; Thomsen, A.R. Sensing of RNA Viruses: a Review of Innate Immune Receptors Involved in Recognizing RNA Virus Invasion. J. Virol. 2012. [Google Scholar] [CrossRef]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef]
- Bryant, C.E.; Gay, N.J.; Heymans, S.; Sacre, S.; Schaefer, L.; Midwood, K.S. Advances in Toll-like receptor biology: Modes of activation by diverse stimuli. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 359–379. [Google Scholar] [CrossRef]
- Brubaker, S.W.; Bonham, K.S.; Zanoni, I.; Kagan, J.C. Innate Immune Pattern Recognition: A Cell Biological Perspective. Annu. Rev. Immunol. 2015, 33, 257–290. [Google Scholar] [CrossRef]
- Alexopoulou, L.; Czopik Holt, A.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-kappa B by Toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef]
- Xu, J.; Yang, A.Y.; Wang, A.C.; Jiang, A.B. Rotavirus and coxsackievirus infection activated different profiles of toll-like receptors and chemokines in intestinal epithelial cells. Inflamm. Res. 2009, 58, 585–592. [Google Scholar] [CrossRef]
- Abe, Y.; Fujii, K.; Nagata, N.; Takeuchi, O.; Akira, S.; Oshiumi, H.; Matsumoto, M.; Seya, T.; Koike, S. The Toll-Like Receptor 3-Mediated Antiviral Response Is Important for Protection against Poliovirus Infection in Poliovirus Receptor Transgenic Mice. J. Virol. 2012, 185–194. [Google Scholar] [CrossRef]
- Yang, J.; Yang, C.; Guo, N.; Zhu, K.; Luo, K.; Zhang, N.; Zhao, H.; Cui, Y.; Chen, L.; Wang, H.; et al. Type I Interferons Triggered through the Toll-Like Receptor 3-TRIF Pathway Control Coxsackievirus A16 Infection in Young Mice. J. Virol. 2015, 89, 10860–10867. [Google Scholar] [CrossRef]
- Negishi, H.; Osawa, T.; Ogami, K.; Ouyang, X.; Sakaguchi, S.; Koshiba, R.; Yanai, H.; Seko, Y.; Shitara, H.; Bishop, K.; et al. A critical link between Toll-like receptor 3 and type II interferon signaling pathways in antiviral innate immunity. Proc. Natl. Acad. Sci. USA 2008, 105, 20446–20451. [Google Scholar] [CrossRef]
- Oshiumi, H.; Okamoto, M.; Fujii, K.; Kawanishi, T.; Matsumoto, M.; Koike, S.; Seya, T. The TLR3/TICAM-1 Pathway Is Mandatory for Innate Immune Responses to Poliovirus Infection. J. Immunol. 2011, 187, 5320–5327. [Google Scholar] [CrossRef]
- Wang, C.; Ji, L.; Yuan, X.; Jin, Y.; Cardona, C.J.; Xing, Z. Differential Regulation of TLR Signaling on the Induction of Antiviral Interferons in Human Intestinal Epithelial Cells Infected with Enterovirus 71. PLoS ONE 2016, 11. [Google Scholar] [CrossRef]
- Gorbea, C.; Makar, K.A.; Pauschinger, M.; Pratt, G.; Bersola, J.L.F.; Varela, J.; David, R.M.; Banks, L.; Huang, C.H.; Li, H.; et al. A role for toll-like receptor 3 variants in host susceptibility to enteroviral myocarditis and dilated cardiomyopathy. J. Biol. Chem. 2010, 285, 23208–23223. [Google Scholar] [CrossRef]
- Belkaya, S.; Kontorovich, A.R.; Byun, M.; Mulero-Navarro, S.; Bajolle, F.; Cobat, A.; Josowitz, R.; Itan, Y.; Quint, R.; Lorenzo, L.; et al. Autosomal Recessive Cardiomyopathy Presenting as Acute Myocarditis. J. Am. Coll. Cardiol. 2017, 69, 1653–1665. [Google Scholar] [CrossRef]
- Satoh, M.; Nakamura, M.; Akatsu, T.; Iwasaka, J.; Shimoda, Y.; Segawa, I.; Hiramori, K. Expression of Toll-like receptor 4 is associated with enteroviral replication in human myocarditis. Clin. Sci. 2003, 104, 577–584. [Google Scholar] [CrossRef]
- Satoh, M.; Nakamura, M.; Akatsu, T.; Shimoda, Y.; Segawa, I.; Hiramori, K. Toll-like receptor 4 is expressed with enteroviral replication in myocardium from patients with dilated cardiomyopathy. Lab. Investig. 2004, 84, 173–181. [Google Scholar] [CrossRef]
- Trianta, K.; Trianta, M. Coxsackievirus B4-Induced Cytokine Production in Pancreatic Cells Is Mediated through Toll-Like Receptor 4. J. Virol. 2004, 78, 11313–11320. [Google Scholar] [CrossRef]
- Hornung, V.; Guenthner-Biller, M.; Bourquin, C.; Ablasser, A.; Schlee, M.; Uematsu, S.; Noronha, A.; Manoharan, M.; Akira, S.; De Fougerolles, A.; et al. Sequence-specific potent induction of IFN-α by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat. Med. 2005, 11, 263–270. [Google Scholar] [CrossRef]
- Li, B.; Ma, H.M.; Wang, X.X.; Li, Y.Q.; Liu, H.B.; Hong, L.Z.; Li, X.; Zheng, W.H.; Ou, W.L. Expression and significance of toll-like receptors 7 and 8 in brain and lung tissues of death cases caused by EV71 infection. Chinese J. Contemp. Pediatr. 2015, 17, 1051–1055. [Google Scholar] [CrossRef]
- Triantafilou, K.; Orthopoulos, G.; Vakakis, E.; Ahmed, M.A.E.; Golenbock, D.T.; Lepper, P.M.; Triantafilou, M. Human cardiac inflammatory responses triggered by Coxsackie B viruses are mainly Toll-like receptor (TLR) 8-dependent. Cell. Microbiol. 2005, 7, 1117–1126. [Google Scholar] [CrossRef]
- Satoh, M.; Akatsu, T.; Ishikawa, Y.; Minami, Y.; Takahashi, Y.; Nakamura, M. Association between toll-like receptor 8 expression and adverse clinical outcomes in patients with enterovirus-associated dilated cardiomyopathy. Am. Heart J. 2007, 154, 581–588. [Google Scholar] [CrossRef]
- Yoneyama, M.; Kikuchi, M.; Matsumoto, K.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Foy, E.; Loo, Y.-M.; Gale, M.; Akira, S.; et al. Shared and Unique Functions of the DExD/H-Box Helicases RIG-I, MDA5, and LGP2 in Antiviral Innate Immunity. J. Immunol. 2005, 175, 2851–2858. [Google Scholar] [CrossRef]
- Hornung, V.; Kato, H.; Poeck, H.; Akira, S.; Conzelmann, K.; Schlee, M. 5’-Triphosphate RNA is the Ligand for RIG-I. Science. 2006, 314. [Google Scholar] [CrossRef]
- Francisco, E.; Suthar, M.; Gale, M.; Rosenfeld, A.B.; Racaniello, V.R. Cell-type specificity and functional redundancy of RIG-I-like receptors in innate immune sensing of Coxsackievirus B3 and encephalomyocarditis virus. Virology 2019, 528, 7–18. [Google Scholar] [CrossRef]
- Feng, Q.; Langereis, M.A.; Olagnier, D.; Chiang, C.; Van De Winkel, R. Coxsackievirus Cloverleaf RNA Containing a 59 Triphosphate Triggers an Antiviral Response via RIG-I Activation. PLoS ONE 2014, 9, 95927. [Google Scholar] [CrossRef]
- Pichlmair, A.; Schulz, O.; Tan, C.-P.; Rehwinkel, J.; Kato, H.; Takeuchi, O.; Akira, S.; Way, M.; Schiavo, G.; Reis e Sousa, C. Activation of MDA5 Requires Higher-Order RNA Structures Generated during Virus Infection. J. Virol. 2009, 83, 10761–10769. [Google Scholar] [CrossRef]
- Kato, H.; Takeuchi, O.; Mikamo-Satoh, E.; Hirai, R.; Kawai, T.; Matsushita, K.; Hiiragi, A.; Dermody, T.S.; Fujita, T.; Akira, S. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid–inducible gene-I and melanoma differentiation–associated gene 5. J. Exp. Med. 2008, 205, 1601–1610. [Google Scholar] [CrossRef]
- Wu, B.; Peisley, A.; Richards, C.; Yao, H.; Zeng, X.; Lin, C.; Chu, F.; Walz, T.; Hur, S. Structural basis for dsRNA recognition, filament formation, and antiviral signal activation by MDA5. Cell 2013, 152, 276–289. [Google Scholar] [CrossRef]
- Triantafilou, K.; Vakakis, E.; Kar, S.; Richer, E.; Evans, G.L.; Triantafilou, M. Visualisation of direct interaction of MDA5 and the dsRNA replicative intermediate form of positive strand RNA viruses. J. Cell Sci. 2012, 125, 4761–4769. [Google Scholar] [CrossRef]
- Kuo, R.L.; Kao, L.T.; Lin, S.J.; Wang, R.Y.L.; Shih, S.R. MDA5 Plays a Crucial Role in Enterovirus 71 RNA-Mediated IRF3 Activation. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Wang, J.P.; Cerny, A.; Asher, D.R.; Kurt-Jones, E.A.; Bronson, R.T.; Finberg, R.W. MDA5 and MAVS Mediate Type I Interferon Responses to Coxsackie B Virus. J. Virol. 2010, 84, 254–260. [Google Scholar] [CrossRef]
- Feng, Q.; Hato, S.V.; Langereis, M.A.; Zoll, J.; Virgen-Slane, R.; Peisley, A.; Hur, S.; Semler, B.L.; van Rij, R.P.; van Kuppeveld, F.J.M. MDA5 Detects the Double-Stranded RNA Replicative Form in Picornavirus-Infected Cells. Cell Rep. 2012, 2, 1187–1196. [Google Scholar] [CrossRef]
- Pang, L.; Gong, X.; Liu, N.; Xie, G.; Gao, W.; Kong, G.; Li, X.; Zhang, J.; Jin, Y.; Duan, Z. A polymorphism in melanoma differentiation-associated gene 5 may be a risk factor for enterovirus 71 infection. Clin. Microbiol. Infect. 2014, 20, O711–O717. [Google Scholar] [CrossRef]
- Hühn, M.H.; McCartney, S.A.; Lind, K.; Svedin, E.; Colonna, M.; Flodström-Tullberg, M. Melanoma differentiation-associated protein-5 (MDA-5) limits early viral replication but is not essential for the induction of type 1 interferons after Coxsackievirus infection. Virology 2010, 401, 42–48. [Google Scholar] [CrossRef]
- Gack, M.U.; Shin, Y.C.; Joo, C.H.; Urano, T.; Liang, C.; Sun, L.; Takeuchi, O.; Akira, S.; Chen, Z.; Inoue, S.; et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 2007, 446, 916–920. [Google Scholar] [CrossRef]
- Oshiumi, H.; Matsumoto, M.; Hatakeyama, S.; Seya, T. Riplet/RNF135, a RING finger protein, ubiquitinates RIG-I to promote interferon-beta induction during the early phase of viral infection. J. Biol. Chem. 2009, 284, 807–817. [Google Scholar] [CrossRef]
- Hou, F.; Sun, L.; Zheng, H.; Skaug, B.; Jiang, Q.X.; Chen, Z.J. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell 2011, 146, 448–461. [Google Scholar] [CrossRef]
- Patel, J.R.; García-Sastre, A. Activation and regulation of pathogen sensor RIG-I. Cytokine Growth Factor Rev. 2014, 25, 513–523. [Google Scholar] [CrossRef]
- Zhang, Q.M.; Song, W.Q.; Li, Y.J.; Qian, J.; Zhai, A.X.; Wu, J.; Li, A.M.; He, J.M.; Zhao, J.Y.; Yu, X.; et al. Over-expression of mitochondrial antiviral signaling protein inhibits coxsackievirus B3 infection by enhancing type-I interferons production. Virol. J. 2012, 9, 2–11. [Google Scholar] [CrossRef]
- Mukherjee, A.; Morosky, S.A.; Delorme-Axford, E.; Dybdahl-Sissoko, N.; Oberste, M.S.; Wang, T.; Coyne, C.B. The Coxsackievirus B 3C pro Protease Cleaves MAVS and TRIF to Attenuate Host Type I Interferon and Apoptotic Signaling. PLoS Pathog. 2011, 7. [Google Scholar] [CrossRef]
- Lei, X.; Sun, Z.; Liu, X.; Jin, Q.; He, B.; Wang, J. Cleavage of the Adaptor Protein TRIF by Enterovirus 71 3C Inhibits Antiviral Responses Mediated by Toll-Like Receptor 3. J. Virol. 2011, 85, 8811–8818. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Z.; Li, L.; Lei, X.; Zhou, H.; Zhou, Z.; He, B.; Wang, J. Enterovirus 68 3C Protease Cleaves TRIF To Attenuate Antiviral Responses Mediated by Toll-Like Receptor 3. J. Virol. 2014, 88, 6650–6659. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Li, X.; Li, P.; Pan, Z.; Ding, Y.; Zou, D.; Zheng, L.; Zhang, Y.; Li, L.; Xiao, L.; et al. Enterovirus 71 inhibits cellular type I interferon signaling by inhibiting host RIG-I ubiquitination. Microb. Pathog. 2016, 100, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Maelfait, J.; Beyaert, R. Emerging Role of Ubiquitination in Antiviral RIG-I Signaling. Microbiol. Mol. Biol. Rev. 2012, 76, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Barral, P.M.; Sarkar, D.; Fisher, P.B.; Racaniello, V.R. RIG-I is cleaved during picornavirus infection. Virology 2009, 391, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Rui, Y.; Su, J.; Wang, H.; Chang, J.; Wang, S.; Zheng, W.; Cai, Y.; Wei, W.; Gordy, J.T.; Markham, R.; et al. Disruption of MDA5-Mediated Innate Immune Responses by the 3C Proteins Coxsackievirus A16, Coxsackievirus A6, and Enterovirus D68. J. Virol. 2017, 91, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Langereis, M.A.; Lork, M.; Nguyen, M.; Hato, S.V.; Lanke, K.; Emdad, L.; Bhoopathi, P.; Fisher, P.B.; Lloyd, R.E.; et al. Enterovirus 2Apro Targets MDA5 and MAVS in Infected Cells. J. Virol. 2014, 88, 3369–3378. [Google Scholar] [CrossRef]
- Barral, P.M.; Morrison, J.M.; Drahos, J.; Gupta, P.; Sarkar, D.; Fisher, P.B.; Racaniello, V.R. MDA-5 Is Cleaved in Poliovirus-Infected Cells. J. Virol. 2007, 81, 3677–3684. [Google Scholar] [CrossRef]
- Wang, B.; Xi, X.; Lei, X.; Zhang, X.; Cui, S.; Wang, J.; Jin, Q.; Zhao, Z. Enterovirus 71 Protease 2Apro Targets MAVS to Inhibit Anti-Viral Type I Interferon Responses. PLoS Pathog. 2013, 9. [Google Scholar] [CrossRef]
- Alirezaei, M.; Flynn, C.T.; Whitton, J.L. Interactions between enteroviruses and autophagy in vivo. Autophagy 2012, 8, 973–975. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Hu, Y.; Li, J.; Zheng, H.; Wang, J.; Guo, L.; Shi, H.; Liu, L. Suppression of the toll-like receptor 7-dependent type I interferon production pathway by autophagy resulting from enterovirus 71 and coxsackievirus A16 infections facilitates their replication. Arch. Virol. 2017, 163, 135–144. [Google Scholar] [CrossRef]
- Friedman, C.S.; O’Donnell, M.A.; Legarda-Addison, D.; Ng, A.; Cárdenas, W.B.; Yount, J.S.; Moran, T.M.; Basler, C.F.; Komuro, A.; Horvath, C.M.; et al. The tumour suppressor CYLD is a negative regulator of RIG-I-mediated antiviral response. EMBO Rep. 2008, 9, 930–936. [Google Scholar] [CrossRef]
- Xu, C.; He, X.; Zheng, Z.; Zhang, Z.; Wei, C.; Guan, K.; Hou, L.; Zhang, B.; Zhu, L.; Cao, Y.; et al. Downregulation of MicroRNA miR-526a by Enterovirus Inhibits RIG-I-Dependent Innate Immune Response. J. Virol. 2014, 88, 11356–11368. [Google Scholar] [CrossRef]
- Sambuy, Y.; De Angelis, I.; Ranaldi, G.; Scarino, M.L.; Stammati, A.; Zucco, F. The Caco-2 cell line as a model of the intestinal barrier: Influence of cell and culture-related factors on Caco-2 cell functional characteristics. Cell Biol. Toxicol. 2005, 21, 1–26. [Google Scholar] [CrossRef]
- Drummond, C.G.; Nickerson, C.A.; Coyne, C.B. A Three-Dimensional Cell Culture Model To Study Enterovirus Infection of Polarized Intestinal Epithelial Cells. mSphere 2015, 1. [Google Scholar] [CrossRef]
- McCracken, K.W.; Howell, J.C.; Wells, J.M.; Spence, J.R. Generating human intestinal tissue from pluripotent stem cells in vitro. Nat. Protoc. 2011, 6, 1920–1928. [Google Scholar] [CrossRef]
- Spence, J.R.; Mayhew, C.N.; Rankin, S.A.; Kuhar, M.F.; Vallance, J.E.; Tolle, K.; Hoskins, E.E.; Kalinichenko, V.V.; Wells, S.I.; Zorn, A.M.; et al. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature 2011, 470, 105–110. [Google Scholar] [CrossRef]
- Stelzner, M.; Helmrath, M.; Dunn, J.C.Y.; Henning, S.J.; Houchen, C.W.; Kuo, C.; Lynch, J.; Li, L.; Magness, S.T.; Martin, M.G.; Wong, M.H.; et al. A nomenclature for intestinal in vitro cultures. AJP Gastrointest. Liver Physiol. 2012, 302, G1359–G1363. [Google Scholar] [CrossRef]
- Yin, X.; Mead, B.E.; Safaee, H.; Langer, R.; Karp, J.M.; Levy, O. Engineering Stem Cell Organoids. Cell Stem Cell 2016, 18, 25–38. [Google Scholar] [CrossRef]
- Kretzschmar, K.; Clevers, H. Organoids: Modeling Development and the Stem Cell Niche in a Dish. Dev. Cell 2016, 38, 590–600. [Google Scholar] [CrossRef]
- Saxena, K.; Blutt, S.E.; Ettayebi, K.; Zeng, X.-L.; Broughman, J.R.; Crawford, S.E.; Karandikar, U.C.; Sastri, N.P.; Conner, M.E.; Opekun, A.R.; et al. Human Intestinal Enteroids: a New Model To Study Human Rotavirus Infection, Host Restriction, and Pathophysiology. J. Virol. 2016, 90, 43–56. [Google Scholar] [CrossRef]
- Ettayebi, K.; Crawford, S.E.; Murakami, K.; Broughman, J.R.; Karandikar, U.; Tenge, V.R.; Neill, F.H.; Blutt, S.E.; Zeng, X.-L.; Qu, L.; et al. Replication of human noroviruses in stem cell-derived human enteroids. Science 2016, 353, 1387–1394. [Google Scholar] [CrossRef]
- Moon, C.; VanDussen, K.; Miyoshi, H.; Stappenbeck, T. Development of a primary mouse intestinal epithelial cell monolayer culture system to evaluate factors that modeulate IgA transcytosis. Mucosal Immunol. 2014, 7, 818–828. [Google Scholar] [CrossRef]
- Erickson, A.K.; Jesudhasan, P.R.; Mayer, M.J.; Narbad, A.; Winter, S.E.; Pfeiffer, J.K. Bacteria Facilitate Enteric Virus Co-infection of Mammalian Cells and Promote Genetic Recombination. Cell Host Microbe 2018, 23, 77–88. [Google Scholar] [CrossRef]
- Aguilera, E.R.; Nguyen, Y.; Sasaki, J.; Pfeiffer, K. Bacterial Stabilization of a Panel of Picornaviruses. mSphere 2019, 4, e00183-19. [Google Scholar] [CrossRef]
- Ren, R.; Costantini, F.; Gorgacz, E.J.; Lee, J.J.; Racaniello, V.R. Transgenic mice expressing a human poliovirus receptor: A new model for poliomyelitis. Cell 1990, 63, 353–362. [Google Scholar] [CrossRef]
- Sato, Y.; Koike, S.; Ong, K.C.; Fujii, K.; Yamayoshi, S.; Taya, C.; Shitara, H.; Nagata, N.; Shimanuki, M.; Wong, K.T. Transgenic mouse model for the study of enterovirus 71 neuropathogenesis. Proc. Natl. Acad. Sci. USA 2013, 110, 14753–14758. [Google Scholar] [CrossRef]
- Wang, Y.; Pfeiffer, J.K. Emergence of a large-plaque variant in mice infected with coxsackievirus B3. MBio 2016, 7, 1–10. [Google Scholar] [CrossRef]
- Ohka, S.; Igarashi, H.; Nagata, N.; Sakai, M.; Koike, S.; Nochi, T.; Kiyono, H.; Nomoto, A. Establishment of a Poliovirus Oral Infection System in Human Poliovirus Receptor-Expressing Transgenic Mice That Are Deficient in Alpha/Beta Interferon Receptor. J. Virol. 2007, 81, 7902–7912. [Google Scholar] [CrossRef]
- Robinson, C.M.; Wang, Y.; Pfeiffer, J.K. Sex-Dependent Intestinal Replication of an Enteric Virus. J. Virol. 2017, 91, e02101-16. [Google Scholar] [CrossRef]
- Bopegamage, S.; Kovacova, J.; Vargova, A.; Motusova, J.; Petrovicova, A.; Benkovicova, M.; Gomolcak, P.; Bakkers, J.; van Kuppeveld, F.; Melchers, W.J.G.; et al. Coxsackie B virus infection of mice: Inoculation by the oral route protects the pancreas from damage, but not from infection. J. Gen. Virol. 2005, 86, 3271–3280. [Google Scholar] [CrossRef]
- Yang, C.-H.; Liang, C.-T.; Jiang, S.-T.; Chen, K.-H.; Yang, C.-C.; Cheng, M.-L.; Ho, H.-Y. A Novel Murine Model Expressing a Chimeric mSCARB2/hSCARB2 Receptor is Highly Susceptible to Oral Infection with Clinical Isolates of EV71. J. Virol. 2019. [Google Scholar] [CrossRef]
- Chen, Y.C.; Yu, C.K.; Wang, Y.F.; Liu, C.C.; Su, I.J.; Lei, H.Y. A murine oral enterovirus 71 infection model with central nervous system involvement. J. Gen. Virol. 2004, 85, 69–77. [Google Scholar] [CrossRef]
- Zhang, Y.; Cui, W.; Liu, L.; Wang, J.; Zhao, H.; Liao, Y.; Na, R.; Dong, C.; Wang, L.; Xie, Z.; et al. Pathogenesis study of enterovirus 71 infection in rhesus monkeys. Lab. Investig. 2011, 91, 1337–1350. [Google Scholar] [CrossRef]
- Hashimoto, I.; Hagiwara, A.N.D.A. Pathogenicity of a Poliomyelitis-like disease in monkeys infected orally with Enterovirus 71: A model for human infection. Neuropathol. Appl. Neurobiol. 1982, 8, 149–156. [Google Scholar] [CrossRef]
- Liu, L.; Zhao, H.; Zhang, Y.; Wang, J.; Che, Y.; Dong, C.; Zhang, X.; Na, R.; Shi, H.; Jiang, L.; et al. Neonatal rhesus monkey is a potential animal model for studying pathogenesis of EV71 infection. Virology 2011, 412, 91–100. [Google Scholar] [CrossRef]
- Cammock, C.E.; Halnon, N.J.; Skoczylas, J.; Blanchard, J.; Bohm, R.; Miller, C.J.; Lai, C.; Krogstad, P.A. Myocarditis, Disseminated Infection, and Early Viral Persistence Following Experimental Coxsackievirus B Infection of Cynomolgus Monkeys. PLoS ONE 2013, 8, 1–8. [Google Scholar] [CrossRef]
- London, W.T.; Curfman, B.L.; Brown, R.L.; Notkins, A.L. Coxsackie Virus B4 Produces Transient Diabetes in Nonhuman Primates. Diabetes 1986, 35, 712–716. [Google Scholar]
- Bachtold, J.G.; Gebhardt, L.P.; Bubel, H.C. Cellular Immunity of Monkeys versus Antibody Titers Following Oral Inoculation with Poliomyelitis Virus. J. Immunol. 1955, 75, 475–477. [Google Scholar]
- Coid, C.; Beswick, T. The sensitivity of monkeys to subcutaneous inoculation of the Brunenders strain of type-I poliomyelitis virus. J. Pathol. Bacteriol. 1960, 79, 325–330. [Google Scholar] [CrossRef]
- Coid, C.R.; Beswick, T.S.L.; Tobin, J.O.H. The virulence of strains of poliovirus for cynomolgus monkeys after subcutaneous injection. J. Hyg. (Lond). 1961, 59, 387–394. [Google Scholar] [CrossRef]
- Shen, L.; Chen, C.Y.; Huang, D.; Wang, R.; Zhang, M.; Qian, L.; Zhu, Y.; Zhang, A.Z.; Yang, E.; Qaqish, A.; et al. Pathogenic Events in a Nonhuman Primate Model of Oral Poliovirus Infection Leading to Paralytic Poliomyelitis. J. Virol. 2017, 91, 1–15. [Google Scholar] [CrossRef]
- Nagata, N.; Iwasaki, T.; Ami, Y.; Tano, Y.; Harashima, A.; Suzaki, Y.; Sato, Y.; Hasegawa, H.; Sata, T.; Miyamura, T.; et al. Differential localization of neurons susceptible to enterovirus 71 and poliovirus type 1 in the central nervous system of cynomolgus monkeys after intravenous inoculation. J. Gen. Virol. 2004, 85, 2981–2989. [Google Scholar] [CrossRef]
- Sabin, A.B. Immunization of Chimpanzees and Human Beings With Avirulent Strains of Poliomyelitis Virus. Ann. N. Y. Acad. Sci. 1955, 61, 1050–1056. [Google Scholar] [CrossRef]
- Horstmann, D.M.; Melnick, J.L.; Ward, R.; José Sá Fleitas, M. The susceptibility of infant rhesus monkeys to poliomyelitis virus administered by mouth. J. Exp. Med. 1947, 86, 309–323. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Host Protein | EV Serotype | Role | Reference |
|---|---|---|---|
| PVR (CD155) | PV | Binding, entry, uncoating | [52] |
| CAR | CVB1, CVB2, CVB3, CVB4, CVB5, and CVB6 | Binding, entry, uncoating | [49,50,51] |
| DAF | CVA21, CVB1, CVB3 (some isolates), CVB5, E3, E6, E7, E11, E12, E13, E19, E19, E20, E21, E25, E29, and E30 | Attachment | [55,56] |
| SCARB2 | EV71, CVA7, CVA14, and CVA16 | Binding, entry, uncoating | [46] |
| PSGL1 | EV71, CVA2, CVA7, CVA10, CVA14, and CVA16 | Attachment | [47,59] |
| KREMEN1 | CVA10 | Binding, entry | [60] |
| Sialic acid | EV71 | Attachment | [61] |
| ICAM5 | EV-D68 | Binding, entry | [62] |
| Integrin ⍺2β1 (VLA-2) | E1 | Binding, entry, uncoating | [63] |
| FcRn | Echoviruses | Binding, entry, uncoating | [53,54] |
| Host Protein | EV Serotype | Mechanism of Cleavage | Reference |
|---|---|---|---|
| TRIF | CVB3 | 3Cpro | [114] |
| EV-D68 | 3Cpro | [115] | |
| EV71 | 3Cpro | [116] | |
| RIG-I | EV71 | Decreases ubiquitination of RIG-I inhibiting recruitment to MAVS | [117,118] |
| PV | 3Cpro | [119] | |
| MDA5 | CVA6 | 3Cpro | [120] |
| CVA16 | 3Cpro | [120] | |
| CVB3 | 2Apro | [121] | |
| EV-D68 | 3Cpro | [120] | |
| EV71 | Unknown | [104] | |
| PV | Caspase Dependent | [122] | |
| MAVS | CVB3 | 2Apro, 3Cpro | [114,121] |
| EV71 | 2Apro | [121,123] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wells, A.I.; Coyne, C.B. Enteroviruses: A Gut-Wrenching Game of Entry, Detection, and Evasion. Viruses 2019, 11, 460. https://doi.org/10.3390/v11050460
Wells AI, Coyne CB. Enteroviruses: A Gut-Wrenching Game of Entry, Detection, and Evasion. Viruses. 2019; 11(5):460. https://doi.org/10.3390/v11050460
Chicago/Turabian StyleWells, Alexandra I., and Carolyn B. Coyne. 2019. "Enteroviruses: A Gut-Wrenching Game of Entry, Detection, and Evasion" Viruses 11, no. 5: 460. https://doi.org/10.3390/v11050460
APA StyleWells, A. I., & Coyne, C. B. (2019). Enteroviruses: A Gut-Wrenching Game of Entry, Detection, and Evasion. Viruses, 11(5), 460. https://doi.org/10.3390/v11050460