The Antigenic Topology of Norovirus as Defined by B and T Cell Epitope Mapping: Implications for Universal Vaccines and Therapeutics

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
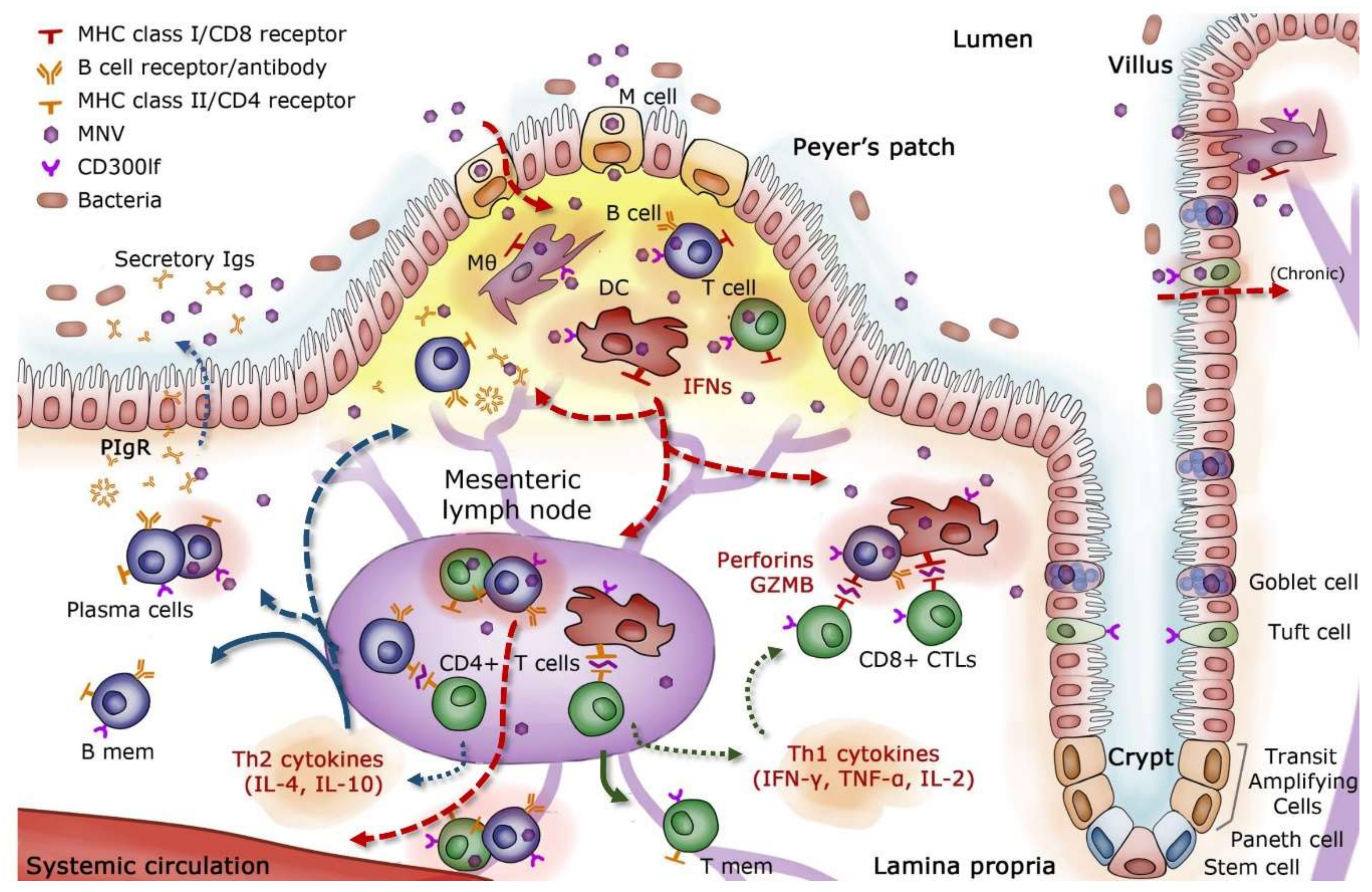
Abstract
1. Introduction
2. Antigenic Diversity of Noroviruses
3. Adaptive Immunity to Noroviruses
4. Isolation and Characterization of Norovirus Monoclonal Antibodies
4.1. Recombinant Norovirus Capsid Antigens
4.2. Origins and Types of Norovirus Monoclonal Antibodies
4.3. Binding, Blockade, and Neutralization Assays
4.4. Precision B Cell Epitope Mapping
5. HBGA Blockade and Human Norovirus Monoclonal Antibodies
5.1. Mechanisms of Antibody Blockade
5.2. Blockade Epitopes of Genogroup I Noroviruses
5.3. Blockade Epitopes of Genogroup II Noroviruses
6. Beyond Blockade: Virus Neutralization
6.1. Neutralization of Murine and Human Noroviruses
6.2. Beyond Receptor Binding Inhibition: Other Mechanisms of Neutralization
7. T Cell Epitope Mapping
8. Applications of Norovirus Epitope Studies
8.1. Improved Human Norovirus Diagnostics
8.2. Therapeutic Potential
8.3. Design of Universal Vaccines
9. Summary
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Patel, M.M.; Widdowson, M.A.; Glass, R.I.; Akazawa, K.; Vinje, J.; Parashar, U.D. Systematic literature review of role of noroviruses in sporadic gastroenteritis. Emerg. Infect. Dis. 2008, 14, 1224–1231. [Google Scholar] [CrossRef] [PubMed]
- Hoa Tran, T.N.; Trainor, E.; Nakagomi, T.; Cunliffe, N.A.; Nakagomi, O. Molecular epidemiology of noroviruses associated with acute sporadic gastroenteritis in children: Global distribution of genogroups, genotypes and GII.4 variants. J. Clin. Virol. 2013, 56, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.D.; Goulter, R.M.; Jaykus, L.A. Human norovirus as a foodborne pathogen: Challenges and developments. Annu. Rev. Food Sci. Technol. 2015, 6, 411–433. [Google Scholar] [CrossRef]
- Dolin, R.; Blacklow, N.R.; DuPont, H.; Buscho, R.F.; Wyatt, R.G.; Kasel, J.A.; Hornick, R.; Chanock, R.M. Biological properties of Norwalk agent of acute infectious nonbacterial gastroenteritis. Proc. Soc. Exp. Biol. Med. 1972, 140, 578–583. [Google Scholar] [CrossRef]
- Cutler, A.J.; Oliveira, J.; Ferreira, R.C.; Challis, B.; Walker, N.M.; Caddy, S.; Lu, J.; Stevens, H.E.; Smyth, D.J.; Pekalski, M.L.; et al. Capturing the systemic immune signature of a norovirus infection: An n-of-1 case study within a clinical trial—Wellcome Open Research. Wellcome Open Res. 2017, 2, 28. [Google Scholar] [CrossRef]
- Saito, M.; Goel-Apaza, S.; Espetia, S.; Velasquez, D.; Cabrera, L.; Loli, S.; Crabtree, J.E.; Black, R.E.; Kosek, M.; Checkley, W.; et al. Multiple norovirus infections in a birth cohort in a Peruvian Periurban community. Clin. Infect. Dis. 2014, 58, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Chan, M.C.; Wong, B.; Choi, K.W.; Sin, W.; Lui, G.; Chan, P.K.; Lai, R.W.; Cockram, C.S.; Sung, J.J.; et al. Fecal viral concentration and diarrhea in norovirus gastroenteritis. Emerg. Infect. Dis. 2007, 13, 1399–1401. [Google Scholar] [CrossRef] [PubMed]
- Van Asten, L.; Siebenga, J.; van den Wijngaard, C.; Verheij, R.; van Vliet, H.; Kretzschmar, M.; Boshuizen, H.; van Pelt, W.; Koopmans, M. Unspecified gastroenteritis illness and deaths in the elderly associated with norovirus epidemics. Epidemiology 2011, 22, 336–343. [Google Scholar] [CrossRef]
- Bok, K.; Green, K.Y. Norovirus gastroenteritis in immunocompromised patients. N. Engl. J. Med. 2012, 367, 2126–2132. [Google Scholar] [CrossRef]
- Payne, D.C.; Vinje, J.; Szilagyi, P.G.; Edwards, K.M.; Staat, M.A.; Weinberg, G.A.; Hall, C.B.; Chappell, J.; Bernstein, D.I.; Curns, A.T.; et al. Norovirus and medically attended gastroenteritis in U.S. children. N. Engl. J. Med. 2013, 368, 1121–1130. [Google Scholar] [CrossRef]
- Hickman, D.; Jones, M.K.; Zhu, S.; Kirkpatrick, E.; Ostrov, D.A.; Wang, X.; Ukhanova, M.; Sun, Y.; Mai, V.; Salemi, M.; et al. The effect of malnutrition on norovirus infection. MBio 2014, 5, e01032-13. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, S.M.; Lopman, B.A.; Ozawa, S.; Hall, A.J.; Lee, B.Y. Global Economic Burden of Norovirus Gastroenteritis. PLoS ONE 2016, 11, e0151219. [Google Scholar] [CrossRef] [PubMed]
- Scallan, E.; Hoekstra, R.M.; Angulo, F.J.; Tauxe, R.V.; Widdowson, M.A.; Roy, S.L.; Jones, J.L.; Griffin, P.M. Foodborne illness acquired in the United States—Major pathogens. Emerg. Infect. Dis. 2011, 17, 7–15. [Google Scholar] [CrossRef]
- Hoffmann, S.; Batz, M.B.; Morris, J.G., Jr. Annual cost of illness and quality-adjusted life year losses in the United States due to 14 foodborne pathogens. J. Food Prot. 2012, 75, 1292–1302. [Google Scholar] [CrossRef] [PubMed]
- Netzler, N.E.; Enosi Tuipulotu, D.; White, P.A. Norovirus antivirals: Where are we now? Med. Res. Rev. 2019, 39, 860–886. [Google Scholar] [CrossRef]
- Hardy, M.E. Norovirus protein structure and function. FEMS Microbiol. Lett. 2005, 253, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Prasad, B.V.; Hardy, M.E.; Dokland, T.; Bella, J.; Rossmann, M.G.; Estes, M.K. X-ray crystallographic structure of the Norwalk virus capsid. Science 1999, 286, 287–290. [Google Scholar] [CrossRef]
- Shanker, S.; Czako, R.; Sankaran, B.; Atmar, R.L.; Estes, M.K.; Prasad, B.V. Structural analysis of determinants of histo-blood group antigen binding specificity in genogroup I noroviruses. J. Virol. 2014, 88, 6168–6180. [Google Scholar] [CrossRef]
- Glass, P.J.; Zeng, C.Q.; Estes, M.K. Two nonoverlapping domains on the Norwalk virus open reading frame 3 (ORF3) protein are involved in the formation of the phosphorylated 35K protein and in ORF3-capsid protein interactions. J. Virol. 2003, 77, 3569–3577. [Google Scholar] [CrossRef]
- Smith, T.J. Structural studies on antibody recognition and neutralization of viruses. Curr. Opin. Virol. 2011, 1, 150–156. [Google Scholar] [CrossRef]
- Tan, M.; Jiang, X. Norovirus and its histo-blood group antigen receptors: An answer to a historical puzzle. Trends Microbiol. 2005, 13, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.K.; Leuthold, M.M.; Hansman, G.S. Human noroviruses’ fondness for histo-blood group antigens. J. Virol. 2015, 89, 2024–2040. [Google Scholar] [CrossRef]
- Zheng, D.P.; Ando, T.; Fankhauser, R.L.; Beard, R.S.; Glass, R.I.; Monroe, S.S. Norovirus classification and proposed strain nomenclature. Virology 2006, 346, 312–323. [Google Scholar] [CrossRef]
- Kroneman, A.; Vega, E.; Vennema, H.; Vinje, J.; White, P.A.; Hansman, G.; Green, K.; Martella, V.; Katayama, K.; Koopmans, M. Proposal for a unified norovirus nomenclature and genotyping. Arch. Virol. 2013, 158, 2059–2068. [Google Scholar] [CrossRef]
- Vinje, J. Advances in laboratory methods for detection and typing of norovirus. J. Clin. Microbiol. 2015, 53, 373–381. [Google Scholar] [CrossRef]
- Schneider, W.L.; Roossinck, M.J. Genetic diversity in RNA virus quasispecies is controlled by host-virus interactions. J. Virol. 2001, 75, 6566–6571. [Google Scholar] [CrossRef] [PubMed]
- Lauring, A.S.; Frydman, J.; Andino, R. The role of mutational robustness in RNA virus evolution. Nat. Rev. Microbiol. 2013, 11, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Bull, R.A.; Tu, E.T.; McIver, C.J.; Rawlinson, W.D.; White, P.A. Emergence of a new norovirus genotype II.4 variant associated with global outbreaks of gastroenteritis. J. Clin. Microbiol. 2006, 44, 327–333. [Google Scholar] [CrossRef]
- Patel, M.M.; Hall, A.J.; Vinje, J.; Parashar, U.D. Noroviruses: A comprehensive review. J. Clin. Virol. 2009, 44, 1–8. [Google Scholar] [CrossRef]
- Bok, K.; Abente, E.J.; Realpe-Quintero, M.; Mitra, T.; Sosnovtsev, S.V.; Kapikian, A.Z.; Green, K.Y. Evolutionary dynamics of GII.4 noroviruses over a 34-year period. J. Virol. 2009, 83, 11890–11901. [Google Scholar] [CrossRef] [PubMed]
- Zakikhany, K.; Allen, D.J.; Brown, D.; Iturriza-Gomara, M. Molecular evolution of GII-4 Norovirus strains. PLoS ONE 2012, 7, e41625. [Google Scholar] [CrossRef]
- Siebenga, J.J.; Vennema, H.; Renckens, B.; de Bruin, E.; van der Veer, B.; Siezen, R.J.; Koopmans, M. Epochal evolution of GGII.4 norovirus capsid proteins from 1995 to 2006. J. Virol. 2007, 81, 9932–9941. [Google Scholar] [CrossRef]
- Lindesmith, L.C.; Donaldson, E.F.; Lobue, A.D.; Cannon, J.L.; Zheng, D.P.; Vinje, J.; Baric, R.S. Mechanisms of GII.4 norovirus persistence in human populations. PLoS Med. 2008, 5, e31. [Google Scholar] [CrossRef]
- Allen, D.J.; Noad, R.; Samuel, D.; Gray, J.J.; Roy, P.; Iturriza-Gomara, M. Characterisation of a GII-4 norovirus variant-specific surface-exposed site involved in antibody binding. Virol. J. 2009, 6, 150. [Google Scholar] [CrossRef]
- Lindesmith, L.C.; Donaldson, E.F.; Baric, R.S. Norovirus GII.4 strain antigenic variation. J. Virol. 2011, 85, 231–242. [Google Scholar] [CrossRef]
- Lindesmith, L.C.; Beltramello, M.; Donaldson, E.F.; Corti, D.; Swanstrom, J.; Debbink, K.; Lanzavecchia, A.; Baric, R.S. Immunogenetic mechanisms driving norovirus GII.4 antigenic variation. PLoS Pathog. 2012, 8, e1002705. [Google Scholar] [CrossRef]
- Lindesmith, L.C.; Costantini, V.; Swanstrom, J.; Debbink, K.; Donaldson, E.F.; Vinje, J.; Baric, R.S. Emergence of a norovirus GII.4 strain correlates with changes in evolving blockade epitopes. J. Virol. 2013, 87, 2803–2813. [Google Scholar] [CrossRef]
- Debbink, K.; Lindesmith, L.C.; Donaldson, E.F.; Costantini, V.; Beltramello, M.; Corti, D.; Swanstrom, J.; Lanzavecchia, A.; Vinje, J.; Baric, R.S. Emergence of new pandemic GII.4 Sydney norovirus strain correlates with escape from herd immunity. J. Infect. Dis. 2013, 208, 1877–1887. [Google Scholar] [CrossRef]
- De Graaf, M.; van Beek, J.; Vennema, H.; Podkolzin, A.T.; Hewitt, J.; Bucardo, F.; Templeton, K.; Mans, J.; Nordgren, J.; Reuter, G.; et al. Emergence of a novel GII.17 norovirus—End of the GII.4 era? Euro Surveill. 2015, 20, 21178. [Google Scholar] [CrossRef]
- Zhang, X.F.; Huang, Q.; Long, Y.; Jiang, X.; Zhang, T.; Tan, M.; Zhang, Q.L.; Huang, Z.Y.; Li, Y.H.; Ding, Y.Q.; et al. An outbreak caused by GII.17 norovirus with a wide spectrum of HBGA-associated susceptibility. Sci. Rep. 2015, 5, 17687. [Google Scholar] [CrossRef]
- Singh, B.K.; Koromyslova, A.; Hefele, L.; Gurth, C.; Hansman, G.S. Structural Evolution of the Emerging 2014-2015 GII.17 Noroviruses. J. Virol. 2015, 90, 2710–2715. [Google Scholar] [CrossRef]
- Lu, J.; Fang, L.; Zheng, H.; Lao, J.; Yang, F.; Sun, L.; Xiao, J.; Lin, J.; Song, T.; Ni, T.; et al. The Evolution and Transmission of Epidemic GII.17 Noroviruses. J. Infect. Dis. 2016, 214, 556–564. [Google Scholar] [CrossRef]
- Parra, G.I.; Squires, R.B.; Karangwa, C.K.; Johnson, J.A.; Lepore, C.J.; Sosnovtsev, S.V.; Green, K.Y. Static and Evolving Norovirus Genotypes: Implications for Epidemiology and Immunity. PLoS Pathog. 2017, 13, e1006136. [Google Scholar] [CrossRef]
- Todd, K.V.; Tripp, R.A. Human Norovirus: Experimental Models of Infection. Viruses 2019, 11, 151. [Google Scholar] [CrossRef]
- Chachu, K.A.; Strong, D.W.; LoBue, A.D.; Wobus, C.E.; Baric, R.S.; Virgin, H.W.t. Antibody is critical for the clearance of murine norovirus infection. J. Virol. 2008, 82, 6610–6617. [Google Scholar] [CrossRef]
- Grau, K.R.; Roth, A.N.; Zhu, S.; Hernandez, A.; Colliou, N.; DiVita, B.B.; Philip, D.T.; Riffe, C.; Giasson, B.; Wallet, S.M.; et al. The major targets of acute norovirus infection are immune cells in the gut-associated lymphoid tissue. Nat. Microbiol. 2017, 2, 1586–1591. [Google Scholar] [CrossRef]
- Gonzalez-Hernandez, M.B.; Liu, T.; Payne, H.C.; Stencel-Baerenwald, J.E.; Ikizler, M.; Yagita, H.; Dermody, T.S.; Williams, I.R.; Wobus, C.E. Efficient norovirus and reovirus replication in the mouse intestine requires microfold (M) cells. J. Virol. 2014, 88, 6934–6943. [Google Scholar] [CrossRef]
- Perry, J.W.; Taube, S.; Wobus, C.E. Murine norovirus-1 entry into permissive macrophages and dendritic cells is pH-independent. Virus Res. 2009, 143, 125–129. [Google Scholar] [CrossRef]
- Wobus, C.E.; Karst, S.M.; Thackray, L.B.; Chang, K.O.; Sosnovtsev, S.V.; Belliot, G.; Krug, A.; Mackenzie, J.M.; Green, K.Y.; Virgin, H.W. Replication of Norovirus in cell culture reveals a tropism for dendritic cells and macrophages. PLoS Biol. 2004, 2, e432. [Google Scholar] [CrossRef]
- Jones, M.K.; Watanabe, M.; Zhu, S.; Graves, C.L.; Keyes, L.R.; Grau, K.R.; Gonzalez-Hernandez, M.B.; Iovine, N.M.; Wobus, C.E.; Vinje, J.; et al. Enteric bacteria promote human and mouse norovirus infection of B cells. Science 2014, 346, 755–759. [Google Scholar] [CrossRef]
- Karst, S.M.; Wobus, C.E.; Lay, M.; Davidson, J.; Virgin, H.W.T. STAT1-dependent innate immunity to a Norwalk-like virus. Science 2003, 299, 1575–1578. [Google Scholar] [CrossRef] [PubMed]
- Mumphrey, S.M.; Changotra, H.; Moore, T.N.; Heimann-Nichols, E.R.; Wobus, C.E.; Reilly, M.J.; Moghadamfalahi, M.; Shukla, D.; Karst, S.M. Murine norovirus 1 infection is associated with histopathological changes in immunocompetent hosts, but clinical disease is prevented by STAT1-dependent interferon responses. J. Virol. 2007, 81, 3251–3263. [Google Scholar] [CrossRef]
- Changotra, H.; Jia, Y.; Moore, T.N.; Liu, G.; Kahan, S.M.; Sosnovtsev, S.V.; Karst, S.M. Type I and type II interferons inhibit the translation of murine norovirus proteins. J. Virol. 2009, 83, 5683–5692. [Google Scholar] [CrossRef] [PubMed]
- Chachu, K.A.; LoBue, A.D.; Strong, D.W.; Baric, R.S.; Virgin, H.W. Immune mechanisms responsible for vaccination against and clearance of mucosal and lymphatic norovirus infection. PLoS Pathog. 2008, 4, e1000236. [Google Scholar] [CrossRef]
- Waugh, E.; Chen, A.; Baird, M.A.; Brown, C.M.; Ward, V.K. Characterization of the chemokine response of RAW264.7 cells to infection by murine norovirus. Virus Res. 2014, 181, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Groom, J.R.; Luster, A.D. CXCR3 ligands: Redundant, collaborative and antagonistic functions. Immunol. Cell Biol. 2011, 89, 207–215. [Google Scholar] [CrossRef]
- Park, K.; Cha, K.E.; Myung, H. Observation of inflammatory responses in mice orally fed with bacteriophage T7. J. Appl. Microbiol. 2014, 117, 627–633. [Google Scholar] [CrossRef]
- Tomov, V.T.; Osborne, L.C.; Dolfi, D.V.; Sonnenberg, G.F.; Monticelli, L.A.; Mansfield, K.; Virgin, H.W.; Artis, D.; Wherry, E.J. Persistent enteric murine norovirus infection is associated with functionally suboptimal virus-specific CD8 T cell responses. J. Virol. 2013, 87, 7015–7031. [Google Scholar] [CrossRef]
- Nice, T.J.; Baldridge, M.T.; McCune, B.T.; Norman, J.M.; Lazear, H.M.; Artyomov, M.; Diamond, M.S.; Virgin, H.W. Interferon-lambda cures persistent murine norovirus infection in the absence of adaptive immunity. Science 2015, 347, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Turula, H.; Bragazzi Cunha, J.; Mainou, B.A.; Ramakrishnan, S.K.; Wilke, C.A.; Gonzalez-Hernandez, M.B.; Pry, A.; Fava, J.; Bassis, C.M.; Edelman, J.; et al. Natural Secretory Immunoglobulins Promote Enteric Viral Infections. J. Virol. 2018, 92, e00826-18. [Google Scholar] [CrossRef]
- Zhu, S.; Regev, D.; Watanabe, M.; Hickman, D.; Moussatche, N.; Jesus, D.M.; Kahan, S.M.; Napthine, S.; Brierley, I.; Hunter, R.N., 3rd; et al. Identification of immune and viral correlates of norovirus protective immunity through comparative study of intra-cluster norovirus strains. PLoS Pathog. 2013, 9, e1003592. [Google Scholar] [CrossRef]
- Wilen, C.B.; Lee, S.; Hsieh, L.L.; Orchard, R.C.; Desai, C.; Hykes, B.L., Jr.; McAllaster, M.R.; Balce, D.R.; Feehley, T.; Brestoff, J.R.; et al. Tropism for tuft cells determines immune promotion of norovirus pathogenesis. Science 2018, 360, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Tomov, V.T.; Palko, O.; Lau, C.W.; Pattekar, A.; Sun, Y.; Tacheva, R.; Bengsch, B.; Manne, S.; Cosma, G.L.; Eisenlohr, L.C.; et al. Differentiation and Protective Capacity of Virus-Specific CD8(+) T Cells Suggest Murine Norovirus Persistence in an Immune-Privileged Enteric Niche. Immunity 2017, 47, 723–738.e5. [Google Scholar] [CrossRef] [PubMed]
- Mora, J.R. Homing imprinting and immunomodulation in the gut: Role of dendritic cells and retinoids. Inflamm. Bowel Dis. 2008, 14, 275–289. [Google Scholar] [CrossRef] [PubMed]
- Kolawole, A.O.; Xia, C.; Li, M.; Gamez, M.; Yu, C.; Rippinger, C.M.; Yucha, R.E.; Smith, T.J.; Wobus, C.E. Newly isolated mAbs broaden the neutralizing epitope in murine norovirus. J. Gen. Virol. 2014, 95, 1958–1968. [Google Scholar] [CrossRef] [PubMed]
- Wobus, C.E.; Thackray, L.B.; Virgin, H.W. Murine Norovirus: A Model System To Study Norovirus Biology and Pathogenesis. J. Virol. 2006, 80, 5104–5112. [Google Scholar] [CrossRef] [PubMed]
- Thackray, L.B.; Wobus, C.E.; Chachu, K.A.; Liu, B.; Alegre, E.R.; Henderson, K.S.; Kelley, S.T.; Virgin, H.W., 4th. Murine noroviruses comprising a single genogroup exhibit biological diversity despite limited sequence divergence. J. Virol. 2007, 81, 10460–10473. [Google Scholar] [CrossRef] [PubMed]
- Ettayebi, K.; Crawford, S.E.; Murakami, K.; Broughman, J.R.; Karandikar, U.; Tenge, V.R.; Neill, F.H.; Blutt, S.E.; Zeng, X.-L.; Qu, L.; et al. Replication of human noroviruses in stem cell–derived human enteroids. Science 2016, 353, 1387–1393. [Google Scholar] [CrossRef] [PubMed]
- Karandikar, U.C.; Crawford, S.E.; Ajami, N.J.; Murakami, K.; Kou, B.; Ettayebi, K.; Papanicolaou, G.A.; Jongwutiwes, U.; Perales, M.A.; Shia, J.; et al. Detection of human norovirus in intestinal biopsies from immunocompromised transplant patients. J. Gen. Virol. 2016, 97, 2291–2300. [Google Scholar] [CrossRef]
- Taube, S.; Perry, J.W.; Yetming, K.; Patel, S.P.; Auble, H.; Shu, L.; Nawar, H.F.; Lee, C.H.; Connell, T.D.; Shayman, J.A.; et al. Ganglioside-linked terminal sialic acid moieties on murine macrophages function as attachment receptors for murine noroviruses. J. Virol. 2009, 83, 4092–4101. [Google Scholar] [CrossRef]
- Hutson, A.M.; Atmar, R.L.; Marcus, D.M.; Estes, M.K. Norwalk virus-like particle hemagglutination by binding to h histo-blood group antigens. J. Virol. 2003, 77, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Orchard, R.C.; Wilen, C.B.; Doench, J.G.; Baldridge, M.T.; McCune, B.T.; Lee, Y.C.; Lee, S.; Pruett-Miller, S.M.; Nelson, C.A.; Fremont, D.H.; et al. Discovery of a proteinaceous cellular receptor for a norovirus. Science 2016, 353, 933–936. [Google Scholar] [CrossRef] [PubMed]
- Newman, K.L.; Moe, C.L.; Kirby, A.E.; Flanders, W.D.; Parkos, C.A.; Leon, J.S. Human norovirus infection and the acute serum cytokine response. Clin. Exp. Immunol. 2015, 182, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, D.S.; Blacklow, N.R.; Trier, J.S. The mucosal lesion of the proximal small intestine in acute infectious nonbacterial gastroenteritis. N. Engl. J. Med. 1973, 288, 1318–1323. [Google Scholar] [CrossRef] [PubMed]
- Long, K.Z.; Garcia, C.; Ko, G.; Santos, J.I.; Al Mamun, A.; Rosado, J.L.; DuPont, H.L.; Nathakumar, N. Vitamin A modifies the intestinal chemokine and cytokine responses to norovirus infection in Mexican children. J. Nutr. 2011, 141, 957–963. [Google Scholar] [CrossRef] [PubMed]
- Lindesmith, L.; Moe, C.; Lependu, J.; Frelinger, J.A.; Treanor, J.; Baric, R.S. Cellular and humoral immunity following Snow Mountain virus challenge. J. Virol. 2005, 79, 2900–2909. [Google Scholar] [CrossRef]
- Ponterio, E.; Petrizzo, A.; Di Bartolo, I.; Buonaguro, F.M.; Buonaguro, L.; Ruggeri, F.M. Pattern of activation of human antigen presenting cells by genotype GII.4 norovirus virus-like particles. J. Transl. Med. 2013, 11, 127. [Google Scholar] [CrossRef] [PubMed]
- Lindesmith, L.C.; Donaldson, E.; Leon, J.; Moe, C.L.; Frelinger, J.A.; Johnston, R.E.; Weber, D.J.; Baric, R.S. Heterotypic humoral and cellular immune responses following Norwalk virus infection. J. Virol. 2010, 84, 1800–1815. [Google Scholar] [CrossRef]
- Chen, S.M.; Lin, C.P.; Tsai, J.D.; Chao, Y.H.; Sheu, J.N. The significance of serum and fecal levels of interleukin-6 and interleukin-8 in hospitalized children with acute rotavirus and norovirus gastroenteritis. Pediatr. Neonatol. 2014, 55, 120–126. [Google Scholar] [CrossRef][Green Version]
- Malm, M.; Hyoty, H.; Knip, M.; Vesikari, T.; Blazevic, V. Development of T cell immunity to norovirus and rotavirus in children under five years of age. Sci. Rep. 2019, 9, 3199. [Google Scholar] [CrossRef]
- Malm, M.; Tamminen, K.; Vesikari, T.; Blazevic, V. Norovirus-Specific Memory T Cell Responses in Adult Human Donors. Front. Microbiol. 2016, 7, 1570. [Google Scholar] [CrossRef] [PubMed]
- Tacket, C.O.; Sztein, M.B.; Losonsky, G.A.; Wasserman, S.S.; Estes, M.K. Humoral, mucosal, and cellular immune responses to oral Norwalk virus-like particles in volunteers. Clin. Immunol. 2003, 108, 241–247. [Google Scholar] [CrossRef]
- Koo, H.L.; Ajami, N.; Atmar, R.L.; DuPont, H.L. Noroviruses: The leading cause of gastroenteritis worldwide. Discov. Med. 2010, 10, 61–70. [Google Scholar]
- Johnson, P.C.; Mathewson, J.J.; DuPont, H.L.; Greenberg, H.B. Multiple-challenge study of host susceptibility to Norwalk gastroenteritis in US adults. J. Infect. Dis. 1990, 161, 18–21. [Google Scholar] [CrossRef] [PubMed]
- Okhuysen, P.C.; Jiang, X.; Ye, L.; Johnson, P.C.; Estes, M.K. Viral shedding and fecal IgA response after Norwalk virus infection. J. Infect. Dis. 1995, 171, 566–569. [Google Scholar] [CrossRef]
- Lindesmith, L.C.; Ferris, M.T.; Mullan, C.W.; Ferreira, J.; Debbink, K.; Swanstrom, J.; Richardson, C.; Goodwin, R.R.; Baehner, F.; Mendelman, P.M.; et al. Broad Blockade Antibody Responses in Human Volunteers after Immunization with a Multivalent Norovirus VLP Candidate Vaccine: Immunological Analyses from a Phase I Clinical Trial. PLoS Med. 2015, 12, e1001807. [Google Scholar] [CrossRef]
- Bok, K.; Parra, G.I.; Mitra, T.; Abente, E.; Shaver, C.K.; Boon, D.; Engle, R.; Yu, C.; Kapikian, A.Z.; Sosnovtsev, S.V.; et al. Chimpanzees as an animal model for human norovirus infection and vaccine development. Proc. Natl. Acad. Sci. USA 2011, 108, 325–330. [Google Scholar] [CrossRef]
- Chen, Z.; Sosnovtsev, S.V.; Bok, K.; Parra, G.I.; Makiya, M.; Agulto, L.; Green, K.Y.; Purcell, R.H. Development of Norwalk virus-specific monoclonal antibodies with therapeutic potential for the treatment of Norwalk virus gastroenteritis. J. Virol. 2013, 87, 9547–9557. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Woodward, J.; Gkrania-Klotsas, E.; Kumararatne, D. Chronic norovirus infection and common variable immunodeficiency. Clin. Exp. Immunol. 2017, 188, 363–370. [Google Scholar] [CrossRef]
- Knoll, B.M.; Lindesmith, L.C.; Yount, B.L.; Baric, R.S.; Marty, F.M. Resolution of diarrhea in an immunocompromised patient with chronic norovirus gastroenteritis correlates with constitution of specific antibody blockade titer. Infection 2016, 44, 551–554. [Google Scholar] [CrossRef]
- Green, K.Y.; Lew, J.F.; Jiang, X.; Kapikian, A.Z.; Estes, M.K. Comparison of the reactivities of baculovirus-expressed recombinant Norwalk virus capsid antigen with those of the native Norwalk virus antigen in serologic assays and some epidemiologic observations. J. Clin. Microbiol. 1993, 31, 2185–2191. [Google Scholar] [PubMed]
- White, L.J.; Ball, J.M.; Hardy, M.E.; Tanaka, T.N.; Kitamoto, N.; Estes, M.K. Attachment and entry of recombinant Norwalk virus capsids to cultured human and animal cell lines. J. Virol. 1996, 70, 6589–6597. [Google Scholar] [PubMed]
- Jiang, X.; Wang, M.; Graham, D.Y.; Estes, M.K. Expression, self-assembly, and antigenicity of the Norwalk virus capsid protein. J. Virol. 1992, 66, 6527–6532. [Google Scholar]
- Harrington, P.R.; Yount, B.; Johnston, R.E.; Davis, N.; Moe, C.; Baric, R.S. Systemic, mucosal, and heterotypic immune induction in mice inoculated with Venezuelan equine encephalitis replicons expressing Norwalk virus-like particles. J. Virol. 2002, 76, 730–742. [Google Scholar] [CrossRef] [PubMed]
- Taube, S.; Kurth, A.; Schreier, E. Generation of recombinant Norovirus-like particles (VLP) in the human endothelial kidney cell line 293T. Arch. Virol. 2005, 150, 1425–1431. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Buehner, N.A.; Hutson, A.M.; Estes, M.K.; Mason, H.S. Tomato is a highly effective vehicle for expression and oral immunization with Norwalk virus capsid protein. Plant Biotechnol. J. 2006, 4, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Santi, L.; Batchelor, L.; Huang, Z.; Hjelm, B.; Kilbourne, J.; Arntzen, C.J.; Chen, Q.; Mason, H.S. An efficient plant viral expression system generating orally immunogenic Norwalk virus-like particles. Vaccine 2008, 26, 1846–1854. [Google Scholar] [CrossRef]
- Lindesmith, L.C.; Donaldson, E.F.; Beltramello, M.; Pintus, S.; Corti, D.; Swanstrom, J.; Debbink, K.; Jones, T.A.; Lanzavecchia, A.; Baric, R.S. Particle conformation regulates antibody access to a conserved GII.4 norovirus blockade epitope. J. Virol. 2014, 88, 8826–8842. [Google Scholar] [CrossRef]
- Tian, P.; Yang, D.; Jiang, X.; Zhong, W.; Cannon, J.L.; Burkhardt, W., 3rd; Woods, J.W.; Hartman, G.; Lindesmith, L.; Baric, R.S.; et al. Specificity and kinetics of norovirus binding to magnetic bead-conjugated histo-blood group antigens. J. Appl. Microbiol. 2010, 109, 1753–1762. [Google Scholar] [CrossRef]
- Kou, B.; Crawford, S.E.; Ajami, N.J.; Czako, R.; Neill, F.H.; Tanaka, T.N.; Kitamoto, N.; Palzkill, T.G.; Estes, M.K.; Atmar, R.L. Characterization of cross-reactive norovirus-specific monoclonal antibodies. Clin. Vaccine Immunol. 2015, 22, 160–167. [Google Scholar] [CrossRef]
- Rydell, G.E.; Nilsson, J.; Rodriguez-Diaz, J.; Ruvoen-Clouet, N.; Svensson, L.; Le Pendu, J.; Larson, G. Human noroviruses recognize sialyl Lewis x neoglycoprotein. Glycobiology 2009, 19, 309–320. [Google Scholar] [CrossRef]
- Garaicoechea, L.; Aguilar, A.; Parra, G.I.; Bok, M.; Sosnovtsev, S.V.; Canziani, G.; Green, K.Y.; Bok, K.; Parreño, V. Llama Nanoantibodies with Therapeutic Potential against Human Norovirus Diarrhea. PLoS ONE 2015, 10, e0133665. [Google Scholar] [CrossRef] [PubMed]
- Debbink, K.; Donaldson, E.F.; Lindesmith, L.C.; Baric, R.S. Genetic mapping of a highly variable norovirus GII.4 blockade epitope: Potential role in escape from human herd immunity. J. Virol. 2012, 86, 1214–1226. [Google Scholar] [CrossRef]
- Parra, G.I.; Abente, E.J.; Sandoval-Jaime, C.; Sosnovtsev, S.V.; Bok, K.; Green, K.Y. Multiple antigenic sites are involved in blocking the interaction of GII.4 norovirus capsid with ABH histo-blood group antigens. J. Virol. 2012, 86, 7414–7426. [Google Scholar] [CrossRef]
- Debbink, K.; Lindesmith, L.C.; Donaldson, E.F.; Swanstrom, J.; Baric, R.S. Chimeric GII.4 norovirus virus-like-particle-based vaccines induce broadly blocking immune responses. J. Virol. 2014, 88, 7256–7266. [Google Scholar] [CrossRef] [PubMed]
- Parker, T.D.; Kitamoto, N.; Tanaka, T.; Hutson, A.M.; Estes, M.K. Identification of Genogroup I and Genogroup II broadly reactive epitopes on the norovirus capsid. J. Virol. 2005, 79, 7402–7409. [Google Scholar] [CrossRef] [PubMed]
- Swanstrom, J.; Lindesmith, L.C.; Donaldson, E.F.; Yount, B.; Baric, R.S. Characterization of blockade antibody responses in GII.2.1976 Snow Mountain virus-infected subjects. J. Virol. 2014, 88, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Hegde, R.S.; Jiang, X. The P domain of norovirus capsid protein forms dimer and binds to histo-blood group antigen receptors. J. Virol. 2004, 78, 6233–6242. [Google Scholar] [CrossRef]
- Tan, M.; Fang, P.; Chachiyo, T.; Xia, M.; Huang, P.; Fang, Z.; Jiang, W.; Jiang, X. Noroviral P particle: Structure, function and applications in virus-host interaction. Virology 2008, 382, 115–123. [Google Scholar] [CrossRef]
- Tan, M.; Fang, P.A.; Xia, M.; Chachiyo, T.; Jiang, W.; Jiang, X. Terminal modifications of norovirus P domain resulted in a new type of subviral particles, the small P particles. Virology 2011, 410, 345–352. [Google Scholar] [CrossRef]
- Tan, M.; Jiang, X. The p domain of norovirus capsid protein forms a subviral particle that binds to histo-blood group antigen receptors. J. Virol. 2005, 79, 14017–14030. [Google Scholar] [CrossRef]
- Tamminen, K.; Huhti, L.; Koho, T.; Lappalainen, S.; Hytonen, V.P.; Vesikari, T.; Blazevic, V. A comparison of immunogenicity of norovirus GII-4 virus-like particles and P-particles. Immunology 2012, 135, 89–99. [Google Scholar] [CrossRef]
- Fang, H.; Tan, M.; Xia, M.; Wang, L.; Jiang, X. Norovirus P particle efficiently elicits innate, humoral and cellular immunity. PLoS ONE 2013, 8, e63269. [Google Scholar] [CrossRef]
- Batten, C.A.; Clarke, I.N.; Kempster, S.L.; Oliver, S.L.; Bridger, J.C.; Lambden, P.R. Characterization of a cross-reactive linear epitope in human genogroup I and bovine genogroup III norovirus capsid proteins. Virology 2006, 356, 179–187. [Google Scholar] [CrossRef][Green Version]
- Oliver, S.L.; Batten, C.A.; Deng, Y.; Elschner, M.; Otto, P.; Charpilienne, A.; Clarke, I.N.; Bridger, J.C.; Lambden, P.R. Genotype 1 and genotype 2 bovine noroviruses are antigenically distinct but share a cross-reactive epitope with human noroviruses. J. Clin. Microbiol. 2006, 44, 992–998. [Google Scholar] [CrossRef]
- Li, X.; Zhou, R.; Tian, X.; Li, H.; Zhou, Z. Characterization of a cross-reactive monoclonal antibody against Norovirus genogroups I, II, III and V. Virus Res. 2010, 151, 142–147. [Google Scholar] [CrossRef]
- Gray, J.J.; Cunliffe, C.; Ball, J.; Graham, D.Y.; Desselberger, U.; Estes, M.K. Detection of immunoglobulin M (IgM), IgA, and IgG Norwalk virus-specific antibodies by indirect enzyme-linked immunosorbent assay with baculovirus-expressed Norwalk virus capsid antigen in adult volunteers challenged with Norwalk virus. J. Clin. Microbiol. 1994, 32, 3059–3063. [Google Scholar]
- Alvarado, G.; Ettayebi, K.; Atmar, R.L.; Bombardi, R.G.; Kose, N.; Estes, M.K.; Crowe, J.E., Jr. Human Monoclonal Antibodies That Neutralize Pandemic GII.4 Noroviruses. Gastroenterology 2018, 155, 1898–1907. [Google Scholar] [CrossRef]
- Koromyslova, A.D.; Morozov, V.A.; Hefele, L.; Hansman, G.S. Human Norovirus Neutralized by a Monoclonal Antibody Targeting the HBGA Pocket. J. Virol. 2019, 93, e02174-18. [Google Scholar] [CrossRef]
- Tamminen, K.; Malm, M.; Vesikari, T.; Blazevic, V. Norovirus-specific mucosal antibodies correlate to systemic antibodies and block norovirus virus-like particles binding to histo-blood group antigens. Clin. Immunol. 2018, 197, 110–117. [Google Scholar] [CrossRef]
- Lindesmith, L.C.; Beltramello, M.; Swanstrom, J.; Jones, T.A.; Corti, D.; Lanzavecchia, A.; Baric, R.S. Serum Immunoglobulin A Cross-Strain Blockade of Human Noroviruses. Open Forum Infect. Dis. 2015, 2, ofv084. [Google Scholar] [CrossRef] [PubMed]
- Lindesmith, L.; Moe, C.; Marionneau, S.; Ruvoen, N.; Jiang, X.; Lindblad, L.; Stewart, P.; LePendu, J.; Baric, R. Human susceptibility and resistance to Norwalk virus infection. Nat. Med. 2003, 9, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Ramani, S.; Neill, F.H.; Opekun, A.R.; Gilger, M.A.; Graham, D.Y.; Estes, M.K.; Atmar, R.L. Mucosal and Cellular Immune Responses to Norwalk Virus. J. Infect. Dis. 2015, 212, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Sapparapu, G.; Czako, R.; Alvarado, G.; Shanker, S.; Prasad, B.V.; Atmar, R.L.; Estes, M.K.; Crowe, J.E., Jr. Frequent Use of the IgA Isotype in Human B Cells Encoding Potent Norovirus-Specific Monoclonal Antibodies That Block HBGA Binding. PLoS Pathog. 2016, 12, e1005719. [Google Scholar] [CrossRef] [PubMed]
- Shanker, S.; Czako, R.; Sapparapu, G.; Alvarado, G.; Viskovska, M.; Sankaran, B.; Atmar, R.L.; Crowe, J.E., Jr.; Estes, M.K.; Prasad, B.V. Structural basis for norovirus neutralization by an HBGA blocking human IgA antibody. Proc. Natl. Acad. Sci. USA 2016, 113, E5830–E5837. [Google Scholar] [CrossRef]
- Muyldermans, S. Nanobodies: Natural single-domain antibodies. Annu. Rev. Biochem. 2013, 82, 775–797. [Google Scholar] [CrossRef] [PubMed]
- Koromyslova, A.D.; Hansman, G.S. Nanobody binding to a conserved epitope promotes norovirus particle disassembly. J. Virol. 2015, 89, 2718–2730. [Google Scholar] [CrossRef]
- Doerflinger, S.Y.; Tabatabai, J.; Schnitzler, P.; Farah, C.; Rameil, S.; Sander, P.; Koromyslova, A.; Hansman, G.S. Development of a Nanobody-Based Lateral Flow Immunoassay for Detection of Human Norovirus. mSphere 2016, 1, e00219-16. [Google Scholar] [CrossRef]
- Koromyslova, A.D.; Hansman, G.S. Nanobodies targeting norovirus capsid reveal functional epitopes and potential mechanisms of neutralization. PLoS Pathog. 2017, 13, e1006636. [Google Scholar] [CrossRef]
- Ruoff, K.; Kilic, T.; Devant, J.; Koromyslova, A.; Ringel, A.; Hempelmann, A.; Geiss, C.; Graf, J.; Haas, M.; Roggenbach, I.; et al. Structural Basis of Nanobodies Targeting the Prototype Norovirus. J. Virol. 2019, 93, e02005-18. [Google Scholar] [CrossRef]
- Huston, J.S.; McCartney, J.; Tai, M.S.; Mottola-Hartshorn, C.; Jin, D.; Warren, F.; Keck, P.; Oppermann, H. Medical applications of single-chain antibodies. Int. Rev. Immunol. 1993, 10, 195–217. [Google Scholar] [CrossRef]
- Parra, G.I.; Sosnovtsev, S.V.; Abente, E.J.; Sandoval-Jaime, C.; Bok, K.; Dolan, M.A.; Green, K.Y. Mapping and modeling of a strain-specific epitope in the Norwalk virus capsid inner shell. Virology 2016, 492, 232–241. [Google Scholar] [CrossRef]
- Burton-MacLeod, J.A.; Kane, E.M.; Beard, R.S.; Hadley, L.A.; Glass, R.I.; Ando, T. Evaluation and comparison of two commercial enzyme-linked immunosorbent assay kits for detection of antigenically diverse human noroviruses in stool samples. J. Clin. Microbiol. 2004, 42, 2587–2595. [Google Scholar] [CrossRef]
- Kou, B.; Huang, W.; Neill, F.H.; Palzkill, T.; Estes, M.K.; Atmar, R.L. Norovirus Antigen Detection with a Combination of Monoclonal and Single-Chain Antibodies. J. Clin. Microbiol. 2015, 53, 3916–3918. [Google Scholar] [CrossRef]
- De Rougemont, A.; Ruvoen-Clouet, N.; Simon, B.; Estienney, M.; Elie-Caille, C.; Aho, S.; Pothier, P.; Le Pendu, J.; Boireau, W.; Belliot, G. Qualitative and Quantitative Analysis of the Binding of GII.4 Norovirus Variants onto Human Blood Group Antigens. J. Virol. 2011, 85, 4057–4070. [Google Scholar] [CrossRef]
- Heggelund, J.E.; Varrot, A.; Imberty, A.; Krengel, U. Histo-blood group antigens as mediators of infections. Curr. Opin. Struct. Biol. 2017, 44, 190–200. [Google Scholar] [CrossRef]
- Koromyslova, A.D.; Leuthold, M.M.; Bowler, M.W.; Hansman, G.S. The sweet quartet: Binding of fucose to the norovirus capsid. Virology 2015, 483, 203–208. [Google Scholar] [CrossRef]
- Le Pendu, J.; Ruvoen-Clouet, N.; Kindberg, E.; Svensson, L. Mendelian resistance to human norovirus infections. Semin. Immunol. 2006, 18, 375–386. [Google Scholar] [CrossRef]
- Reeck, A.; Kavanagh, O.; Estes, M.K.; Opekun, A.R.; Gilger, M.A.; Graham, D.Y.; Atmar, R.L. Serological correlate of protection against norovirus-induced gastroenteritis. J. Infect. Dis. 2010, 202, 1212–1218. [Google Scholar] [CrossRef]
- Tan, M.; Jiang, X. Norovirus-host interaction: Multi-selections by human histo-blood group antigens. Trends Microbiol. 2011, 19, 382–388. [Google Scholar] [CrossRef]
- Shanker, S.; Choi, J.M.; Sankaran, B.; Atmar, R.L.; Estes, M.K.; Prasad, B.V. Structural analysis of histo-blood group antigen binding specificity in a norovirus GII.4 epidemic variant: Implications for epochal evolution. J. Virol. 2011, 85, 8635–8645. [Google Scholar] [CrossRef]
- Nordgren, J.; Kindberg, E.; Lindgren, P.E.; Matussek, A.; Svensson, L. Norovirus gastroenteritis outbreak with a secretor-independent susceptibility pattern, Sweden. Emerg. Infect. Dis. 2010, 16, 81–87. [Google Scholar] [CrossRef]
- Donaldson, E.F.; Lindesmith, L.C.; Lobue, A.D.; Baric, R.S. Viral shape-shifting: Norovirus evasion of the human immune system. Nat. Rev. Microbiol. 2010, 8, 231–241. [Google Scholar] [CrossRef]
- Frenck, R.; Bernstein, D.I.; Xia, M.; Huang, P.; Zhong, W.; Parker, S.; Dickey, M.; McNeal, M.; Jiang, X. Predicting susceptibility to norovirus GII.4 by use of a challenge model involving humans. J. Infect. Dis. 2012, 206, 1386–1393. [Google Scholar] [CrossRef]
- Czako, R.; Atmar, R.L.; Opekun, A.R.; Gilger, M.A.; Graham, D.Y.; Estes, M.K. Serum hemagglutination inhibition activity correlates with protection from gastroenteritis in persons infected with Norwalk virus. Clin. Vaccine Immunol. 2012, 19, 284–287. [Google Scholar] [CrossRef]
- Lindesmith, L.C.; Debbink, K.; Swanstrom, J.; Vinje, J.; Costantini, V.; Baric, R.S.; Donaldson, E.F. Monoclonal antibody-based antigenic mapping of norovirus GII.4-2002. J. Virol. 2012, 86, 873–883. [Google Scholar] [CrossRef]
- Atmar, R.L.; Bernstein, D.I.; Harro, C.D.; Al-Ibrahim, M.S.; Chen, W.H.; Ferreira, J.; Estes, M.K.; Graham, D.Y.; Opekun, A.R.; Richardson, C.; et al. Norovirus vaccine against experimental human Norwalk Virus illness. N. Engl. J. Med. 2011, 365, 2178–2187. [Google Scholar] [CrossRef]
- Green, K.Y.; Belliot, G.; Taylor, J.L.; Valdesuso, J.; Lew, J.F.; Kapikian, A.Z.; Lin, F.Y. A predominant role for Norwalk-like viruses as agents of epidemic gastroenteritis in Maryland nursing homes for the elderly. J. Infect. Dis. 2002, 185, 133–146. [Google Scholar] [CrossRef]
- Carmona-Vicente, N.; Allen, D.J.; Rodríguez-Díaz, J.; Iturriza-Gómara, M.; Buesa, J. Antibodies against Lewis antigens inhibit the binding of human norovirus GII.4 virus-like particles to saliva but not to intestinal Caco-2 cells. Virol. J. 2016, 13, 82. [Google Scholar] [CrossRef]
- Debbink, K.; Lindesmith, L.C.; Ferris, M.T.; Swanstrom, J.; Beltramello, M.; Corti, D.; Lanzavecchia, A.; Baric, R.S. Within-host evolution results in antigenically distinct GII.4 noroviruses. J. Virol. 2014, 88, 7244–7255. [Google Scholar] [CrossRef]
- Karst, S.M.; Baric, R.S. What is the reservoir of emergent human norovirus strains? J. Virol. 2015, 89, 5756–5759. [Google Scholar] [CrossRef]
- Van Beek, J.; de Graaf, M.; Smits, S.; Schapendonk, C.M.E.; Verjans, G.; Vennema, H.; van der Eijk, A.A.; Phan, M.V.T.; Cotten, M.; Koopmans, M. Whole-Genome Next-Generation Sequencing to Study Within-Host Evolution of Norovirus (NoV) Among Immunocompromised Patients With Chronic NoV Infection. J. Infect. Dis. 2017, 216, 1513–1524. [Google Scholar] [CrossRef]
- Schorn, R.; Hohne, M.; Meerbach, A.; Bossart, W.; Wuthrich, R.P.; Schreier, E.; Muller, N.J.; Fehr, T. Chronic norovirus infection after kidney transplantation: Molecular evidence for immune-driven viral evolution. Clin. Infect. Dis. 2010, 51, 307–314. [Google Scholar] [CrossRef]
- Lindesmith, L.C.; Brewer-Jensen, P.D.; Mallory, M.L.; Yount, B.; Collins, M.H.; Debbink, K.; Graham, R.L.; Baric, R.S. Human Norovirus Epitope D Plasticity Allows Escape from Antibody Immunity without Loss of Capacity for Binding Cellular Ligands. J. Virol. 2019, 93, e01813-18. [Google Scholar] [CrossRef]
- Choi, J.M.; Hutson, A.M.; Estes, M.K.; Prasad, B.V. Atomic resolution structural characterization of recognition of histo-blood group antigens by Norwalk virus. Proc. Natl. Acad. Sci. USA 2008, 105, 9175–9180. [Google Scholar] [CrossRef]
- Mallagaray, A.; Lockhauserbaumer, J.; Hansman, G.; Uetrecht, C.; Peters, T. Attachment of norovirus to histo blood group antigens: A cooperative multistep process. Angew. Chem. Int. Ed. Engl. 2015, 54, 12014–12019. [Google Scholar] [CrossRef]
- Tan, M.; Xia, M.; Chen, Y.; Bu, W.; Hegde, R.S.; Meller, J.; Li, X.; Jiang, X. Conservation of carbohydrate binding interfaces: Evidence of human HBGA selection in norovirus evolution. PLoS ONE 2009, 4, e5058. [Google Scholar] [CrossRef]
- Allen, D.J.; Gray, J.J.; Gallimore, C.I.; Xerry, J.; Iturriza-Gomara, M. Analysis of amino acid variation in the P2 domain of the GII-4 norovirus VP1 protein reveals putative variant-specific epitopes. PLoS ONE 2008, 3, e1485. [Google Scholar] [CrossRef]
- Siebenga, J.J.; Lemey, P.; Kosakovsky Pond, S.L.; Rambaut, A.; Vennema, H.; Koopmans, M. Phylodynamic reconstruction reveals norovirus GII.4 epidemic expansions and their molecular determinants. PLoS Pathog. 2010, 6, e1000884. [Google Scholar] [CrossRef]
- Lindesmith, L.C.; Mallory, M.L.; Debbink, K.; Donaldson, E.F.; Brewer-Jensen, P.D.; Swann, E.W.; Sheahan, T.P.; Graham, R.L.; Beltramello, M.; Corti, D.; et al. Conformational Occlusion of Blockade Antibody Epitopes, a Novel Mechanism of GII.4 Human Norovirus Immune Evasion. mSphere 2018, 3, e00518-17. [Google Scholar] [CrossRef]
- Kolawole, A.O.; Smith, H.Q.; Svoboda, S.A.; Lewis, M.S.; Sherman, M.B.; Lynch, G.C.; Pettitt, B.M.; Smith, T.J.; Wobus, C.E. Norovirus Escape from Broadly Neutralizing Antibodies Is Limited to Allostery-Like Mechanisms. mSphere 2017, 2, e00334-17. [Google Scholar] [CrossRef]
- Kolawole, A.O.; Li, M.; Xia, C.; Fischer, A.E.; Giacobbi, N.S.; Rippinger, C.M.; Proescher, J.B.; Wu, S.K.; Bessling, S.L.; Gamez, M.; et al. Flexibility in surface-exposed loops in a virus capsid mediates escape from antibody neutralization. J. Virol. 2014, 88, 4543–4557. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lochridge, V.P.; Jutila, K.L.; Graff, J.W.; Hardy, M.E. Epitopes in the P2 domain of norovirus VP1 recognized by monoclonal antibodies that block cell interactions. J. Gen. Virol. 2005, 86, 2799–2806. [Google Scholar] [CrossRef] [PubMed]
- Ettayebi, K.; Hardy, M.E. Recombinant norovirus-specific scFv inhibit virus-like particle binding to cellular ligands. Virol. J. 2008, 5, 21. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yildiz, M.; Kocak, A. Molecular Dynamics Studies of Histo-Blood Group Antigen Blocking Human Immunoglobulin A Antibody and Escape Mechanism in Noroviruses Upon Mutation. J. Comput. Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Debbink, K.; Lindesmith, L.C.; Donaldson, E.F.; Baric, R.S. Norovirus immunity and the great escape. PLoS Pathog. 2012, 8, e1002921. [Google Scholar] [CrossRef] [PubMed]
- Lindesmith, L.C.; Kocher, J.F.; Donaldson, E.F.; Debbink, K.; Mallory, M.L.; Swann, E.W.; Brewer-Jensen, P.D.; Baric, R.S. Emergence of Novel Human Norovirus GII.17 Strains Correlates With Changes in Blockade Antibody Epitopes. J. Infect. Dis. 2017, 216, 1227–1234. [Google Scholar] [CrossRef]
- Lindesmith, L.C.; Brewer-Jensen, P.D.; Mallory, M.L.; Debbink, K.; Swann, E.W.; Vinje, J.; Baric, R.S. Antigenic characterization of a novel recombinant GII.P16-GII.4 Sydney norovirus strain with minor sequence variation leading to antibody escape. J. Infect. Dis. 2018, 217, 1145–1152. [Google Scholar] [CrossRef]
- Carmona-Vicente, N.; Vila-Vicent, S.; Allen, D.; Gozalbo-Rovira, R.; Iturriza-Gómara, M.; Buesa, J.; Rodríguez-Díaz, J.; López, S. Characterization of a Novel Conformational GII.4 Norovirus Epitope: Implications for Norovirus-Host Interactions. J. Virol. 2016, 90, 7703–7714. [Google Scholar] [CrossRef]
- Cannon, J.P.; O’Driscoll, M.; Litman, G.W. Specific lipid recognition is a general feature of CD300 and TREM molecules. Immunogenetics 2012, 64, 39–47. [Google Scholar] [CrossRef]
- Kilic, T.; Koromyslova, A.; Malak, V.; Hansman, G.S. Atomic Structure of the Murine Norovirus Protruding Domain and Soluble CD300lf Receptor Complex. J. Virol. 2018, 92, e00413-18. [Google Scholar] [CrossRef]
- Nelson, C.A.; Wilen, C.B.; Dai, Y.N.; Orchard, R.C.; Kim, A.S.; Stegeman, R.A.; Hsieh, L.L.; Smith, T.J.; Virgin, H.W.; Fremont, D.H. Structural basis for murine norovirus engagement of bile acids and the CD300lf receptor. Proc. Natl. Acad. Sci. USA 2018, 115, E9201–E9210. [Google Scholar] [CrossRef]
- Kilic, T.; Koromyslova, A.; Hansman, G.S. Structural Basis for Human Norovirus Capsid Binding to Bile Acids. J. Virol. 2019, 93, e01581-18. [Google Scholar] [CrossRef]
- Taube, S.; Rubin, J.R.; Katpally, U.; Smith, T.J.; Kendall, A.; Stuckey, J.A.; Wobus, C.E. High-resolution x-ray structure and functional analysis of the murine norovirus 1 capsid protein protruding domain. J. Virol. 2010, 84, 5695–5705. [Google Scholar] [CrossRef]
- Lochridge, V.P.; Hardy, M.E. A single-amino-acid substitution in the P2 domain of VP1 of murine norovirus is sufficient for escape from antibody neutralization. J. Virol. 2007, 81, 12316–12322. [Google Scholar] [CrossRef]
- Huang, W.; Samanta, M.; Crawford, S.E.; Estes, M.K.; Neill, F.H.; Atmar, R.L.; Palzkill, T. Identification of human single-chain antibodies with broad reactivity for noroviruses. Protein Eng. Des. Sel. 2014, 27, 339–349. [Google Scholar] [CrossRef]
- Malm, M.; Tamminen, K.; Vesikari, T.; Blazevic, V. Type-specific and cross-reactive antibodies and T cell responses in norovirus VLP immunized mice are targeted both to conserved and variable domains of capsid VP1 protein. Mol. Immunol. 2016, 78, 27–37. [Google Scholar] [CrossRef]
- Malm, M.; Uusi-Kerttula, H.; Vesikari, T.; Blazevic, V. High serum levels of norovirus genotype-specific blocking antibodies correlate with protection from infection in children. J. Infect. Dis. 2014, 210, 1755–1762. [Google Scholar] [CrossRef]
- Blazevic, V.; Malm, M.; Vesikari, T. Induction of homologous and cross-reactive GII.4-specific blocking antibodies in children after GII.4 New Orleans norovirus infection. J. Med. Virol. 2015, 87, 1656–1661. [Google Scholar] [CrossRef]
- Czakó, R.; Atmar, R.L.; Opekun, A.R.; Gilger, M.A.; Graham, D.Y.; Estes, M.K. Experimental Human Infection with Norwalk Virus Elicits a Surrogate Neutralizing Antibody Response with Cross-Genogroup Activity. Clin. Vaccine Immunol. 2015, 22, 221–228. [Google Scholar] [CrossRef]
- LoBue, A.D.; Lindesmith, L.; Yount, B.; Harrington, P.R.; Thompson, J.M.; Johnston, R.E.; Moe, C.L.; Baric, R.S. Multivalent norovirus vaccines induce strong mucosal and systemic blocking antibodies against multiple strains. Vaccine 2006, 24, 5220–5234. [Google Scholar] [CrossRef] [PubMed]
- Rockx, B.; Baric, R.S.; de Grijs, I.; Duizer, E.; Koopmans, M.P. Characterization of the homo- and heterotypic immune responses after natural norovirus infection. J. Med. Virol. 2005, 77, 439–446. [Google Scholar] [CrossRef]
- Strong, D.W.; Thackray, L.B.; Smith, T.J.; Virgin, H.W. Protruding domain of capsid protein is necessary and sufficient to determine murine norovirus replication and pathogenesis in vivo. J. Virol. 2012, 86, 2950–2958. [Google Scholar] [CrossRef]
- Kocher, J.; Bui, T.; Giri-Rachman, E.; Wen, K.; Li, G.; Yang, X.; Liu, F.; Tan, M.; Xia, M.; Zhong, W.; et al. Intranasal P Particle Vaccine Provided Partial Cross-Variant Protection against Human GII.4 Norovirus Diarrhea in Gnotobiotic Pigs. J. Virol. 2014, 88, 9728–9743. [Google Scholar] [CrossRef]
- LoBue, A.D.; Lindesmith, L.C.; Baric, R.S. Identification of cross-reactive norovirus CD4+ T cell epitopes. J. Virol. 2010, 84, 8530–8538. [Google Scholar] [CrossRef]
- Ellis, J.M.; Henson, V.; Slack, R.; Ng, J.; Hartzman, R.J.; Katovich Hurley, C. Frequencies of HLA-A2 alleles in five U.S. population groups. Predominance Of A*02011 and identification of HLA-A*0231. Hum. Immunol. 2000, 61, 334–340. [Google Scholar] [CrossRef]
- Cao, K.; Hollenbach, J.; Shi, X.; Shi, W.; Chopek, M.; Fernandez-Vina, M.A. Analysis of the frequencies of HLA-A, B, and C alleles and haplotypes in the five major ethnic groups of the United States reveals high levels of diversity in these loci and contrasting distribution patterns in these populations. Hum. Immunol. 2001, 62, 1009–1030. [Google Scholar] [CrossRef]
- Malm, M.; Vesikari, T.; Blazevic, V. Identification of a First Human Norovirus CD8(+) T Cell Epitope Restricted to HLA-A(*)0201 Allele. Front. Immunol. 2018, 9, 2782. [Google Scholar] [CrossRef] [PubMed]
- Kirby, A.; Iturriza-Gomara, M. Norovirus diagnostics: Options, applications and interpretations. Expert Rev. Anti-Infect. Ther. 2012, 10, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Kirby, A.; Gurgel, R.Q.; Dove, W.; Vieira, S.C.; Cunliffe, N.A.; Cuevas, L.E. An evaluation of the RIDASCREEN and IDEIA enzyme immunoassays and the RIDAQUICK immunochromatographic test for the detection of norovirus in faecal specimens. J. Clin. Virol. 2010, 49, 254–257. [Google Scholar] [CrossRef]
- Ambert-Balay, K.; Pothier, P. Evaluation of 4 immunochromatographic tests for rapid detection of norovirus in faecal samples. J. Clin. Virol. 2013, 56, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Bruggink, L.D.; Dunbar, N.L.; Marshall, J.A. Evaluation of the updated RIDA(R)QUICK (Version N1402) immunochromatographic assay for the detection of norovirus in clinical specimens. J. Virol. Methods 2015, 223, 82–87. [Google Scholar] [CrossRef]
- De Bruin, E.; Duizer, E.; Vennema, H.; Koopmans, M.P. Diagnosis of Norovirus outbreaks by commercial ELISA or RT-PCR. J. Virol. Methods 2006, 137, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Beersma, M.F.; Sukhrie, F.H.; Bogerman, J.; Verhoef, L.; Mde Melo, M.; Vonk, A.G.; Koopmans, M. Unrecognized norovirus infections in health care institutions and their clinical impact. J. Clin. Microbiol. 2012, 50, 3040–3045. [Google Scholar] [CrossRef]
- Sakamaki, N.; Ohiro, Y.; Ito, M.; Makinodan, M.; Ohta, T.; Suzuki, W.; Takayasu, S.; Tsuge, H. Bioluminescent enzyme immunoassay for the detection of norovirus capsid antigen. Clin. Vaccine Immunol. 2012, 19, 1949–1954. [Google Scholar] [CrossRef] [PubMed]
- Yasuura, M.; Fujimaki, M. Detection of Extremely Low Concentrations of Biological Substances Using Near-Field Illumination. Sci. Rep. 2016, 6, 39241. [Google Scholar] [CrossRef] [PubMed]
- Gairard-Dory, A.C.; Degot, T.; Hirschi, S.; Schuller, A.; Leclercq, A.; Renaud-Picard, B.; Gourieux, B.; Kessler, R. Clinical usefulness of oral immunoglobulins in lung transplant recipients with norovirus gastroenteritis: A case series. Transplant. Proc. 2014, 46, 3603–3605. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.C.; Wang, Y.Y.; Zhang, X.F.; Tan, M.; Xia, M.; Wu, X.B.; Jiang, X.; Nie, J. Evaluation of anti-norovirus IgY from egg yolk of chickens immunized with norovirus P particles. J. Virol. Methods 2012, 186, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.C.; Zhang, X.F.; Tan, M.; Huang, P.; Lei, W.; Fang, H.; Zhong, W.; Jiang, X. A dual chicken IgY against rotavirus and norovirus. Antivir. Res. 2013, 97, 293–300. [Google Scholar] [CrossRef]
- Vega, C.G.; Bok, M.; Vlasova, A.N.; Chattha, K.S.; Fernandez, F.M.; Wigdorovitz, A.; Parreno, V.G.; Saif, L.J. IgY antibodies protect against human Rotavirus induced diarrhea in the neonatal gnotobiotic piglet disease model. PLoS ONE 2012, 7, e42788. [Google Scholar] [CrossRef]
- Ehrlich, P.H.; Moustafa, Z.A.; Harfeldt, K.E.; Isaacson, C.; Ostberg, L. Potential of primate monoclonal antibodies to substitute for human antibodies: Nucleotide sequence of chimpanzee Fab fragments. Hum. Antib. Hybrid. 1990, 1, 23–26. [Google Scholar] [CrossRef]
- Ogata, N.; Ostberg, L.; Ehrlich, P.H.; Wong, D.C.; Miller, R.H.; Purcell, R.H. Markedly prolonged incubation period of hepatitis B in a chimpanzee passively immunized with a human monoclonal antibody to the a determinant of hepatitis B surface antigen. Proc. Natl. Acad. Sci. USA 1993, 90, 3014–3018. [Google Scholar] [CrossRef]
- Coppieters, K.; Dreier, T.; Silence, K.; de Haard, H.; Lauwereys, M.; Casteels, P.; Beirnaert, E.; Jonckheere, H.; Van de Wiele, C.; Staelens, L.; et al. Formatted anti-tumor necrosis factor alpha VHH proteins derived from camelids show superior potency and targeting to inflamed joints in a murine model of collagen-induced arthritis. Arthritis Rheum. 2006, 54, 1856–1866. [Google Scholar] [CrossRef]
- Vincke, C.; Loris, R.; Saerens, D.; Martinez-Rodriguez, S.; Muyldermans, S.; Conrath, K. General strategy to humanize a camelid single-domain antibody and identification of a universal humanized nanobody scaffold. J. Biol. Chem. 2009, 284, 3273–3284. [Google Scholar] [CrossRef]
- Holliger, P.; Hudson, P.J. Engineered antibody fragments and the rise of single domains. Nat. Biotechnol. 2005, 23, 1126–1136. [Google Scholar] [CrossRef]
- Pant, N.; Hultberg, A.; Zhao, Y.; Svensson, L.; Pan-Hammarstrom, Q.; Johansen, K.; Pouwels, P.H.; Ruggeri, F.M.; Hermans, P.; Frenken, L.; et al. Lactobacilli expressing variable domain of llama heavy-chain antibody fragments (lactobodies) confer protection against rotavirus-induced diarrhea. J. Infect. Dis. 2006, 194, 1580–1588. [Google Scholar] [CrossRef]
- Van der Vaart, J.M.; Pant, N.; Wolvers, D.; Bezemer, S.; Hermans, P.W.; Bellamy, K.; Sarker, S.A.; van der Logt, C.P.; Svensson, L.; Verrips, C.T.; et al. Reduction in morbidity of rotavirus induced diarrhoea in mice by yeast produced monovalent llama-derived antibody fragments. Vaccine 2006, 24, 4130–4137. [Google Scholar] [CrossRef] [PubMed]
- Tokuhara, D.; Alvarez, B.; Mejima, M.; Hiroiwa, T.; Takahashi, Y.; Kurokawa, S.; Kuroda, M.; Oyama, M.; Kozuka-Hata, H.; Nochi, T.; et al. Rice-based oral antibody fragment prophylaxis and therapy against rotavirus infection. J. Clin. Investig. 2013, 123, 3829–3838. [Google Scholar] [CrossRef] [PubMed]
- Vega, C.G.; Bok, M.; Vlasova, A.N.; Chattha, K.S.; Gomez-Sebastian, S.; Nunez, C.; Alvarado, C.; Lasa, R.; Escribano, J.M.; Garaicoechea, L.L.; et al. Recombinant monovalent llama-derived antibody fragments (VHH) to rotavirus VP6 protect neonatal gnotobiotic piglets against human rotavirus-induced diarrhea. PLoS Pathog. 2013, 9, e1003334. [Google Scholar] [CrossRef] [PubMed]
- Chagla, Z.; Quirt, J.; Woodward, K.; Neary, J.; Rutherford, C. Chronic norovirus infection in a transplant patient successfully treated with enterally administered immune globulin. J. Clin. Virol. 2013, 58, 306–308. [Google Scholar] [CrossRef] [PubMed]
- Ronchetti, A.M.; Henry, B.; Ambert-Balay, K.; Pothier, P.; Decroocq, J.; Leblond, V.; Roos-Weil, D. Norovirus-related chronic diarrhea in a patient treated with alemtuzumab for chronic lymphocytic leukemia. BMC Infect. Dis. 2014, 14, 239. [Google Scholar] [CrossRef]
- Nilsson, M.; Hedlund, K.O.; Thorhagen, M.; Larson, G.; Johansen, K.; Ekspong, A.; Svensson, L. Evolution of human calicivirus RNA in vivo: Accumulation of mutations in the protruding P2 domain of the capsid leads to structural changes and possibly a new phenotype. J. Virol. 2003, 77, 13117–13124. [Google Scholar] [CrossRef]
- Koromyslova, A.; Tripathi, S.; Morozov, V.; Schroten, H.; Hansman, G.S. Human norovirus inhibition by a human milk oligosaccharide. Virology 2017, 508, 81–89. [Google Scholar] [CrossRef]
- Weichert, S.; Koromyslova, A.; Singh, B.K.; Hansman, S.; Jennewein, S.; Schroten, H.; Hansman, G.S. Structural Basis for Norovirus Inhibition by Human Milk Oligosaccharides. J. Virol. 2016, 90, 4843–4848. [Google Scholar] [CrossRef]
- Schroten, H.; Hanisch, F.G.; Hansman, G.S. Human Norovirus Interactions with Histo-Blood Group Antigens and Human Milk Oligosaccharides. J. Virol. 2016, 90, 5855–5859. [Google Scholar] [CrossRef]
- Dolin, R.; Blacklow, N.R.; DuPont, H.; Formal, S.; Buscho, R.F.; Kasel, J.A.; Chames, R.P.; Hornick, R.; Chanock, R.M. Transmission of acute infectious nonbacterial gastroenteritis to volunteers by oral administration of stool filtrates. J. Infect. Dis. 1971, 123, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Parrino, T.A.; Schreiber, D.S.; Trier, J.S.; Kapikian, A.Z.; Blacklow, N.R. Clinical immunity in acute gastroenteritis caused by Norwalk agent. N. Engl. J. Med. 1977, 297, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, R.G.; Dolin, R.; Blacklow, N.R.; DuPont, H.L.; Buscho, R.F.; Thornhill, T.S.; Kapikian, A.Z.; Chanock, R.M. Comparison of three agents of acute infectious nonbacterial gastroenteritis by cross-challenge in volunteers. J. Infect. Dis. 1974, 129, 709–714. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.L.; Lindesmith, L.C.; Donaldson, E.F.; Saxe, L.; Baric, R.S.; Vinje, J. Herd immunity to GII.4 noroviruses is supported by outbreak patient sera. J. Virol. 2009, 83, 5363–5374. [Google Scholar] [CrossRef]
- Simmons, K.; Gambhir, M.; Leon, J.; Lopman, B. Duration of immunity to norovirus gastroenteritis. Emerg. Infect. Dis. 2013, 19, 1260–1267. [Google Scholar] [CrossRef] [PubMed]
- Hogle, J.M. Poliovirus cell entry: Common structural themes in viral cell entry pathways. Annu. Rev. Microbiol. 2002, 56, 677–702. [Google Scholar] [CrossRef]
- Adeyemi, O.O.; Nicol, C.; Stonehouse, N.J.; Rowlands, D.J. Increasing Type 1 Poliovirus Capsid Stability by Thermal Selection. J. Virol. 2017, 91, e01586-16. [Google Scholar] [CrossRef] [PubMed]
- Singharoy, A.; Polavarapu, A.; Joshi, H.; Baik, M.H.; Ortoleva, P. Epitope fluctuations in the human papillomavirus are under dynamic allosteric control: A computational evaluation of a new vaccine design strategy. J. Am. Chem. Soc. 2013, 135, 18458–18468. [Google Scholar] [CrossRef]
- LoBue, A.D.; Thompson, J.M.; Lindesmith, L.; Johnston, R.E.; Baric, R.S. Alphavirus-adjuvanted norovirus-like particle vaccines: Heterologous, humoral, and mucosal immune responses protect against murine norovirus challenge. J. Virol. 2009, 83, 3212–3227. [Google Scholar] [CrossRef] [PubMed]
- Parra, G.I.; Bok, K.; Taylor, R.; Haynes, J.R.; Sosnovtsev, S.V.; Richardson, C.; Green, K.Y. Immunogenicity and specificity of norovirus Consensus GII.4 virus-like particles in monovalent and bivalent vaccine formulations. Vaccine 2012, 30, 3580–3586. [Google Scholar] [CrossRef] [PubMed]
- Ramani, S.; Atmar, R.L.; Estes, M.K. Epidemiology of human noroviruses and updates on vaccine development. Curr. Opin. Gastroenterol. 2014, 30, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Ramani, S.; Neill, F.H.; Ferreira, J.; Treanor, J.J.; Frey, S.E.; Topham, D.J.; Goodwin, R.R.; Borkowski, A.; Baehner, F.; Mendelman, P.M.; et al. B-Cell Responses to Intramuscular Administration of a Bivalent Virus-Like Particle Human Norovirus Vaccine. Clin. Vaccine Immunol. 2017, 24, e00571-16. [Google Scholar] [CrossRef] [PubMed]
- Ball, J.M.; Graham, D.Y.; Opekun, A.R.; Gilger, M.A.; Guerrero, R.A.; Estes, M.K. Recombinant Norwalk virus-like particles given orally to volunteers: Phase I study. Gastroenterology 1999, 117, 40–48. [Google Scholar] [CrossRef]
- El-Kamary, S.S.; Pasetti, M.F.; Mendelman, P.M.; Frey, S.E.; Bernstein, D.I.; Treanor, J.J.; Ferreira, J.; Chen, W.H.; Sublett, R.; Richardson, C.; et al. Adjuvanted intranasal Norwalk virus-like particle vaccine elicits antibodies and antibody-secreting cells that express homing receptors for mucosal and peripheral lymphoid tissues. J. Infect. Dis. 2010, 202, 1649–1658. [Google Scholar] [CrossRef]
- Treanor, J.J.; Atmar, R.L.; Frey, S.E.; Gormley, R.; Chen, W.H.; Ferreira, J.; Goodwin, R.; Borkowski, A.; Clemens, R.; Mendelman, P.M. A novel intramuscular bivalent norovirus virus-like particle vaccine candidate--reactogenicity, safety, and immunogenicity in a phase 1 trial in healthy adults. J. Infect. Dis. 2014, 210, 1763–1771. [Google Scholar] [CrossRef]
- Periwal, S.B.; Kourie, K.R.; Ramachandaran, N.; Blakeney, S.J.; DeBruin, S.; Zhu, D.; Zamb, T.J.; Smith, L.; Udem, S.; Eldridge, J.H.; et al. A modified cholera holotoxin CT-E29H enhances systemic and mucosal immune responses to recombinant Norwalk virus-virus like particle vaccine. Vaccine 2003, 21, 376–385. [Google Scholar] [CrossRef]
- Parra, G.I.; Green, K.Y. Sequential Gastroenteritis Episodes Caused by 2 Norovirus Genotypes. Emerg. Infect. Dis. 2014, 20, 1016–1018. [Google Scholar] [CrossRef] [PubMed]
- Tamminen, K.; Huhti, L.; Vesikari, T.; Blazevic, V. Pre-existing immunity to norovirus GII-4 virus-like particles does not impair de novo immune responses to norovirus GII-12 genotype. Viral Immunol. 2013, 26, 167–170. [Google Scholar] [CrossRef]
- Kobayashi, M.; Ohfuji, S.; Fukushima, W.; Maeda, A.; Maeda, K.; Fujioka, M.; Hirota, Y. Immunogenicity and reactogenicity of a monovalent inactivated 2009 influenza A vaccine in adolescents: With special reference to pre-existing antibody. J. Pediatr. 2012, 160, 632–637. [Google Scholar] [CrossRef]
- Knuchel, M.C.; Marty, R.R.; Morin, T.N.; Ilter, O.; Zuniga, A.; Naim, H.Y. Relevance of a pre-existing measles immunity prior immunization with a recombinant measles virus vector. Hum. Vaccines Immunother. 2013, 9, 599–606. [Google Scholar] [CrossRef]
- Saif, M.A.; Bonney, D.K.; Bigger, B.; Forsythe, L.; Williams, N.; Page, J.; Babiker, Z.O.; Guiver, M.; Turner, A.J.; Hughes, S.; et al. Chronic norovirus infection in pediatric hematopoietic stem cell transplant recipients: A cause of prolonged intestinal failure requiring intensive nutritional support. Pediatr. Transplant. 2011, 15, 505–509. [Google Scholar] [CrossRef]
- Wingfield, T.; Gallimore, C.I.; Xerry, J.; Gray, J.J.; Klapper, P.; Guiver, M.; Blanchard, T.J. Chronic norovirus infection in an HIV-positive patient with persistent diarrhoea: A novel cause. J. Clin. Virol. 2010, 49, 219–222. [Google Scholar] [CrossRef]
- Nicollier-Jamot, B.; Pico, V.; Pothier, P.; Kohli, E. Molecular cloning, expression, self-assembly, antigenicity, and seroepidemiology of a genogroup II norovirus isolated in France. J. Clin. Microbiol. 2003, 41, 3901–3904. [Google Scholar] [CrossRef] [PubMed][Green Version]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Loben Sels, J.M.; Green, K.Y. The Antigenic Topology of Norovirus as Defined by B and T Cell Epitope Mapping: Implications for Universal Vaccines and Therapeutics. Viruses 2019, 11, 432. https://doi.org/10.3390/v11050432
van Loben Sels JM, Green KY. The Antigenic Topology of Norovirus as Defined by B and T Cell Epitope Mapping: Implications for Universal Vaccines and Therapeutics. Viruses. 2019; 11(5):432. https://doi.org/10.3390/v11050432
Chicago/Turabian Stylevan Loben Sels, Jessica M., and Kim Y. Green. 2019. "The Antigenic Topology of Norovirus as Defined by B and T Cell Epitope Mapping: Implications for Universal Vaccines and Therapeutics" Viruses 11, no. 5: 432. https://doi.org/10.3390/v11050432
APA Stylevan Loben Sels, J. M., & Green, K. Y. (2019). The Antigenic Topology of Norovirus as Defined by B and T Cell Epitope Mapping: Implications for Universal Vaccines and Therapeutics. Viruses, 11(5), 432. https://doi.org/10.3390/v11050432