The Third Annual Meeting of the European Virus Bioinformatics Center


 , ,
, ,  , , , , ,
, , , , ,
Abstract
1. Introduction
2. Sessions and Oral Presentations
2.1. Systems Virology
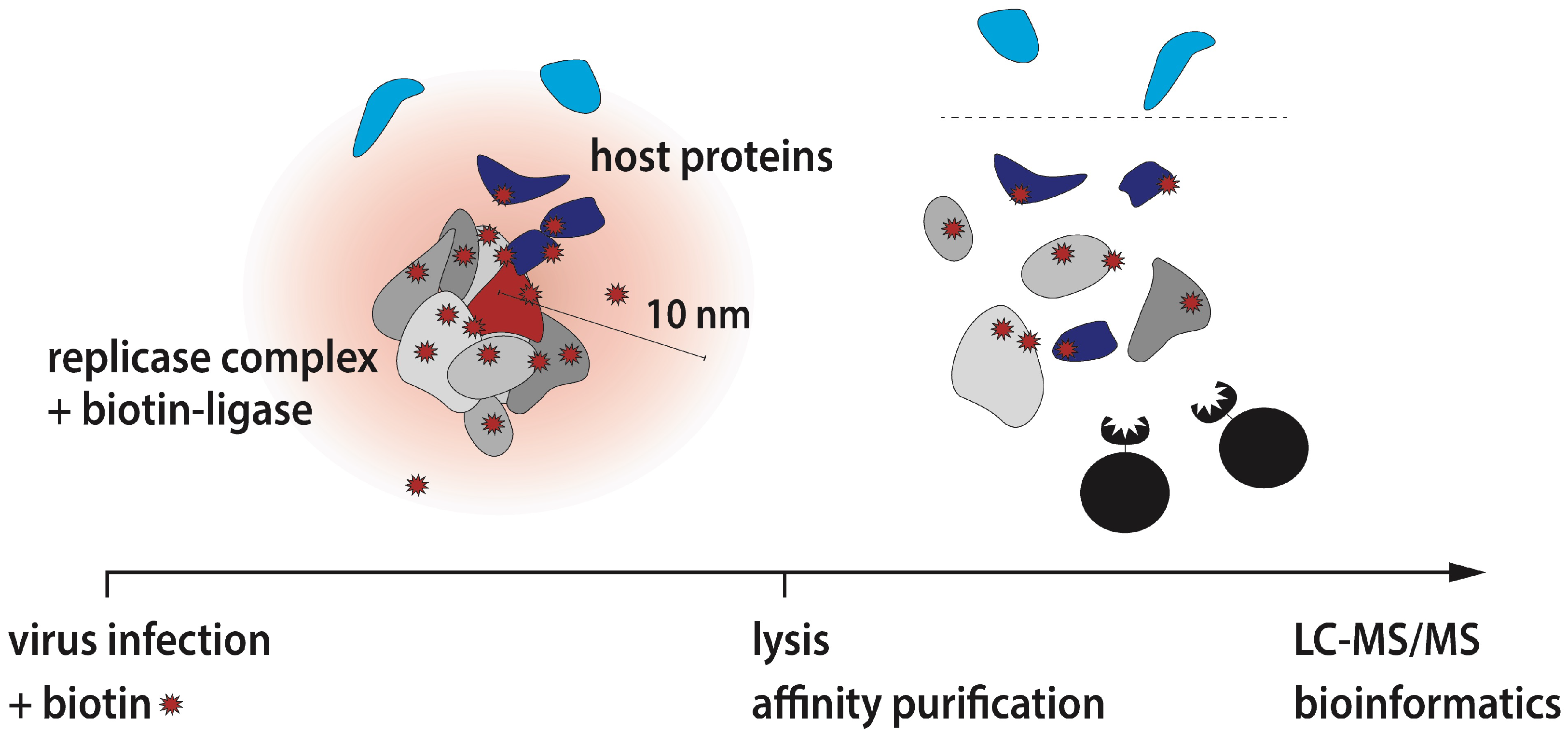
2.1.1. Determination of Host Proteins Composing the Microenvironment of Coronavirus Replicase Complexes, by Volker Thiel
2.1.2. Co-Infection between Staphylococcus aureus and Influenza Virus Reduces Endothelial Barrier Function, by Stefanie Deinhardt-Emmer
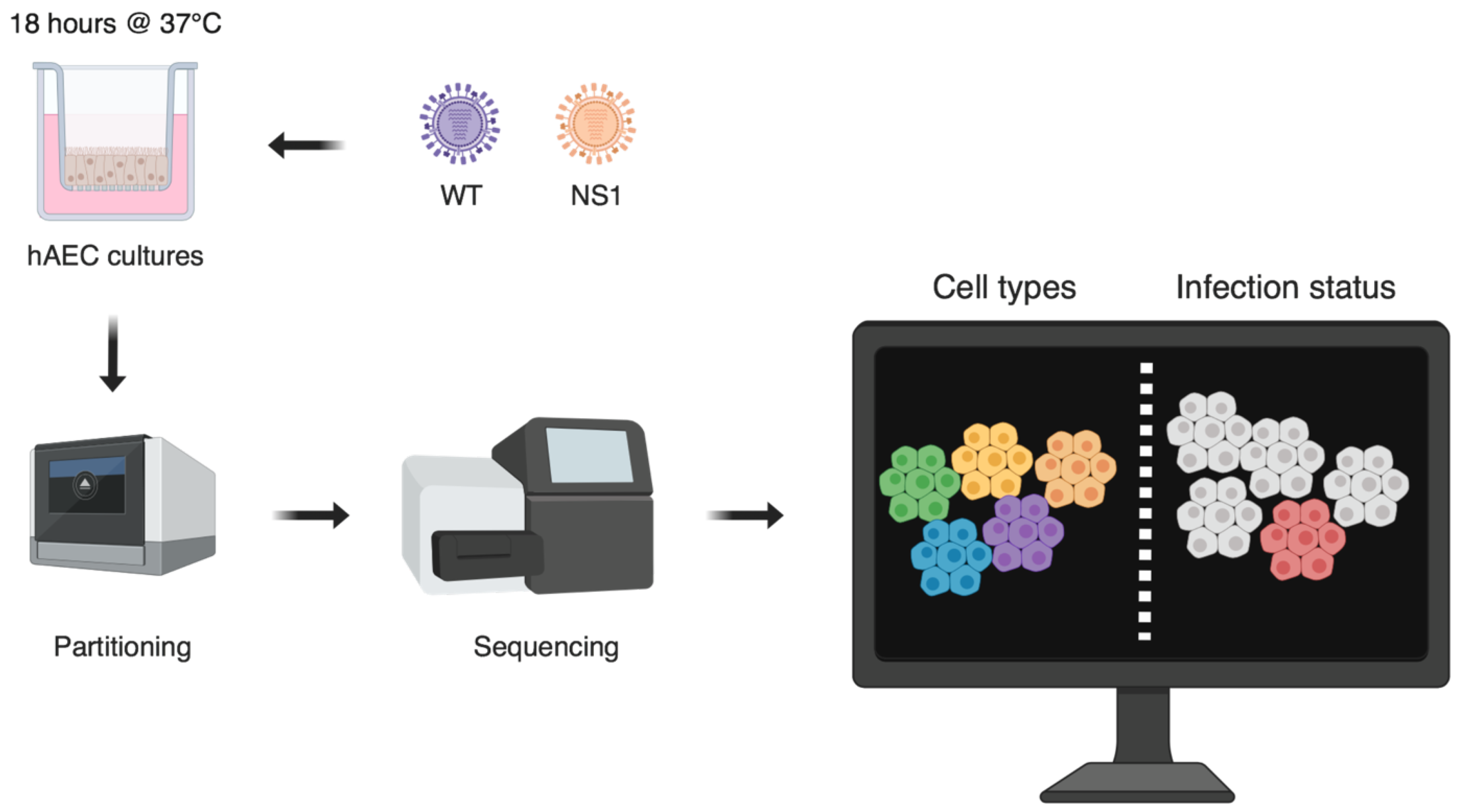
2.1.3. Single Cell Analysis of iNfluenza Virus Infection in Its Natural Target Cells Reveals Cell Type-Specific Host Responses and Disparate Viral Burden, by Jenna Nicole Kelly
2.2. Virus–Host Interactions and the Virome
2.2.1. Roles of Phages in Impacting Infectious Diseases in Human Microbiomes, by R. J. Martha Clokie
2.2.2. Global Phylogeography and Ancient Evolution of the Widespread Human Gut Virus crAssphage, by Bas E. Dutilh
2.2.3. Genome-Resolved Metaviromics for the Detection of Pathogenic Viruses in the Environment: Will Eating Shellfish Make You Ill?, by Evelien M. Adriaenssens
2.3. Virus Classification and Evolution
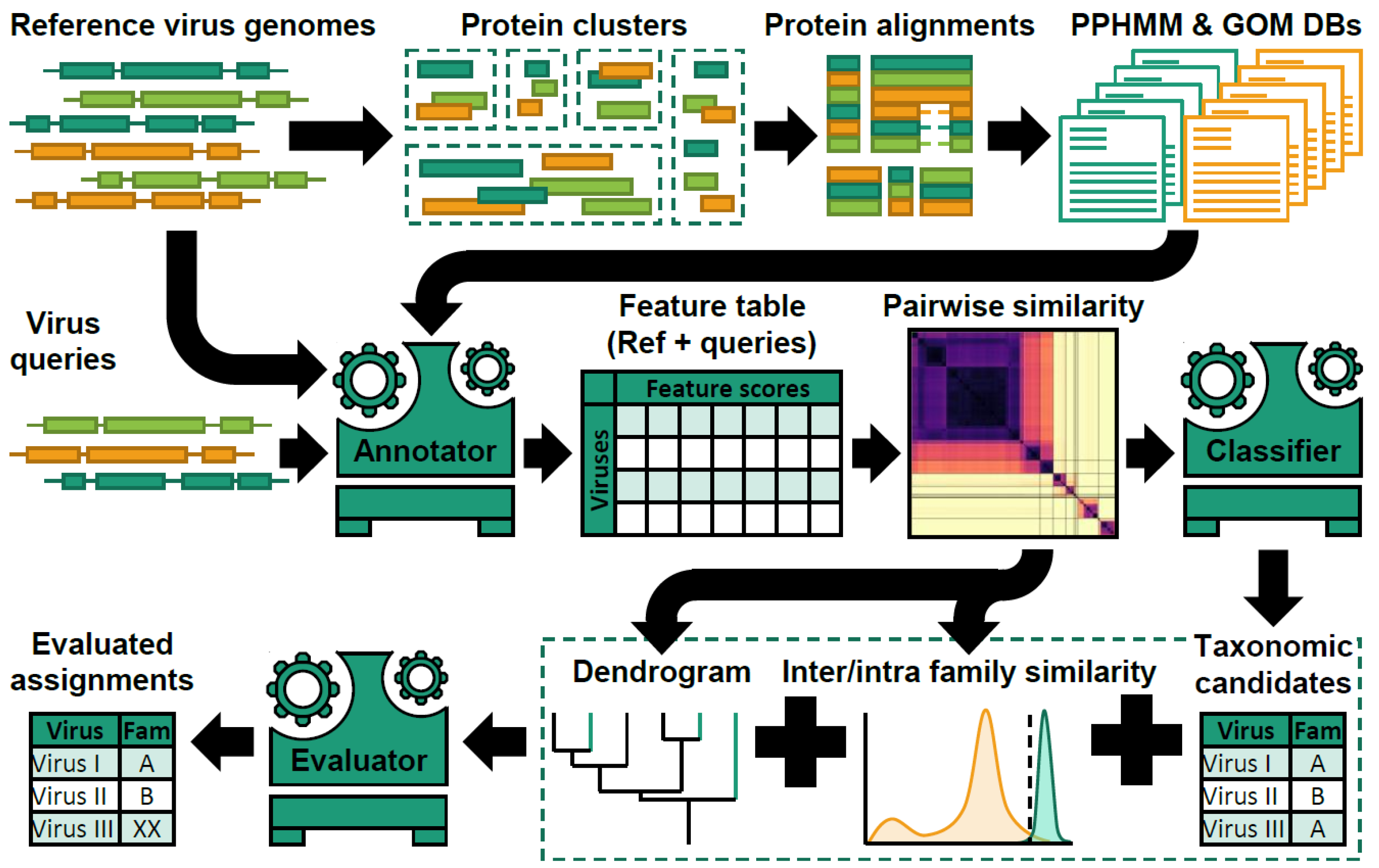
2.3.1. The Classification of Viruses in Metagenomic Datasets—Where Do You Draw the Line?, by Peter Simmonds
2.3.2. Detecting Viruses in Ancient Human Remains, by Julian Susat
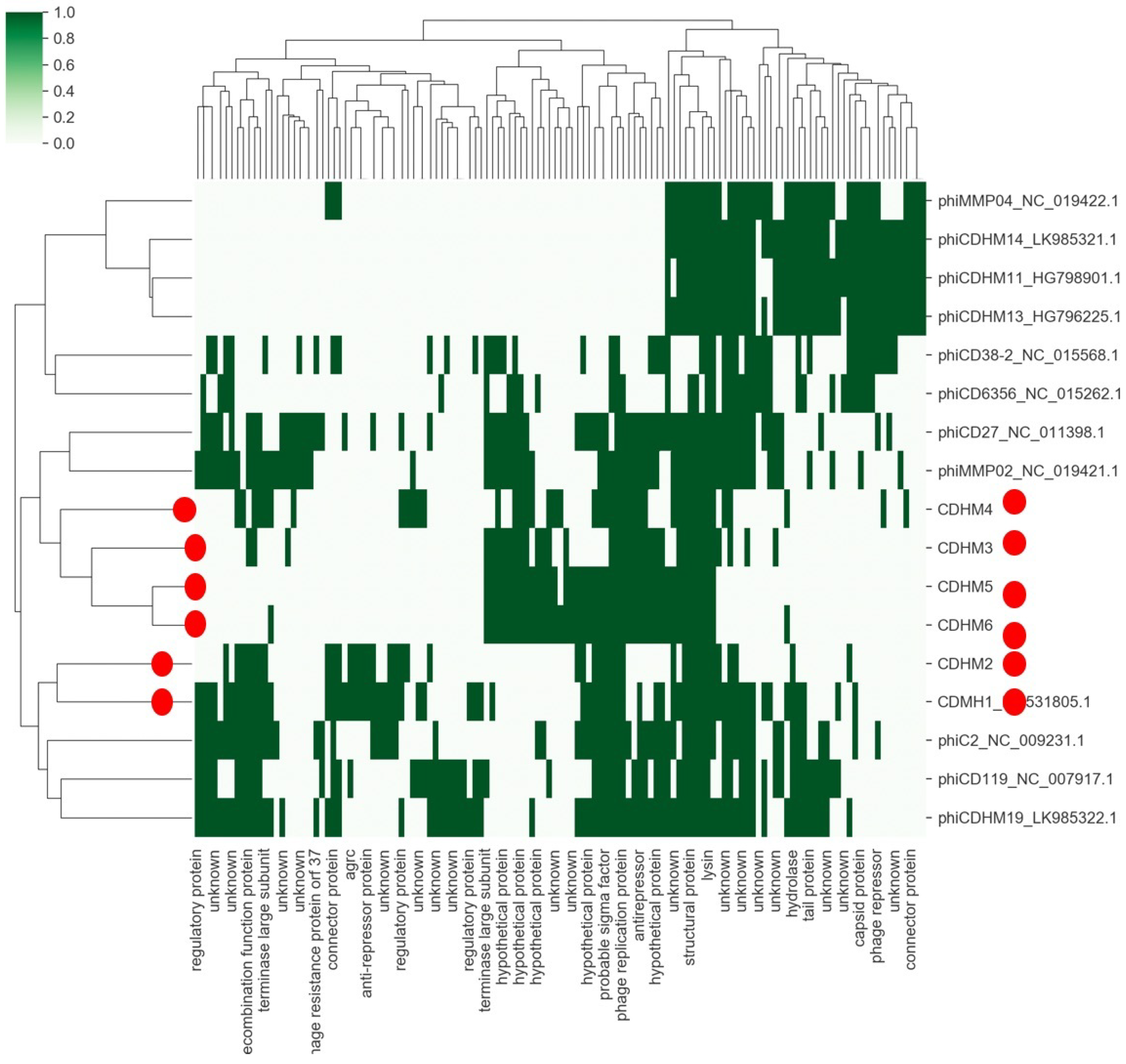
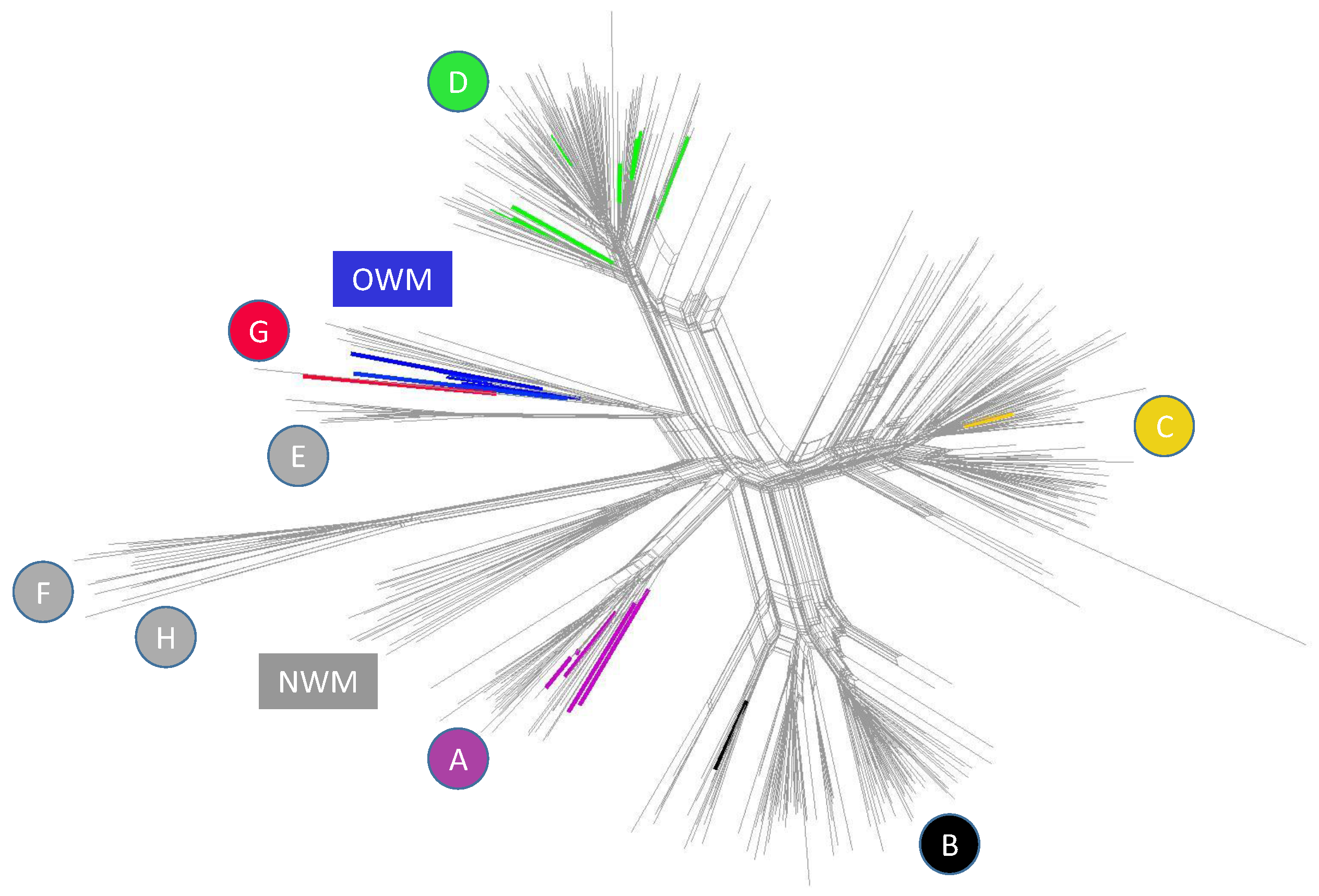
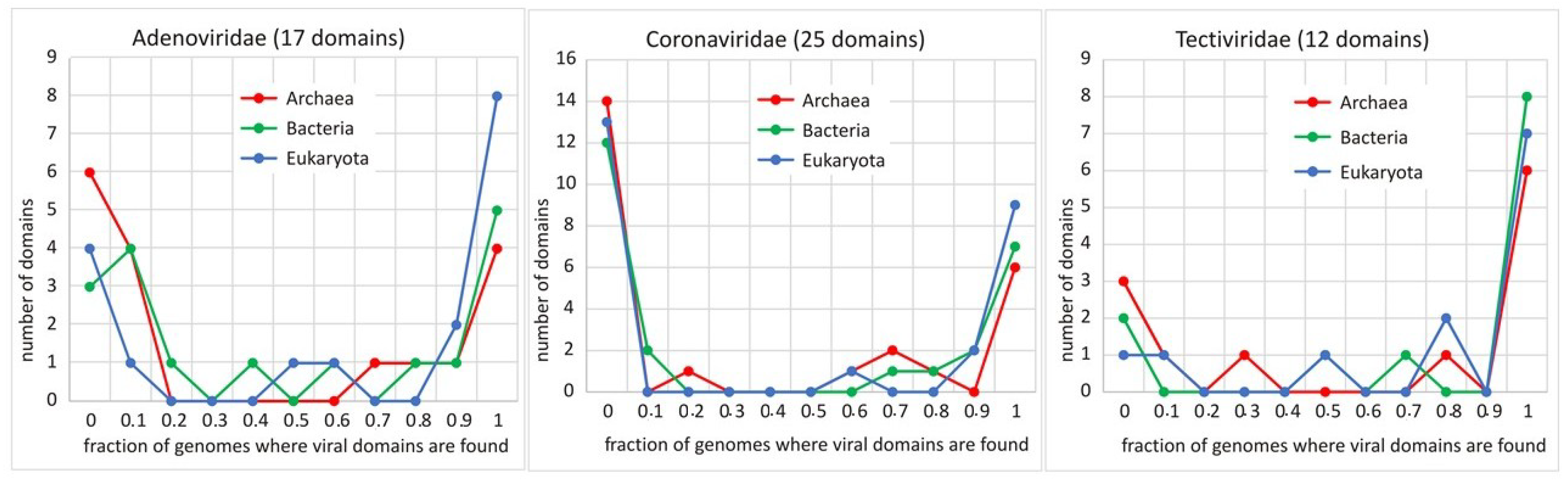
2.3.3. Virosphere and Biosphere—How Related They Are? A Protein (Domain) Based View, by Aare Abroi
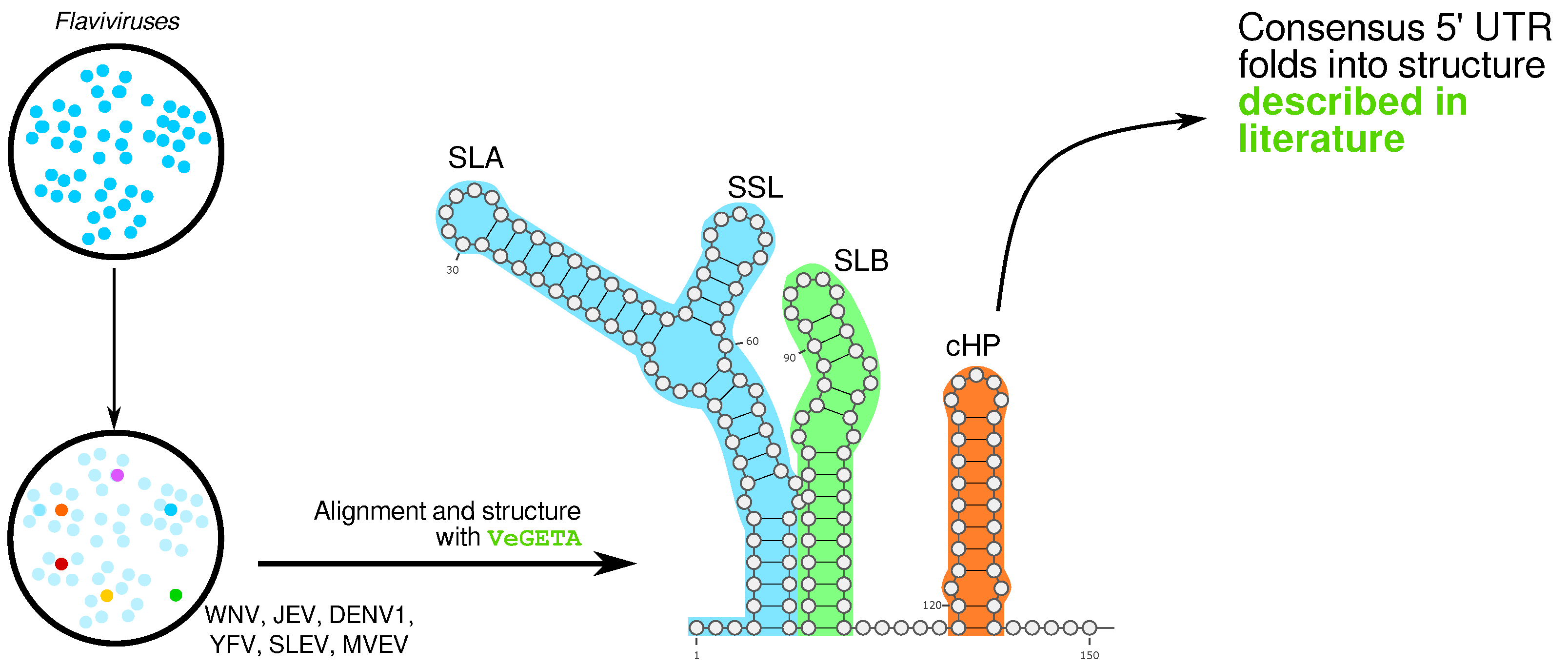
2.3.4. RNA Secondary Structures in Whole Genome Alignments of Viruses, by Kevin Lamkiewicz
2.4. Epidemiology, Surveillance and Evolution
2.4.1. Phylodynamics for Tracking Epidemic, Endemic and Evolving Viral Strains, by Samantha Lycett
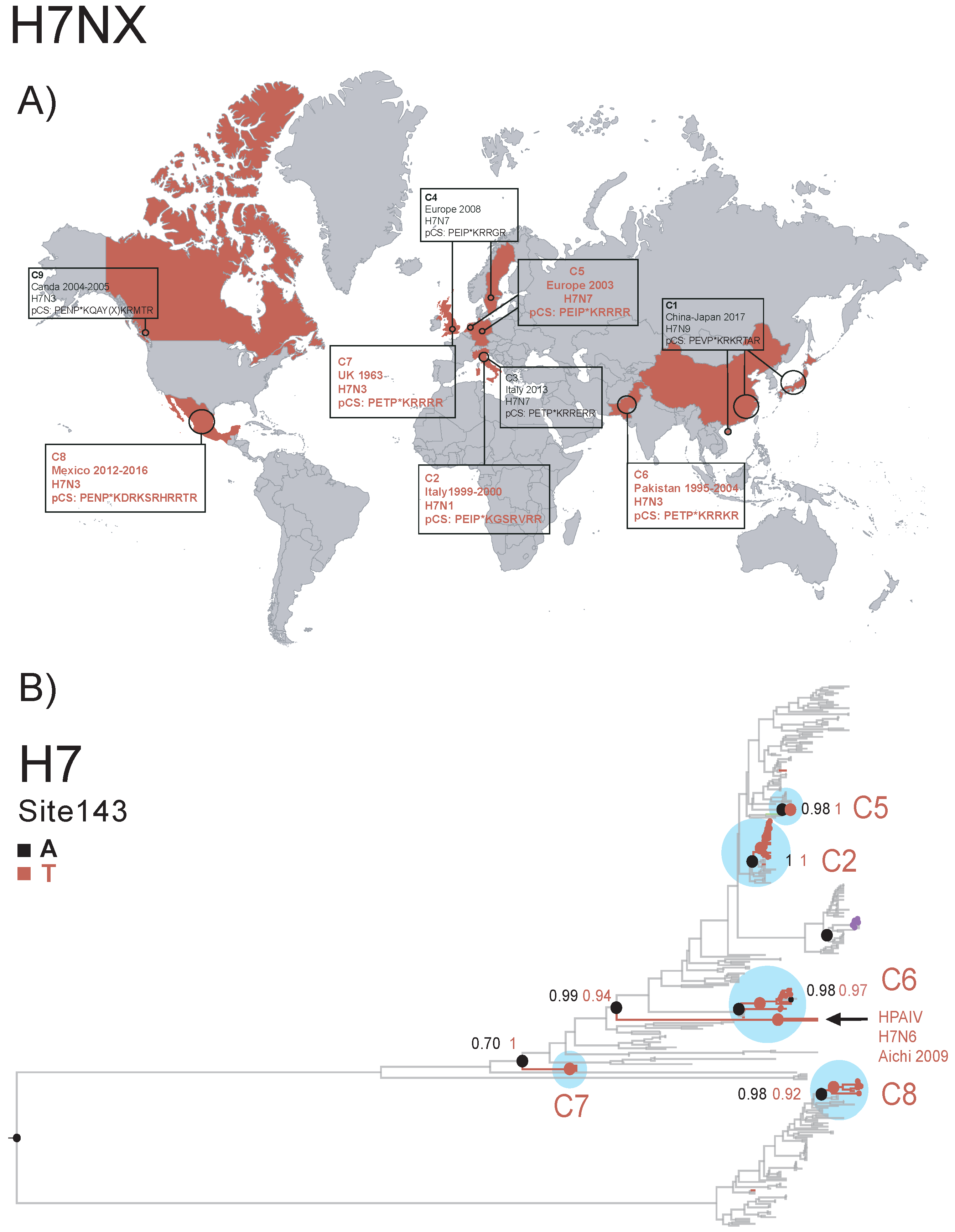
2.4.2. Parallel Evolution and the Emergence of Highly-Pathogenic Avian Influenza A Viruses, by Marina Escalera-Zamudio
2.4.3. Evolutionary Origins of Epidemic Potential among Human RNA Viruses, by Lu Lu
3. Poster Session
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hufsky, F.; Ibrahim, B.; Beer, M.; Deng, L.; Le Mercier, P.; McMahon, D.P.; Palmarini, M.; Thiel, V.; Marz, M. Virologists—Heroes need weapons. PLoS Pathog. 2018, 14, e1006771. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, B.; McMahon, D.P.; Hufsky, F.; Beer, M.; Deng, L.; Le Mercier, P.; Palmarini, M.; Thiel, V.; Marz, M. A new era of virus bioinformatics. Virus Res. 2018, 251, 86–90. [Google Scholar] [CrossRef]
- Ibrahim, B.; Arkhipova, K.; Andeweg, A.; Posada-Céspedes, S.; Enault, F.; Gruber, A.; Koonin, E.; Kupczok, A.; Lemey, P.; McHardy, A.; et al. Bioinformatics Meets Virology: The European Virus Bioinformatics Center’s Second Annual Meeting. Viruses 2018, 10, 256. [Google Scholar] [CrossRef] [PubMed]
- V’kovski, P.; Gerber, M.; Kelly, J.; Pfaender, S.; Ebert, N.; Lagache, S.B.; Simillion, C.; Portmann, J.; Stalder, H.; Gaschen, V.; et al. Determination of host proteins composing the microenvironment of coronavirus replicase complexes by proximity-labeling. eLife 2019, 8, e42037. [Google Scholar] [CrossRef] [PubMed]
- Mayr, F.B.; Yende, S.; Angus, D.C. Epidemiology of severe sepsis. Virulence 2014, 5, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Iuliano, A.D.; Roguski, K.M.; Chang, H.H.; Muscatello, D.J.; Palekar, R.; Tempia, S.; Cohen, C.; Gran, J.M.; Schanzer, D.; Cowling, B.J.; et al. Estimates of global seasonal influenza-associated respiratory mortality: A modelling study. Lancet (London, England) 2018, 391, 1285–1300. [Google Scholar] [CrossRef]
- Papanicolaou, G.A. Severe influenza and S. aureus pneumonia: For whom the bell tolls? Virulence 2013, 4, 666–668. [Google Scholar] [CrossRef] [PubMed]
- Klemm, C.; Bruchhagen, C.; van Krüchten, A.; Niemann, S.; Löffler, B.; Peters, G.; Ludwig, S.; Ehrhardt, C. Mitogen-activated protein kinases (MAPKs) regulate IL-6 over-production during concomitant influenza virus and Staphylococcus aureus infection. Sci. Rep. 2017, 7, 42473. [Google Scholar] [CrossRef]
- van Krüchten, A.; Wilden, J.J.; Niemann, S.; Peters, G.; Löffler, B.; Ludwig, S.; Ehrhardt, C. Staphylococcus aureus triggers a shift from influenza virus-induced apoptosis to necrotic cell death. FASEB J. 2018, 32, 2779–2793. [Google Scholar] [CrossRef] [PubMed]
- Russell, A.B.; Trapnell, C.; Bloom, J.D. Extreme heterogeneity of influenza virus infection in single cells. eLife 2018, 7, e32303. [Google Scholar] [CrossRef] [PubMed]
- Steuerman, Y.; Cohen, M.; Peshes-Yaloz, N.; Valadarsky, L.; Cohn, O.; David, E.; Frishberg, A.; Mayo, L.; Bacharach, E.; Amit, I.; et al. Dissection of Influenza Infection In Vivo by Single-Cell RNA Sequencing. Cell Syst. 2018, 6, 679–691. [Google Scholar] [CrossRef] [PubMed]
- Jonsdottir, H.R.; Dijkman, R. Characterization of Human Coronaviruses on Well-Differentiated Human Airway Epithelial Cell Cultures. In Coronaviruses; Springer: New York, NY, USA, 2015; pp. 73–87. [Google Scholar] [CrossRef]
- James, C.E.; Davies, E.V.; Fothergill, J.L.; Walshaw, M.J.; Beale, C.M.; Brockhurst, M.A.; Winstanley, C. Lytic activity by temperate phages of Pseudomonas aeruginosa in long-term cystic fibrosis chronic lung infections. ISME J. 2015, 9, 1391–1398. [Google Scholar] [CrossRef] [PubMed]
- Shan, J.; Ramachandran, A.; Thanki, A.M.; Vukusic, F.B.I.; Barylski, J.; Clokie, M.R.J. Bacteriophages are more virulent to bacteria with human cells than they are in bacterial culture; insights from HT-29 cells. Sci. Rep. 2018, 8, 5091. [Google Scholar] [CrossRef]
- Nale, J.Y.; Redgwell, T.A.; Millard, A.; Clokie, M.R.J. Efficacy of an Optimised Bacteriophage Cocktail to Clear Clostridium difficile in a Batch Fermentation Model. Antibiotics 2018, 7, 13. [Google Scholar] [CrossRef]
- Nale, J.Y.; Spencer, J.; Hargreaves, K.R.; Buckley, A.M.; Trzepiński, P.; Douce, G.R.; Clokie, M.R.J. Bacteriophage Combinations Significantly Reduce Clostridium difficile Growth In Vitro and Proliferation In Vivo. Antimicrob. Agents Chemother. 2016, 60, 968–981. [Google Scholar] [CrossRef]
- Hendrix, R.W.; Smith, M.C.; Burns, R.N.; Ford, M.E.; Hatfull, G.F. Evolutionary relationships among diverse bacteriophages and prophages: All the world’s a phage. Proc. Natl. Acad. Sci. USA 1999, 96, 2192–2197. [Google Scholar] [CrossRef]
- Sanjuán, R.; Nebot, M.R.; Chirico, N.; Mansky, L.M.; Belshaw, R. Viral mutation rates. J. Virol. 2010, 84, 9733–9748. [Google Scholar] [CrossRef]
- Breitbart, M.; Salamon, P.; Andresen, B.; Mahaffy, J.M.; Segall, A.M.; Mead, D.; Azam, F.; Rohwer, F. Genomic analysis of uncultured marine viral communities. Proc. Natl. Acad. Sci. USA 2002, 99, 14250–14255. [Google Scholar] [CrossRef]
- Roux, S.; Brum, J.R.; Dutilh, B.E.; Sunagawa, S.; Duhaime, M.B.; Loy, A.; Poulos, B.T.; Solonenko, N.; Lara, E.; Poulain, J.; et al. Ecogenomics and potential biogeochemical impacts of globally abundant ocean viruses. Nature 2016, 537, 689. [Google Scholar] [CrossRef]
- Breitbart, M.; Rohwer, F. Here a virus, there a virus, everywhere the same virus? Trends Microbiol. 2005, 13, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Dutilh, B.E.; Cassman, N.; McNair, K.; Sanchez, S.E.; Silva, G.G.Z.; Boling, L.; Barr, J.J.; Speth, D.R.; Seguritan, V.; Aziz, R.K.; et al. A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nat. Commun. 2014, 5, 4498. [Google Scholar] [CrossRef] [PubMed]
- Stern, A.; Mick, E.; Tirosh, I.; Sagy, O.; Sorek, R. CRISPR targeting reveals a reservoir of common phages associated with the human gut microbiome. Genome Res. 2012, 22, 1985–1994. [Google Scholar] [CrossRef] [PubMed]
- Manrique, P.; Bolduc, B.; Walk, S.T.; van der Oost, J.; de Vos, W.M.; Young, M.J. Healthy human gut phageome. Proc. Natl. Acad. Sci. USA 2016, 113, 10400–10405. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudabadi, G.; Phillips, R. A comprehensive and quantitative exploration of thousands of viral genomes. eLife 2018, 7, e31955. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.S.; McNair, K.; Cuevas, D.; Bailey, B.; Segall, A.; Edwards, R.A. Prophage genomics reveals patterns in phage genome organization and replication. bioRxiv 2017, 114819. [Google Scholar] [CrossRef]
- Miller, E.S.; Kutter, E.; Mosig, G.; Arisaka, F.; Kunisawa, T.; Ruger, W. Bacteriophage T4 Genome. Microbiol. Mol. Biol. Rev. 2003, 67, 86–156. [Google Scholar] [CrossRef]
- Brum, J.R.; Schenck, R.O.; Sullivan, M.B. Global morphological analysis of marine viruses shows minimal regional variation and dominance of non-tailed viruses. ISME J. 2013, 7, 1738–1751. [Google Scholar] [CrossRef] [PubMed]
- Mavrich, T.N.; Hatfull, G.F. Bacteriophage evolution differs by host, lifestyle and genome. Nat. Microbiol. 2017, 2. [Google Scholar] [CrossRef]
- Kupczok, A.; Neve, H.; Huang, K.D.; Hoeppner, M.P.; Heller, K.J.; Franz, C.M.A.P.; Dagan, T. Rates of Mutation and Recombination in Siphoviridae Phage Genome Evolution over Three Decades. Mol. Biol. Evol. 2018, 35, 1147–1159. [Google Scholar] [CrossRef]
- Simmonds, P.; Aiewsakun, P.; Katzourakis, A. Prisoners of war — host adaptation and its constraints on virus evolution. Nat. Rev. Microbiol. 2018, 17, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Edwards, R.; Vega, A.; Norman, H.; Ohaeri, M.C.; Levi, K.; Dinsdale, E.; Cinek, O.; Aziz, R.; McNair, K.; Barr, J.; et al. Global phylogeography and ancient evolution of the widespread human gut virus crAssphage. bioRxiv 2019, 527796. [Google Scholar] [CrossRef]
- Mokili, J.L.; Rohwer, F.; Dutilh, B.E. Metagenomics and future perspectives in virus discovery. Curr. Opin. Virol. 2012, 2, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Symonds, E.M.; Breitbart, M. Affordable Enteric Virus Detection Techniques Are Needed to Support Changing Paradigms in Water Quality Management. Clean 2014, 43, 8–12. [Google Scholar] [CrossRef]
- Bibby, K. Metagenomic identification of viral pathogens. Trends Biotechnol. 2013, 31, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Adriaenssens, E.M.; Dutilh, B.E.; Koonin, E.V.; Kropinski, A.M.; Krupovic, M.; Kuhn, J.H.; Lavigne, R.; Brister, J.R.; Varsani, A.; et al. Minimum Information about an Uncultivated Virus Genome (MIUViG). Nat. Biotechnol. 2018. [Google Scholar] [CrossRef]
- Eren, A.M.; Esen, Ö.C.; Quince, C.; Vineis, J.H.; Morrison, H.G.; Sogin, M.L.; Delmont, T.O. Anvi’o: An advanced analysis and visualization platform for ’omics data. PeerJ 2015, 3, e1319. [Google Scholar] [CrossRef]
- Adriaenssens, E.; Farkas, K.; Harrison, C.; Jones, D.; Allison, H.E.; McCarthy, A.J. Viromic analysis of wastewater input to a river catchment reveals a diverse assemblage of RNA viruses. bioRxiv 2018, 248203. [Google Scholar] [CrossRef]
- Simmonds, P.; Adams, M.J.; Benkő, M.; Breitbart, M.; Brister, J.R.; Carstens, E.B.; Davison, A.J.; Delwart, E.; Gorbalenya, A.E.; Harrach, B.; et al. Consensus statement: Virus taxonomy in the age of metagenomics. Nat. Rev. Microbiol. 2017, 15, 161–168. [Google Scholar] [CrossRef]
- Aiewsakun, P.; Adriaenssens, E.M.; Lavigne, R.; Kropinski, A.M.; Simmonds, P. Evaluation of the genomic diversity of viruses infecting bacteria, archaea and eukaryotes using a common bioinformatic platform: Steps towards a unified taxonomy. J Gen Virol 2018, 99, 1331–1343. [Google Scholar] [CrossRef]
- Aiewsakun, P.; Simmonds, P. The genomic underpinnings of eukaryotic virus taxonomy: Creating a sequence-based framework for family-level virus classification. Microbiome 2018, 6, 38. [Google Scholar] [CrossRef] [PubMed]
- Patterson Ross, Z.; Klunk, J.; Fornaciari, G.; Giuffra, V.; Duchêne, S.; Duggan, A.T.; Poinar, D.; Douglas, M.W.; Eden, J.S.; Holmes, E.C.; et al. The paradox of HBV evolution as revealed from a 16th century mummy. PLoS Pathog. 2018, 14, e1006750. [Google Scholar] [CrossRef]
- Krause-Kyora, B.; Susat, J.; Key, F.M.; Kühnert, D.; Bosse, E.; Immel, A.; Rinne, C.; Kornell, S.C.; Yepes, D.; Franzenburg, S.; et al. Neolithic and medieval virus genomes reveal complex evolution of hepatitis B. eLife 2018, 7, e36666. [Google Scholar] [CrossRef] [PubMed]
- Mühlemann, B.; Jones, T.C.; Damgaard, P.d.B.; Allentoft, M.E.; Shevnina, I.; Logvin, A.; Usmanova, E.; Panyushkina, I.P.; Boldgiv, B.; Bazartseren, T.; et al. Ancient hepatitis B viruses from the Bronze Age to the Medieval period. Nature 2018, 557, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Bar-Gal, G.K.; Kim, M.J.; Klein, A.; Shin, D.H.; Oh, C.S.; Kim, J.W.; Kim, T.H.; Kim, S.B.; Grant, P.R.; Pappo, O.; et al. Tracing hepatitis B virus to the 16th century in a Korean mummy. Hepatology 2012, 56, 1671–1680. [Google Scholar] [CrossRef]
- Puustusmaa, M.; Abroi, A. Conservation of the E8 CDS of the E8E2 protein among mammalian papillomaviruses. J. Gen. Virol. 2016, 97, 2333–2345. [Google Scholar] [CrossRef] [PubMed]
- Puustusmaa, M.; Abroi, A. cRegions—a tool for detecting conserved cis-elements in multiple sequence alignment of diverged coding sequences. PeerJ 2019, 6, e6176. [Google Scholar] [CrossRef]
- Nicholson, B.L.; White, K.A. Functional long-range RNA-RNA interactions in positive-strand RNA viruses. Nat. Rev. Microbiol. 2014, 12, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Madhugiri, R.; Karl, N.; Petersen, D.; Lamkiewicz, K.; Fricke, M.; Wend, U.; Scheuer, R.; Marz, M.; Ziebuhr, J. Structural and functional conservation of cis-acting RNA elements in coronavirus 5’-terminal genome regions. Virology 2018, 517, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Kuraku, S.; Zmasek, C.M.; Nishimura, O.; Katoh, K. aLeaves facilitates on-demand exploration of metazoan gene family trees on MAFFT sequence alignment server with enhanced interactivity. Nucleic Acids Res. 2013, 41, W22–W28. [Google Scholar] [CrossRef] [PubMed]
- Will, S.; Reiche, K.; Hofacker, I.L.; Stadler, P.F.; Backofen, R. Inferring noncoding RNA families and classes by means of genome-scale structure-based clustering. PLoS Comput. Biol. 2007, 3, e65. [Google Scholar] [CrossRef]
- Pickett, B.E.; Sadat, E.L.; Zhang, Y.; Noronha, J.M.; Squires, R.B.; Hunt, V.; Liu, M.; Kumar, S.; Zaremba, S.; Gu, Z.; et al. ViPR: An open bioinformatics database and analysis resource for virology research. Nucleic Acids Res. 2012, 40, D593–D598. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Sanlés, A.; Ríos-Marco, P.; Romero-López, C.; Berzal-Herranz, A. Functional Information Stored in the Conserved Structural RNA Domains of Flavivirus Genomes. Front. Microbiol. 2017, 8, 546. [Google Scholar] [CrossRef]
- Filomatori, C.V.; Lodeiro, M.F.; Alvarez, D.E.; Samsa, M.M.; Pietrasanta, L.; Gamarnik, A.V. A 5’ RNA element promotes dengue virus RNA synthesis on a circular genome. Genes Dev. 2006, 20, 2238–2249. [Google Scholar] [CrossRef] [PubMed]
- Clyde, K.; Barrera, J.; Harris, E. The capsid-coding region hairpin element (cHP) is a critical determinant of dengue virus and West Nile virus RNA synthesis. Virology 2008, 379, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Kozak, M. Downstream secondary structure facilitates recognition of initiator codons by eukaryotic ribosomes. Proc. Natl. Acad. Sci. USA 1990, 87, 8301–8305. [Google Scholar] [CrossRef]
- Global Consortium for H5N8 and Related Influenza Viruses. Role for migratory wild birds in the global spread of avian influenza H5N8. Science 2016, 354, 213–217. [Google Scholar] [CrossRef]
- Lycett, S.; Tanya, V.N.; Hall, M.; King, D.; Mazeri, S.; Mioulet, V.; Knowles, N.; Wadsworth, J.; Bachanek-Bankowska, K.; Victor, N.N.; et al. The evolution and phylodynamics of serotype A and SAT2 foot-and-mouth disease viruses in endemic regions of Africa. bioRxiv 2019, 572198. [Google Scholar] [CrossRef]
- Duchatel, F.; Bronsvoort, M.; Lycett, S. Phylogeographic analysis and identification of factors impacting the diffusion of Foot-and-Mouth disease virus in Africa. bioRxiv 2018, 358044. [Google Scholar] [CrossRef]
- Dhingra, M.S.; Artois, J.; Dellicour, S.; Lemey, P.; Dauphin, G.; Von Dobschuetz, S.; Van Boeckel, T.P.; Castellan, D.M.; Morzaria, S.; Gilbert, M. Geographical and Historical Patterns in the Emergences of Novel Highly Pathogenic Avian Influenza (HPAI) H5 and H7 Viruses in Poultry. Front. Vet. Sci. 2018, 5, 84. [Google Scholar] [CrossRef]
- Abdelwhab, E.M.; Veits, J.; Ulrich, R.; Kasbohm, E.; Teifke, J.P.; Mettenleiter, T.C. Composition of the Hemagglutinin Polybasic Proteolytic Cleavage Motif Mediates Variable Virulence of H7N7 Avian Influenza Viruses. Sci. Rep. 2016, 6, 3950. [Google Scholar] [CrossRef] [PubMed][Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Date | Location | # of Participants | Key outcomes |
|---|---|---|---|
| 6–8 March 2017 | Friedrich Schiller University Jena, Germany | ~100 | Founding of the Center; Discussion of the role of EVBC; Election of the first Board of Directors; Insights into EU policy and funding opportunities. |
| 9–10 April 2018 | Utrecht University, Netherlands | ~120 | Extending of the EVBC network to include America and Asia; Discussion and design of joint projects; Insights on first applied European fund among EVBC members [3]. |
| 28–29 March 2019 | University of Glasgow, United Kingdom | ~110 | Inclusion of contributed talks in themed sections in the scientific programme; Establishment of travel, poster and best contributed talk awards for junior scientists; Need for greater coordination and communication within the European virology community. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hufsky, F.; Ibrahim, B.; Modha, S.; Clokie, M.R.J.; Deinhardt-Emmer, S.; Dutilh, B.E.; Lycett, S.; Simmonds, P.; Thiel, V.; Abroi, A.; et al. The Third Annual Meeting of the European Virus Bioinformatics Center. Viruses 2019, 11, 420. https://doi.org/10.3390/v11050420
Hufsky F, Ibrahim B, Modha S, Clokie MRJ, Deinhardt-Emmer S, Dutilh BE, Lycett S, Simmonds P, Thiel V, Abroi A, et al. The Third Annual Meeting of the European Virus Bioinformatics Center. Viruses. 2019; 11(5):420. https://doi.org/10.3390/v11050420
Chicago/Turabian StyleHufsky, Franziska, Bashar Ibrahim, Sejal Modha, Martha R. J. Clokie, Stefanie Deinhardt-Emmer, Bas E. Dutilh, Samantha Lycett, Peter Simmonds, Volker Thiel, Aare Abroi, and et al. 2019. "The Third Annual Meeting of the European Virus Bioinformatics Center" Viruses 11, no. 5: 420. https://doi.org/10.3390/v11050420
APA StyleHufsky, F., Ibrahim, B., Modha, S., Clokie, M. R. J., Deinhardt-Emmer, S., Dutilh, B. E., Lycett, S., Simmonds, P., Thiel, V., Abroi, A., Adriaenssens, E. M., Escalera-Zamudio, M., Kelly, J. N., Lamkiewicz, K., Lu, L., Susat, J., Sicheritz, T., Robertson, D. L., & Marz, M. (2019). The Third Annual Meeting of the European Virus Bioinformatics Center. Viruses, 11(5), 420. https://doi.org/10.3390/v11050420