Modeling of Mutational Events in the Evolution of Viruses



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Virus Evolution Introduction
Time-Dependent Rate Phenomena
2. Materials and Methods
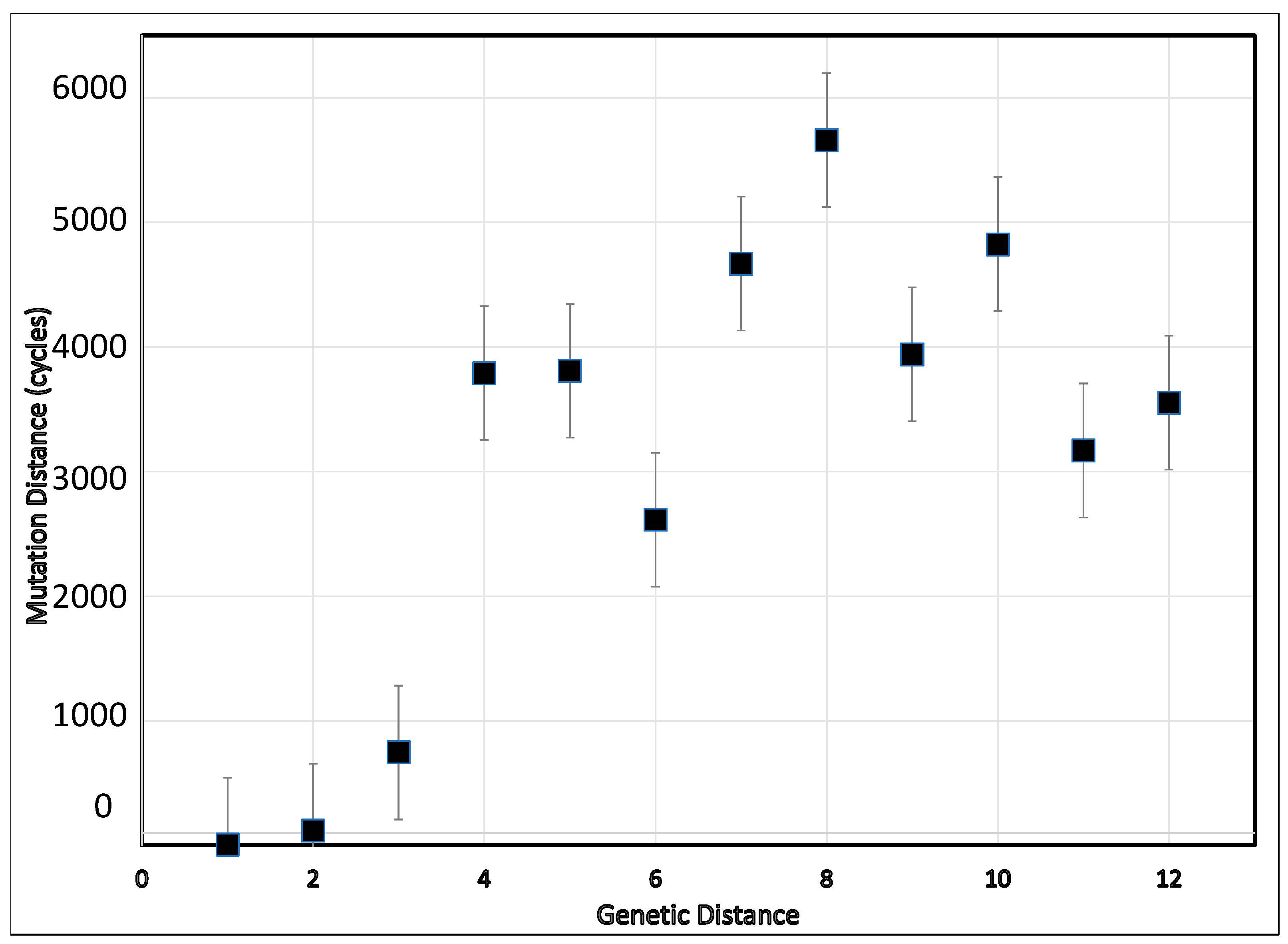
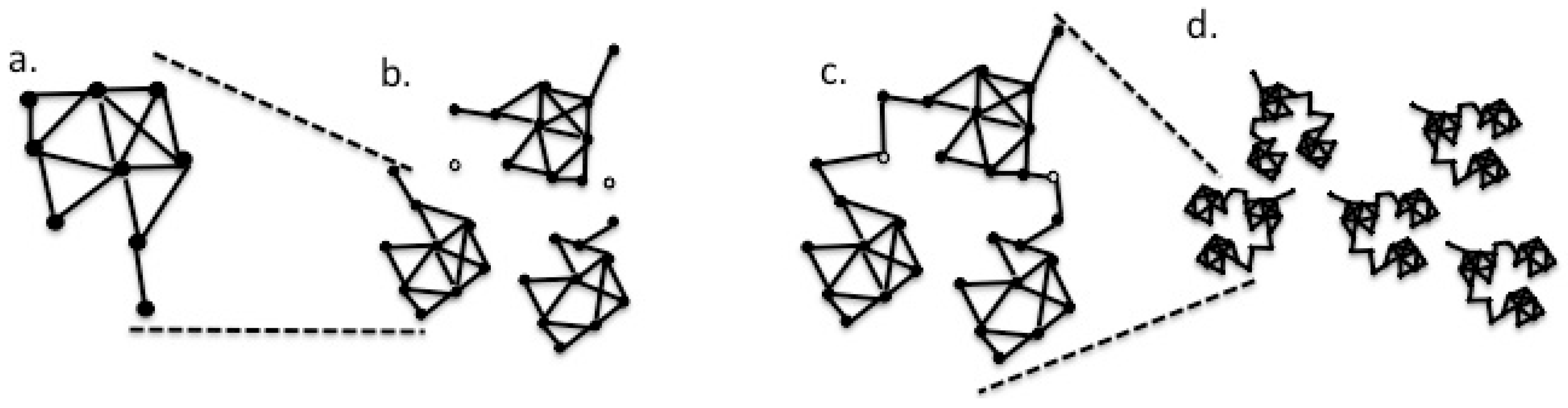
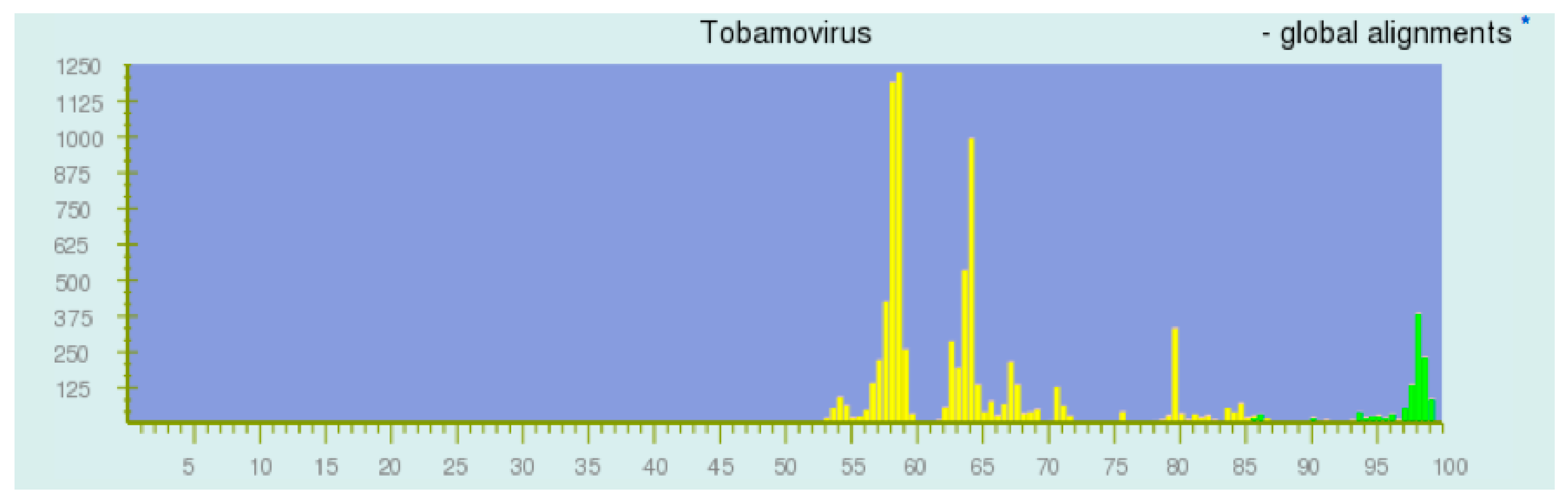
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Bao, X.D.; Roossinck, M.J. A life history view of mutualistic viral symbioses: Quantity or quality for cooperation? Curr. Opin. Microbiol. 2013, 16, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Márquez, L.M.; Redman, R.S.; Rodriguez, R.J.; Roossinck, M.J. A virus in a fungus in a plant: Three-way symbiosis required for thermal tolerance. Science 2007, 315, 513–515. [Google Scholar] [CrossRef] [PubMed]
- Roossinck, M.J. The good viruses: Viral mutualistic symbioses. Nat. Rev. Microbiol. 2011, 9, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Roossinck, M.J. Plants, viruses and the environment: Ecology and mutualism. Virology 2015, 479, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Elena, S.F.; Agudelo-Romero, P.; Carrasco, P.; Codoner, F.M.; Martin, S.; Torres-Barcelo, C.; Sanjuan, R. Experimental evolution of plant RNA viruses. Heredity 2008, 100, 478–483. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Duchêne, S.; Holmes, E.C.; Ho, S.Y.W. Analyses of evolutionary dynamics in viruses are hindered by a time-dependent bias in rate estimates. Proc. R. Soc. B Biol. Sci. 2014, 281, 20140732. [Google Scholar]
- Gibbs, A.J.; Fargette, D.; Garcia-Arenal, F.; Gibbs, M.J. Time--the emerging dimension of plant virus studies. J. Gen. Virol. 2010, 91, 13–22. [Google Scholar]
- Ho, S.Y.W.; Lanfear, R.; Bromham, L.; Phillips, M.J.; Soubrier, J.; Rodrigo, A.G.; Cooper, A. Time-dependent rates of molecular evolution. Mol. Ecol. 2011, 20, 3087–3101. [Google Scholar] [CrossRef]
- Ge, L.; Zhang, J.; Zhou, X.; Li, H. Genetic structure and population variability of tomato yellow leaf curl China virus. J. Virol. 2007, 81, 5902–5907. [Google Scholar] [CrossRef] [PubMed]
- Schneider, W.L.; Roossinck, M.J. Evolutionarily related Sindbis-like plant viruses maintain different levels of population diversity in a common host. J. Virol. 2000, 74, 3130–3134. [Google Scholar] [CrossRef]
- Schneider, W.L.; Roossinck, M.J. Genetic diversity in RNA virus quasispecies is controlled by host-virus interactions. J. Virol. 2001, 75, 6566–6571. [Google Scholar] [CrossRef] [PubMed]
- Emerson, B.C.; Hickerson, M.J. Lack of support for the time-dependent molecular evolution hypothesis. Mol. Ecol. 2015, 24, 702–709. [Google Scholar] [CrossRef] [PubMed]
- Harkins, K.M.; Stone, A.C. Ancient pathogen genomics: Insights into timing and adaptation. J. Hum. Evol. 2015, 79, 137–149. [Google Scholar] [CrossRef]
- Pagan, I.; Firth, C.; Holmes, E.C. Phylogenetic analysis reveals rapid evolutionary dynamics in the plant RNA virus genus tobamovirus. J. Mol. Evol. 2010, 71, 298–307. [Google Scholar] [CrossRef]
- Pagan, I.; Holmes, E.C. Long-term evolution of the Luteoviridae: Time scale and mode of virus speciation. J. Virol. 2010, 84, 6177–6187. [Google Scholar] [CrossRef] [PubMed]
- Harkins, G.; Delport, W.; Duffy, S.; Wood, N.; Monjane, A.; Owor, B.; Donaldson, L.; Saumtally, S.; Triton, G.; Briddon, R.; et al. Experimental evidence indicating that mastreviruses probably did not co-diverge with their hosts. Virol. J. 2009, 6, 104. [Google Scholar] [CrossRef] [PubMed]
- Fraile, A.; Escriu, F.; Aranda, M.A.; Malpica, J.M.; Gibbs, A.J.; García-Arenal, F. A century of tobamovirus evolution in an Australian population of Nicotiana glauca. J. Virol. 1997, 71, 8316–8320. [Google Scholar]
- Malmstrom, C.M.; Shu, R.; Linton, E.W.; Newton, L.A.; Cook, M.A. Barley yellow dwarf viruses (BYDVs) preserved in herbarium specimens illuminate historical disease ecology of invasive and native grasses. J. Ecol. 2007, 95, 1153–1166. [Google Scholar] [CrossRef]
- Smith, O.; Clapham, A.; Rose, P.; Liu, Y.; Wang, J.; Allaby, R.G. A complete ancient RNA genome: Identification, reconstruction and evolutionary history of archaeological Barley Stripe Mosaic Virus. Sci. Rep. 2014, 4, 4003. [Google Scholar] [CrossRef] [PubMed]
- Aiewsakun, P.; Katzourakis, A. Time dependency of foamy virus evolutionary rate estimates. BMC Evol. Biol. 2015, 15, 119. [Google Scholar] [CrossRef]
- Stobbe, A.H.; Melcher, U.; Palmer, M.W.; Roossinck, M.J.; Shen, G. Co-divergence and host-switching in the evolution of tobamoviruses. J. Gen. Virol. 2012, 93, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Biek, R.; Pybus, O.G.; Lloyd-Smith, J.O.; Didelot, X. Measurably evolving pathogens in the genomic era. Trends Ecol. Evol. 2015, 30, 306–313. [Google Scholar] [CrossRef]
- Bielejec, F.; Lemey, P.; Baele, G.; Rambaut, A.; Suchard, M.A. Inferring Heterogeneous Evolutionary Processes Through Time: From Sequence Substitution to Phylogeography. Syst. Biol. 2014, 63, 493–504. [Google Scholar] [CrossRef]
- Snir, S.; Wolf, Y.I.; Koonin, E.V. Universal Pacemaker of Genome Evolution. PLoS Comput. Biol. 2012, 8, e1002785. [Google Scholar] [CrossRef] [PubMed]
- Duchene, D.; Duchene, S.; Ho, S.Y.W. Tree imbalance causes a bias in phylogenetic estimation of evolutionary timescales using heterochronous sequences. Mol. Ecol. Resour. 2015, 15, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Wertheim, J.O.; Smith, M.D.; Smith, D.M.; Scheffler, K.; Pond, S.L.K. Evolutionary Origins of Human Herpes Simplex Viruses 1 and 2. Mol. Biol. Evol. 2014, 31, 2356–2364. [Google Scholar] [CrossRef]
- Manrubia, S.; Cuesta, J.A. Evolution on neutral networks accelerates the ticking rate of the molecular clock. J. R. Soc. Interface 2015, 12, 20141010. [Google Scholar] [CrossRef]
- Park, S.C.; Szendro, I.G.; Neidhart, J.; Krug, J. Phase transition in random adaptive walks on correlated fitness landscapes. Phys. Rev. E 2015, 91, 042707. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, J. Ronald H Chipmunk Basic. Available online: http://www.nicholson.com/rhn/basic/ (accessed on 15 September 2018).
- Jeong, H.; Tombor, B.; Albert, R.; Oltvai, Z.N.; Barabasi, A.L. The large-scale organization of metabolic networks. Nature 2000, 407, 651–654. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.J.; Antoniw, J.F.; Bar-Joseph, M.; Brunt, A.A.; Candresse, T.; Foster, G.D.; Martelli, G.P.; Milne, R.G.; Zavriev, S.K.; Fauquet, C.M. The new plant virus family Flexiviridae and assessment of molecular criteria for species demarcation. Arch. Virol. 2004, 149, 1045–1060. [Google Scholar] [CrossRef]
- Adams, M.J.; Antoniw, J.F.; Fauquet, C.M. Molecular criteria for genus and species discrimination within the family Potyviridae. Arch. Virol. 2005, 150, 459–479. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Kapustin, Y.; Tatusova, T. Virus classification by pairwise sequence comparison (PASC). In Encyclopedia of Virology; Mahy, B.W.J., Regenmortel, M.H.V.V., Eds.; Elsevier: Oxford, UK, 2008; Volume 5, pp. 342–348. [Google Scholar]
- de Villiers, E.M.; Fauquet, C.; Broker, T.R.; Bernard, H.U.; zur Hausen, H. Classification of papillomaviruses. Virology 2004, 324, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Fauquet, C.M.; Stanley, J. Geminivirus classification and nomenclature: Progress and problems. Ann. Appl. Biol. 2003, 142, 165–189. [Google Scholar] [CrossRef]
- Oberste, M.S.; Maher, K.; Kilpatrick, D.R.; Pallansch, M.A. Molecular evolution of the human enteroviruses: Correlation of serotype with VP1 sequence and application to picornavirus classification. J. Virol. 1999, 73, 1941–1948. [Google Scholar] [PubMed]
- Lauber, C.; Gorbalenya, A.E. Partitioning the Genetic Diversity of a Virus Family: Approach and Evaluation through a Case Study of Picornaviruses. J. Virol. 2012, 86, 3890–3904. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, A.; Melcher, U. Modeling of Mutational Events in the Evolution of Viruses. Viruses 2019, 11, 418. https://doi.org/10.3390/v11050418
Ali A, Melcher U. Modeling of Mutational Events in the Evolution of Viruses. Viruses. 2019; 11(5):418. https://doi.org/10.3390/v11050418
Chicago/Turabian StyleAli, Akhtar, and Ulrich Melcher. 2019. "Modeling of Mutational Events in the Evolution of Viruses" Viruses 11, no. 5: 418. https://doi.org/10.3390/v11050418
APA StyleAli, A., & Melcher, U. (2019). Modeling of Mutational Events in the Evolution of Viruses. Viruses, 11(5), 418. https://doi.org/10.3390/v11050418