Extracellular Vesicles and Ebola Virus: A New Mechanism of Immune Evasion

 , and
, and 
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Ebola Virus
1.2. Extracellular Vesicles
2. EVs and VP40
2.1. VP40 and the ESCRT Pathway
2.2. VP40 Influence on the Host Cell Cycle
2.3. EV-Associated VP40 and Recipient Bystander Cell Effects
3. EVs and NP
4. EVs and GP
5. Remaining Questions
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Disclaimer
References
- WHO. Ebola Outbreak 2014–2015. Available online: http://www.who.int/csr/disease/ebola/en/ (accessed on 7 January 2019).
- Ebola. Ebola Situation Reports: Democratic Republic of the Congo. Available online: http://www.who.int/ebola/situation-reports/drc-2018/en/ (accessed on 7 January 2019).
- WHO. Ebola Virus Disease—Democratic Republic of the Congo. Available online: http://www.who.int/csr/don/25-july-2018-ebola-drc/en/ (accessed on 1 August 2018).
- Rougeron, V.; Feldmann, H.; Grard, G.; Becker, S.; Leroy, E.M. Ebola and Marburg haemorrhagic fever. J. Clin. Virol. 2015, 64, 111–119. [Google Scholar] [CrossRef] [PubMed]
- MüHlberger, E. Filovirus replication and transcription. Future Virol. 2007, 2, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.; Kiley, M.P.; Holloway, B.P.; Auperin, D.D. Sequence analysis of the Ebola virus genome: Organization, genetic elements, and comparison with the genome of Marburg virus. Virus Res. 1993, 29, 215–240. [Google Scholar] [CrossRef]
- Volchkov, V.E.; Becker, S.; Volchkova, V.A.; Ternovoj, V.A.; Kotov, A.N.; Netesov, S.V.; Klenk, H.-D. GP mRNA of Ebola Virus Is Edited by the Ebola Virus Polymerase and by T7 and Vaccinia Virus Polymerases1. Virology 1995, 214, 421–430. [Google Scholar] [CrossRef]
- Sanchez, A.; Trappier, S.G.; Mahy, B.W.; Peters, C.J.; Nichol, S.T. The virion glycoproteins of Ebola viruses are encoded in two reading frames and are expressed through transcriptional editing. Proc. Natl. Acad. Sci. USA 1996, 93, 3602–3607. [Google Scholar] [CrossRef]
- Manicassamy, B.; Wang, J.; Rumschlag, E.; Tymen, S.; Volchkova, V.; Volchkov, V.; Rong, L. Characterization of Marburg virus glycoprotein in viral entry. Virology 2007, 358, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Cook, J.D.; Lee, J.E. The Secret Life of Viral Entry Glycoproteins: Moonlighting in Immune Evasion. PLoS Pathog. 2013, 9, e1003258. [Google Scholar] [CrossRef] [PubMed]
- Geisbert, T.W.; Hensley, L.E.; Larsen, T.; Young, H.A.; Reed, D.S.; Geisbert, J.B.; Scott, D.P.; Kagan, E.; Jahrling, P.B.; Davis, K.J. Pathogenesis of Ebola Hemorrhagic Fever in Cynomolgus Macaques: Evidence that Dendritic Cells Are Early and Sustained Targets of Infection. Am. J. Pathol. 2003, 163, 2347–2370. [Google Scholar] [CrossRef]
- Ryabchikova, E.I.; Kolesnikova, L.V.; Luchko, S.V. An Analysis of Features of Pathogenesis in Two Animal Models of Ebola Virus Infection. J. Infect. Dis. 1999, 179, S199–S202. [Google Scholar] [CrossRef]
- Martines, R.B.; Ng, D.L.; Greer, P.W.; Rollin, P.E.; Zaki, S.R. Tissue and cellular tropism, pathology and pathogenesis of Ebola and Marburg viruses. J. Pathol. 2015, 235, 153–174. [Google Scholar] [CrossRef] [PubMed]
- Messaoudi, I.; Amarasinghe, G.K.; Basler, C.F. Filovirus pathogenesis and immune evasion: Insights from Ebola virus and Marburg virus. Nat. Rev. Microbiol. 2015, 13, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Falasca, L.; Agrati, C.; Petrosillo, N.; Di Caro, A.; Capobianchi, M.R.; Ippolito, G.; Piacentini, M. Molecular mechanisms of Ebola virus pathogenesis: Focus on cell death. Cell Death Differ. 2015, 22, 1250–1259. [Google Scholar] [CrossRef] [PubMed]
- Zaki, S.R.; Goldsmith, C.S. Pathologic features of filovirus infections in humans. Curr. Top. Microbiol. Immunol. 1999, 235, 97–116. [Google Scholar]
- Deen, G.F.; Broutet, N.; Xu, W.; Knust, B.; Sesay, F.R.; McDonald, S.L.R.; Ervin, E.; Marrinan, J.E.; Gaillard, P.; Habib, N.; et al. Ebola RNA Persistence in Semen of Ebola Virus Disease Survivors — Final Report. N. Engl. J. Med. 2017, 377, 1428–1437. [Google Scholar] [CrossRef]
- Varkey, J.B.; Shantha, J.G.; Crozier, I.; Kraft, C.S.; Lyon, G.M.; Mehta, A.K.; Kumar, G.; Smith, J.R.; Kainulainen, M.H.; Whitmer, S.; et al. Persistence of Ebola Virus in Ocular Fluid during Convalescence. N. Engl. J. Med. 2015, 372, 2423–2427. [Google Scholar] [CrossRef]
- Sissoko, D.; Keïta, M.; Diallo, B.; Aliabadi, N.; Fitter, D.L.; Dahl, B.A.; Bore, J.A.; Koundouno, F.R.; Singethan, K.; Meisel, S.; et al. Ebola virus persistence in breast milk after no reported illness: A likely source of virus transmission from mother to child. Clin. Infect. Dis. 2017, 64, 513–516. [Google Scholar] [CrossRef]
- Vetter, P.; Fischer, W.A.; Schibler, M.; Jacobs, M.; Bausch, D.G.; Kaiser, L. Ebola Virus Shedding and Transmission: Review of Current Evidence. J. Infect. Dis. 2016, 214, S177–S184. [Google Scholar] [CrossRef]
- Mora-Rillo, M.; Arsuaga, M.; Ramírez-Olivencia, G.; de la Calle, F.; Borobia, A.M.; Sánchez-Seco, P.; Lago, M.; Figueira, J.C.; Fernández-Puntero, B.; Viejo, A.; et al. Acute respiratory distress syndrome after convalescent plasma use: Treatment of a patient with Ebola virus disease contracted in Madrid, Spain. Lancet Respir. Med. 2015, 3, 554–562. [Google Scholar] [CrossRef]
- Liddell, A.M.; Davey, R.T.; Mehta, A.K.; Varkey, J.B.; Kraft, C.S.; Tseggay, G.K.; Badidi, O.; Faust, A.C.; Brown, K.V.; Suffredini, A.F.; et al. Characteristics and Clinical Management of a Cluster of 3 Patients With Ebola Virus Disease, Including the First Domestically Acquired Cases in the United States. Ann. Intern. Med. 2015, 163, 81–90. [Google Scholar] [CrossRef]
- Nordenstedt, H.; Bah, E.I.; de la Vega, M.-A.; Barry, M.; N’Faly, M.; Barry, M.; Crahay, B.; Decroo, T.; Van Herp, M.; Ingelbeen, B. Ebola Virus in Breast Milk in an Ebola Virus–Positive Mother with Twin Babies, Guinea, 2015. Emerg. Infect. Dis. 2016, 22, 759–760. [Google Scholar] [CrossRef]
- Brainard, J.; Pond, K.; Hooper, L.; Edmunds, K.; Hunter, P. Presence and Persistence of Ebola or Marburg Virus in Patients and Survivors: A Rapid Systematic Review. PLoS Negl. Trop. Dis. 2016, 10, e0004475. [Google Scholar] [CrossRef]
- MacIntyre, C.R.; Chughtai, A.A. Recurrence and reinfection—a new paradigm for the management of Ebola virus disease. Int. J. Infect. Dis. 2016, 43, 58–61. [Google Scholar] [CrossRef]
- Overholt, L.; Wohl, D.A.; Fischer, W.A.; Westreich, D.; Tozay, S.; Reeves, E.; Pewu, K.; Adjasso, D.; Hoover, D.; Merenbloom, C.; et al. Stigma and Ebola survivorship in Liberia: Results from a longitudinal cohort study. PLoS ONE 2018, 13, e0206595. [Google Scholar] [CrossRef]
- Chan, J.; Patel, M.; Tobin, S.; Sheppeard, V. Monitoring travellers from Ebola-affected countries in New South Wales, Australia: What is the impact on travellers? BMC Public Health 2017, 17, 113. [Google Scholar] [CrossRef]
- Van Bortel, T.; Basnayake, A.; Wurie, F.; Jambai, M.; Koroma, A.S.; Muana, A.T.; Hann, K.; Eaton, J.; Martin, S.; Nellums, L.B. Psychosocial effects of an Ebola outbreak at individual, community and international levels. Bull. World Health Organ. 2016, 94, 210–214. [Google Scholar] [CrossRef]
- Davtyan, M.; Brown, B.; Folayan, M.O. Addressing Ebola-related Stigma: Lessons Learned from HIV/AIDS. Glob. Health Action 2014, 7, 26058. [Google Scholar] [CrossRef]
- Mate, S.E.; Kugelman, J.R.; Nyenswah, T.G.; Ladner, J.T.; Wiley, M.R.; Cordier-Lassalle, T.; Christie, A.; Schroth, G.P.; Gross, S.M.; Davies-Wayne, G.J.; et al. Molecular Evidence of Sexual Transmission of Ebola Virus. N. Engl. J. Med. 2015, 373, 2448–2454. [Google Scholar] [CrossRef]
- Christie, A.; Davies-Wayne, G.J.; Cordier-Lassalle, T.; Cordier-Lasalle, T.; Blackley, D.J.; Laney, A.S.; Williams, D.E.; Shinde, S.A.; Badio, M.; Lo, T.; et al. Possible sexual transmission of Ebola virus-Liberia, 2015. MMWR Morb. Mortal. Wkly. Rep. 2015, 64, 479–481. [Google Scholar]
- Subissi, L.; Keita, M.; Mesfin, S.; Rezza, G.; Diallo, B.; Van Gucht, S.; Musa, E.O.; Yoti, Z.; Keita, S.; Djingarey, M.H.; et al. Ebola Virus Transmission Caused by Persistently Infected Survivors of the 2014–2016 Outbreak in West Africa. J. Infect. Dis. 2018, 218, S287–S291. [Google Scholar] [CrossRef]
- Arias, A.; Watson, S.J.; Asogun, D.; Tobin, E.A.; Lu, J.; Phan, M.V.T.; Jah, U.; Wadoum, R.E.G.; Meredith, L.; Thorne, L.; et al. Rapid outbreak sequencing of Ebola virus in Sierra Leone identifies transmission chains linked to sporadic cases. Virus Evol. 2016, 2, vew016. [Google Scholar] [CrossRef]
- Keita, M.; Duraffour, S.; Loman, N.J.; Rambaut, A.; Diallo, B.; Magassouba, N.; Carroll, M.W.; Quick, J.; Sall, A.A.; Glynn, J.R.; et al. Unusual Ebola Virus Chain of Transmission, Conakry, Guinea, 2014–2015. Emerg. Infect. Dis. 2016, 22, 2149–2152. [Google Scholar] [CrossRef] [PubMed]
- Diallo, B.; Sissoko, D.; Loman, N.J.; Bah, H.A.; Bah, H.; Worrell, M.C.; Conde, L.S.; Sacko, R.; Mesfin, S.; Loua, A.; et al. Resurgence of Ebola Virus Disease in Guinea Linked to a Survivor With Virus Persistence in Seminal Fluid for More Than 500 Days. Clin. Infect. Dis. 2016, 63, 1353–1356. [Google Scholar] [CrossRef] [PubMed]
- Leroy, E.; Baize, S.; Volchkov, V.; Fisher-Hoch, S.; Georges-Courbot, M.-C.; Lansoud-Soukate, J.; Capron, M.; Debré, P.; Georges, A.; McCormick, J. Human asymptomatic Ebola infection and strong inflammatory response. The Lancet 2000, 355, 2210–2215. [Google Scholar] [CrossRef]
- Mbala, P.; Baguelin, M.; Ngay, I.; Rosello, A.; Mulembakani, P.; Demiris, N.; Edmunds, W.J.; Muyembe, J.-J. Evaluating the frequency of asymptomatic Ebola virus infection. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2017, 372. [Google Scholar] [CrossRef] [PubMed]
- Glynn, J.R.; Bower, H.; Johnson, S.; Houlihan, C.F.; Montesano, C.; Scott, J.T.; Semple, M.G.; Bangura, M.S.; Kamara, A.J.; Kamara, O.; et al. Asymptomatic infection and unrecognised Ebola virus disease in Ebola-affected households in Sierra Leone: A cross-sectional study using a new non-invasive assay for antibodies to Ebola virus. Lancet Infect. Dis. 2017, 17, 645–653. [Google Scholar] [CrossRef]
- Dean, N.E.; Halloran, M.E.; Yang, Y.; Longini, I.M. Transmissibility and Pathogenicity of Ebola Virus: A Systematic Review and Meta-analysis of Household Secondary Attack Rate and Asymptomatic Infection. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2016, 62, 1277–1286. [Google Scholar] [CrossRef] [PubMed]
- Richardson, E.T.; Kelly, J.D.; Barrie, M.B.; Mesman, A.W.; Karku, S.; Quiwa, K.; Marsh, R.H.; Koedoyoma, S.; Daboh, F.; Barron, K.P.; et al. Minimally Symptomatic Infection in an Ebola ‘Hotspot’: A Cross-Sectional Serosurvey. PLoS Negl. Trop. Dis. 2016, 10, e0005087. [Google Scholar] [CrossRef]
- Clark, D.V.; Kibuuka, H.; Millard, M.; Wakabi, S.; Lukwago, L.; Taylor, A.; Eller, M.A.; Eller, L.A.; Michael, N.L.; Honko, A.N.; et al. Long-term sequelae after Ebola virus disease in Bundibugyo, Uganda: A retrospective cohort study. Lancet Infect. Dis. 2015, 15, 905–912. [Google Scholar] [CrossRef]
- Rowe, A.K.; Bertolli, J.; Khan, A.S.; Mukunu, R.; Muyembe-Tamfum, J.J.; Bressler, D.; Williams, A.J.; Peters, C.J.; Rodriguez, L.; Feldmann, H.; et al. Clinical, Virologic, and Immunologic Follow-Up of Convalescent Ebola Hemorrhagic Fever Patients and Their Household Contacts, Kikwit, Democratic Republic of the Congo. J. Infect. Dis. 1999, 179, S28–S35. [Google Scholar] [CrossRef]
- Bower, H.; Glynn, J.R. A systematic review and meta-analysis of seroprevalence surveys of ebolavirus infection. Sci. Data 2017, 4, 160133. [Google Scholar] [CrossRef][Green Version]
- Houlihan, C.F.; McGowan, C.R.; Dicks, S.; Baguelin, M.; Moore, D.A.J.; Mabey, D.; Roberts, C.H.; Kumar, A.; Samuel, D.; Tedder, R.; et al. Ebola exposure, illness experience, and Ebola antibody prevalence in international responders to the West African Ebola epidemic 2014–2016: A cross-sectional study. PLoS Med. 2017, 14, e1002300. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, Secretion, and Intercellular Interactions of Exosomes and Other Extracellular Vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef]
- Abels, E.R.; Breakefield, X.O. Introduction to Extracellular Vesicles: Biogenesis, RNA Cargo Selection, Content, Release, and Uptake. Cell Mol. Neurobiol. 2016, 36, 301–312. [Google Scholar] [CrossRef]
- Xu, R.; Greening, D.W.; Zhu, H.-J.; Takahashi, N.; Simpson, R.J. Extracellular vesicle isolation and characterization: Toward clinical application. J. Clin. Invest. 2016, 126, 1152–1162. [Google Scholar] [CrossRef]
- Akers, J.C.; Gonda, D.; Kim, R.; Carter, B.S.; Chen, C.C. Biogenesis of extracellular vesicles (EV): Exosomes, microvesicles, retrovirus-like vesicles, and apoptotic bodies. J. Neurooncol. 2013, 113, 1–11. [Google Scholar] [CrossRef]
- Keller, S.; Sanderson, M.P.; Stoeck, A.; Altevogt, P. Exosomes: From biogenesis and secretion to biological function. Immunol. Lett. 2006, 107, 102–108. [Google Scholar] [CrossRef]
- Schmidt, O.; Teis, D. The ESCRT machinery. Curr. Biol. 2012, 22, R116–R120. [Google Scholar] [CrossRef]
- Henne, W.M.; Buchkovich, N.J.; Emr, S.D. The ESCRT Pathway. Dev. Cell 2011, 21, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Stuffers, S.; Sem Wegner, C.; Stenmark, H.; Brech, A. Multivesicular endosome biogenesis in the absence of ESCRTs. Traffic Cph. Den. 2009, 10, 925–937. [Google Scholar] [CrossRef]
- van Niel, G.; Charrin, S.; Simoes, S.; Romao, M.; Rochin, L.; Saftig, P.; Marks, M.S.; Rubinstein, E.; Raposo, G. The Tetraspanin CD63 Regulates ESCRT-Independent and -Dependent Endosomal Sorting during Melanogenesis. Dev. Cell 2011, 21, 708–721. [Google Scholar] [CrossRef]
- Edgar, J.R.; Eden, E.R.; Futter, C.E. Hrs- and CD63-Dependent Competing Mechanisms Make Different Sized Endosomal Intraluminal Vesicles. Traffic 2014, 15, 197–211. [Google Scholar] [CrossRef]
- Ghossoub, R.; Lembo, F.; Rubio, A.; Gaillard, C.B.; Bouchet, J.; Vitale, N.; Slavík, J.; Machala, M.; Zimmermann, P. Syntenin-ALIX exosome biogenesis and budding into multivesicular bodies are controlled by ARF6 and PLD2. Nat. Commun. 2014, 5, 3477. [Google Scholar] [CrossRef]
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brügger, B.; Simons, M. Ceramide Triggers Budding of Exosome Vesicles into Multivesicular Endosomes. Science 2008, 319, 1244–1247. [Google Scholar] [CrossRef]
- Alenquer, M.; Amorim, M.J. Exosome Biogenesis, Regulation, and Function in Viral Infection. Viruses 2015, 7, 5066–5083. [Google Scholar] [CrossRef]
- Vitelli, R.; Santillo, M.; Lattero, D.; Chiariello, M.; Bifulco, M.; Bruni, C.B.; Bucci, C. Role of the Small GTPase RAB7 in the Late Endocytic Pathway. J. Biol. Chem. 1997, 272, 4391–4397. [Google Scholar] [CrossRef]
- Feng, Y.; Press, B.; Wandinger-Ness, A. Rab 7: An important regulator of late endocytic membrane traffic. J. Cell Biol. 1995, 131, 1435–1452. [Google Scholar] [CrossRef]
- Baietti, M.F.; Zhang, Z.; Mortier, E.; Melchior, A.; Degeest, G.; Geeraerts, A.; Ivarsson, Y.; Depoortere, F.; Coomans, C.; Vermeiren, E.; et al. Syndecan-syntenin-ALIX regulates the biogenesis of exosomes. Nat. Cell Biol. 2012, 14, 677–685. [Google Scholar] [CrossRef]
- Ostrowski, M.; Carmo, N.B.; Krumeich, S.; Fanget, I.; Raposo, G.; Savina, A.; Moita, C.F.; Schauer, K.; Hume, A.N.; Freitas, R.P.; et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat. Cell Biol. 2010, 12, 19–30. [Google Scholar] [CrossRef]
- Savina, A.; Vidal, M.; Colombo, M.I. The exosome pathway in K562 cells is regulated by Rab11. J. Cell Sci. 2002, 115, 2505–2515. [Google Scholar]
- Beckett, K.; Monier, S.; Palmer, L.; Alexandre, C.; Green, H.; Bonneil, E.; Raposo, G.; Thibault, P.; Le Borgne, R.; Vincent, J.-P. Drosophila S2 cells secrete wingless on exosome-like vesicles but the wingless gradient forms independently of exosomes. Traffic Cph. Den. 2013, 14, 82–96. [Google Scholar] [CrossRef]
- Abrami, L.; Brandi, L.; Moayeri, M.; Brown, M.J.; Krantz, B.A.; Leppla, S.H.; van der Goot, F.G. Hijacking multivesicular bodies enables long-term and exosome-mediated long-distance action of anthrax toxin. Cell Rep. 2013, 5, 986–996. [Google Scholar] [CrossRef]
- Koles, K.; Budnik, V. Exosomes go with the Wnt. Cell. Logist. 2012, 2, 169–173. [Google Scholar] [CrossRef]
- Hsu, C.; Morohashi, Y.; Yoshimura, S.-I.; Manrique-Hoyos, N.; Jung, S.; Lauterbach, M.A.; Bakhti, M.; Grønborg, M.; Möbius, W.; Rhee, J.; et al. Regulation of exosome secretion by Rab35 and its GTPase-activating proteins TBC1D10A-C. J. Cell Biol. 2010, 189, 223–232. [Google Scholar] [CrossRef]
- Peinado, H.; Alečković, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; García-Santos, G.; Ghajar, C.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef]
- Bobrie, A.; Krumeich, S.; Reyal, F.; Recchi, C.; Moita, L.F.; Seabra, M.C.; Ostrowski, M.; Théry, C. Rab27a supports exosome-dependent and -independent mechanisms that modify the tumor microenvironment and can promote tumor progression. Cancer Res. 2012, 72, 4920–4930. [Google Scholar] [CrossRef]
- Anderson, M.R.; Kashanchi, F.; Jacobson, S. Exosomes in Viral Disease. Neurother. J. Am. Soc. Exp. Neurother. 2016, 13, 535–546. [Google Scholar] [CrossRef]
- Schwab, A.; Meyering, S.S.; Lepene, B.; Iordanskiy, S.; van Hoek, M.L.; Hakami, R.M.; Kashanchi, F. Extracellular vesicles from infected cells: Potential for direct pathogenesis. Front. Microbiol. 2015, 6, 1132. [Google Scholar] [CrossRef]
- Crenshaw, B.J.; Gu, L.; Sims, B.; Matthews, Q.L. Exosome Biogenesis and Biological Function in Response to Viral Infections. Open Virol. J. 2018, 12, 134–148. [Google Scholar] [CrossRef]
- Votteler, J.; Sundquist, W.I. Virus Budding and the ESCRT Pathway. Cell Host Microbe 2013, 14, 232–241. [Google Scholar] [CrossRef]
- Ahmed, I.; Akram, Z.; Iqbal, H.M.N.; Munn, A.L. The regulation of Endosomal Sorting Complex Required for Transport and accessory proteins in multivesicular body sorting and enveloped viral budding-An overview. Int. J. Biol. Macromol. 2019, 127, 1–11. [Google Scholar] [CrossRef]
- Christ, L.; Raiborg, C.; Wenzel, E.M.; Campsteijn, C.; Stenmark, H. Cellular Functions and Molecular Mechanisms of the ESCRT Membrane-Scission Machinery. Trends Biochem. Sci. 2017, 42, 42–56. [Google Scholar] [CrossRef]
- Han, Z.; Madara, J.J.; Liu, Y.; Liu, W.; Ruthel, G.; Freedman, B.D.; Harty, R.N. ALIX Rescues Budding of a Double PTAP/PPEY L-Domain Deletion Mutant of Ebola VP40: A Role for ALIX in Ebola Virus Egress. J. Infect. Dis. 2015, 212 Suppl 2, S138–S145. [Google Scholar] [CrossRef]
- Licata, J.M.; Simpson-Holley, M.; Wright, N.T.; Han, Z.; Paragas, J.; Harty, R.N. Overlapping Motifs (PTAP and PPEY) within the Ebola Virus VP40 Protein Function Independently as Late Budding Domains: Involvement of Host Proteins TSG101 and VPS-4. J. Virol. 2003, 77, 1812–1819. [Google Scholar] [CrossRef]
- Martin-Serrano, J.; Zang, T.; Bieniasz, P.D. HIV-1 and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat. Med. 2001, 7, 1313–1319. [Google Scholar] [CrossRef]
- Garrus, J.E.; von Schwedler, U.K.; Pornillos, O.W.; Morham, S.G.; Zavitz, K.H.; Wang, H.E.; Wettstein, D.A.; Stray, K.M.; Côté, M.; Rich, R.L.; et al. Tsg101 and the Vacuolar Protein Sorting Pathway Are Essential for HIV-1 Budding. Cell 2001, 107, 55–65. [Google Scholar] [CrossRef]
- Ku, P.-I.; Bendjennat, M.; Ballew, J.; Landesman, M.B.; Saffarian, S. ALIX Is Recruited Temporarily into HIV-1 Budding Sites at the End of Gag Assembly. PLoS ONE 2014, 9, e96950. [Google Scholar] [CrossRef]
- Carlton, J.G.; Agromayor, M.; Martin-Serrano, J. Differential requirements for Alix and ESCRT-III in cytokinesis and HIV-1 release. Proc. Natl. Acad. Sci. USA 2008, 105, 10541–10546. [Google Scholar] [CrossRef]
- Strack, B.; Calistri, A.; Craig, S.; Popova, E.; Göttlinger, H.G. AIP1/ALIX Is a Binding Partner for HIV-1 p6 and EIAV p9 Functioning in Virus Budding. Cell 2003, 114, 689–699. [Google Scholar] [CrossRef]
- Silvestri, L.S.; Ruthel, G.; Kallstrom, G.; Warfield, K.L.; Swenson, D.L.; Nelle, T.; Iversen, P.L.; Bavari, S.; Aman, M.J. Involvement of Vacuolar Protein Sorting Pathway in Ebola Virus Release Independent of TSG101 Interaction. J. Infect. Dis. 2007, 196, S264–S270. [Google Scholar] [CrossRef]
- Goff, A.; Ehrlich, L.S.; Cohen, S.N.; Carter, C.A. Tsg101 Control of Human Immunodeficiency Virus Type 1 Gag Trafficking and Release. J. Virol. 2003, 77, 9173–9182. [Google Scholar] [CrossRef] [PubMed]
- Dussupt, V.; Javid, M.P.; Abou-Jaoudé, G.; Jadwin, J.A.; Cruz, J. de L.; Nagashima, K.; Bouamr, F. The Nucleocapsid Region of HIV-1 Gag Cooperates with the PTAP and LYPXnL Late Domains to Recruit the Cellular Machinery Necessary for Viral Budding. PLoS Pathog. 2009, 5, e1000339. [Google Scholar] [CrossRef] [PubMed]
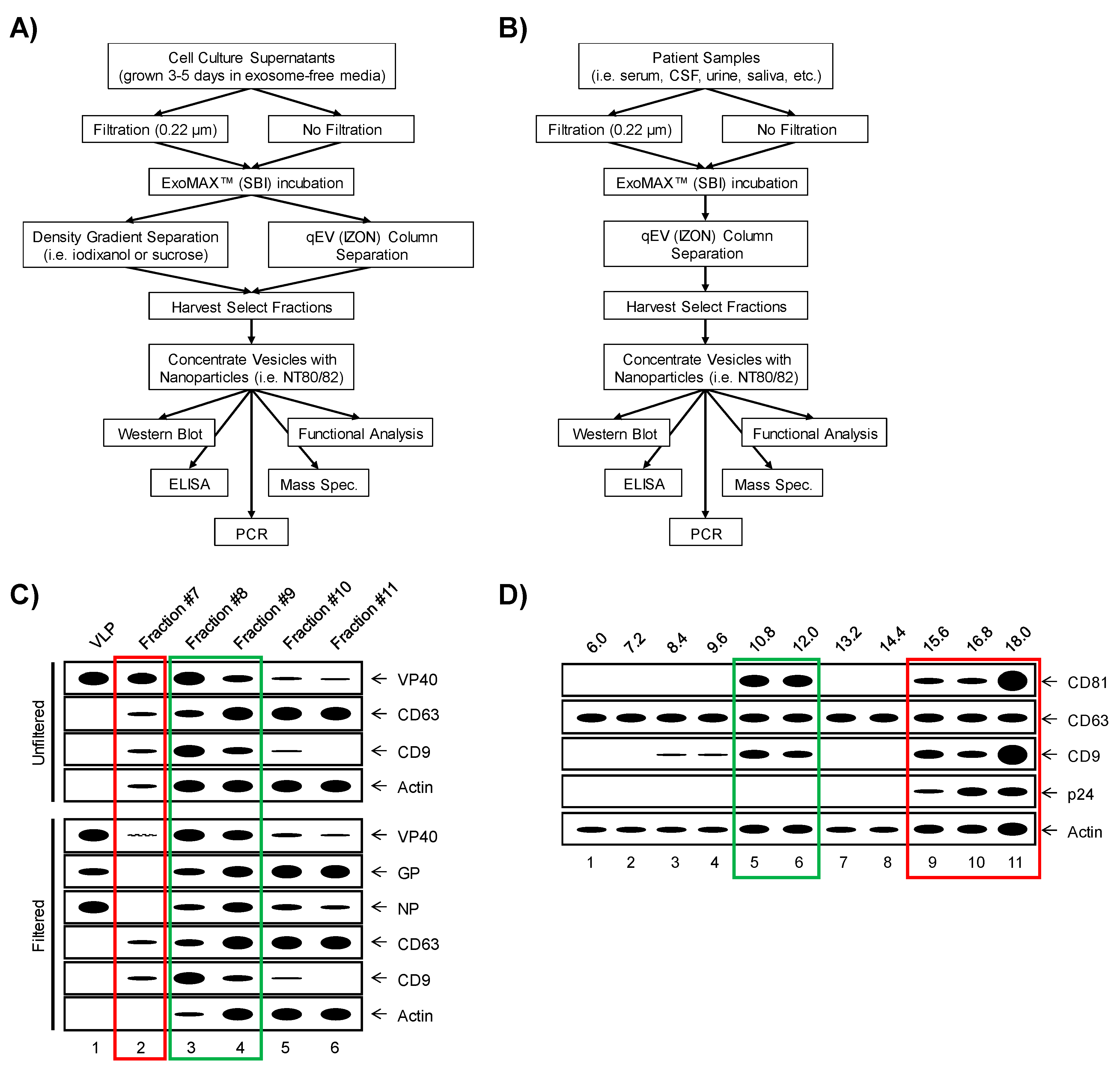
- Greening, D.W.; Xu, R.; Ji, H.; Tauro, B.J.; Simpson, R.J. A protocol for exosome isolation and characterization: Evaluation of ultracentrifugation, density-gradient separation, and immunoaffinity capture methods. Methods Mol. Biol. Clifton NJ 2015, 1295, 179–209. [Google Scholar]
- Konoshenko, M.Y.; Lekchnov, E.A.; Vlassov, A.V.; Laktionov, P.P. Isolation of Extracellular Vesicles: General Methodologies and Latest Trends. BioMed Res. Int. 2018, 2018, 8545347. [Google Scholar] [CrossRef]
- Taylor, D.D.; Shah, S. Methods of isolating extracellular vesicles impact down-stream analyses of their cargoes. Methods 2015, 87, 3–10. [Google Scholar] [CrossRef]
- Momen-Heravi, F.; Balaj, L.; Alian, S.; Mantel, P.-Y.; Halleck, A.E.; Trachtenberg, A.J.; Soria, C.E.; Oquin, S.; Bonebreak, C.M.; Saracoglu, E.; et al. Current methods for the isolation of extracellular vesicles. Biol. Chem. 2013, 394, 1253–1262. [Google Scholar] [CrossRef] [PubMed]
- DeMarino, C.; Pleet, M.L.; Cowen, M.; Barclay, R.A.; Akpamagbo, Y.; Erickson, J.; Ndembe, N.; Charurat, M.; Jumare, J.; Bwala, S.; et al. Antiretroviral Drugs Alter the Content of Extracellular Vesicles from HIV-1-Infected Cells. Sci. Rep. 2018, 8, 7653. [Google Scholar] [CrossRef] [PubMed]
- Timmins, J.; Scianimanico, S.; Schoehn, G.; Weissenhorn, W. Vesicular Release of Ebola Virus Matrix Protein VP40. Virology 2001, 283, 1–6. [Google Scholar] [CrossRef]
- Kallstrom, G.; Warfield, K.L.; Swenson, D.L.; Mort, S.; Panchal, R.G.; Ruthel, G.; Bavari, S.; Aman, M.J. Analysis of Ebola virus and VLP release using an immunocapture assay. J. Virol. Methods 2005, 127, 1–9. [Google Scholar] [CrossRef]
- Bavari, S.; Bosio, C.M.; Wiegand, E.; Ruthel, G.; Will, A.B.; Geisbert, T.W.; Hevey, M.; Schmaljohn, C.; Schmaljohn, A.; Aman, M.J. Lipid Raft Microdomains: A Gateway for Compartmentalized Trafficking of Ebola and Marburg Viruses. J. Exp. Med. 2002, 195, 593–602. [Google Scholar] [CrossRef]
- Beniac, D.R.; Melito, P.L.; deVarennes, S.L.; Hiebert, S.L.; Rabb, M.J.; Lamboo, L.L.; Jones, S.M.; Booth, T.F. The Organisation of Ebola Virus Reveals a Capacity for Extensive, Modular Polyploidy. PLoS ONE 2012, 7, e29608. [Google Scholar] [CrossRef]
- Pleet, M.L.; Mathiesen, A.; DeMarino, C.; Akpamagbo, Y.A.; Barclay, R.A.; Schwab, A.; Iordanskiy, S.; Sampey, G.C.; Lepene, B.; Nekhai, S.; et al. Ebola VP40 in Exosomes Can Cause Immune Cell Dysfunction. Front. Microbiol. 2016, 7, 1765. [Google Scholar] [CrossRef]
- Pleet, M.L.; Erickson, J.; DeMarino, C.; Barclay, R.A.; Cowen, M.; Lepene, B.; Liang, J.; Kuhn, J.H.; Prugar, L.I.; Stonier, S.W.; et al. Ebola Virus VP40 Modulates Cell Cycle and Biogenesis of Extracellular Veiscles. J Infect Dis. 2018, 218, S365–S387. [Google Scholar] [CrossRef]
- Cantin, R.; Diou, J.; Bélanger, D.; Tremblay, A.M.; Gilbert, C. Discrimination between exosomes and HIV-1: Purification of both vesicles from cell-free supernatants. J. Immunol. Methods 2008, 338, 21–30. [Google Scholar] [CrossRef]
- Pleet, M.L.; DeMarino, C.; Lepene, B.; Aman, M.J.; Kashanchi, F. The Role of Exosomal VP40 in Ebola Virus Disease. DNA Cell Biol. 2017, 36, 243–248. [Google Scholar] [CrossRef]
- Timmins, J.; Schoehn, G.; Ricard-Blum, S.; Scianimanico, S.; Vernet, T.; Ruigrok, R.W.H.; Weissenhorn, W. Ebola Virus Matrix Protein VP40 Interaction with Human Cellular Factors Tsg101 and Nedd4. J. Mol. Biol. 2003, 326, 493–502. [Google Scholar] [CrossRef]
- Hartlieb, B.; Weissenhorn, W. Filovirus assembly and budding. Virology 2006, 344, 64–70. [Google Scholar] [CrossRef]
- Jasenosky, L.D.; Neumann, G.; Lukashevich, I.; Kawaoka, Y. Ebola Virus VP40-Induced Particle Formation and Association with the Lipid Bilayer. J. Virol. 2001, 75, 5205–5214. [Google Scholar] [CrossRef]
- Noda, T.; Sagara, H.; Suzuki, E.; Takada, A.; Kida, H.; Kawaoka, Y. Ebola Virus VP40 Drives the Formation of Virus-Like Filamentous Particles Along with GP. J. Virol. 2002, 76, 4855–4865. [Google Scholar] [CrossRef]
- Licata, J.M.; Johnson, R.F.; Han, Z.; Harty, R.N. Contribution of Ebola Virus Glycoprotein, Nucleoprotein, and VP24 to Budding of VP40 Virus-Like Particles. J. Virol. 2004, 78, 7344–7351. [Google Scholar] [CrossRef]
- Noda, T.; Ebihara, H.; Muramoto, Y.; Fujii, K.; Takada, A.; Sagara, H.; Kim, J.H.; Kida, H.; Feldmann, H.; Kawaoka, Y. Assembly and Budding of Ebolavirus. PLoS Pathog. 2006, 2, e99. [Google Scholar] [CrossRef]
- Surjit, M.; Oberoi, R.; Kumar, R.; Lal, S.K. Enhanced α1 Microglobulin Secretion from Hepatitis E Virus ORF3-expressing Human Hepatoma Cells Is Mediated by the Tumor Susceptibility Gene 101. J. Biol. Chem. 2006, 281, 8135–8142. [Google Scholar] [CrossRef]
- Nagashima, S.; Takahashi, M.; Jirintai, S.; Tanaka, T.; Nishizawa, T.; Yasuda, J.; Okamoto, H. Tumour susceptibility gene 101 and the vacuolar protein sorting pathway are required for the release of hepatitis E virions. J. Gen. Virol. 2011, 92, 2838–2848. [Google Scholar] [CrossRef]
- Park, A.; Yun, T.; Vigant, F.; Pernet, O.; Won, S.T.; Dawes, B.E.; Bartkowski, W.; Freiberg, A.N.; Lee, B. Nipah Virus C Protein Recruits Tsg101 to Promote the Efficient Release of Virus in an ESCRT-Dependent Pathway. PLoS Pathog. 2016, 12, e1005659. [Google Scholar] [CrossRef]
- Bissig, C.; Gruenberg, J. ALIX and the multivesicular endosome: ALIX in Wonderland. Trends Cell Biol. 2014, 24, 19–25. [Google Scholar] [CrossRef]
- Zhai, Q.; Fisher, R.D.; Chung, H.-Y.; Myszka, D.G.; Sundquist, W.I.; Hill, C.P. Structural and functional studies of ALIX interactions with YPXnL late domains of HIV-1 and EIAV. Nat. Struct. Mol. Biol. 2008, 15, 43–49. [Google Scholar] [CrossRef]
- Yasuda, J.; Nakao, M.; Kawaoka, Y.; Shida, H. Nedd4 Regulates Egress of Ebola Virus-Like Particles from Host Cells. J. Virol. 2003, 77, 9987–9992. [Google Scholar] [CrossRef]
- Malakhova, O.A.; Zhang, D.-E. ISG15 Inhibits Nedd4 Ubiquitin E3 Activity and Enhances the Innate Antiviral Response. J. Biol. Chem. 2008, 283, 8783–8787. [Google Scholar] [CrossRef]
- Han, Z.; Sagum, C.A.; Bedford, M.T.; Sidhu, S.S.; Sudol, M.; Harty, R.N. ITCH E3 Ubiquitin Ligase Interacts with Ebola Virus VP40 To Regulate Budding. J. Virol. 2016, 90, 9163–9171. [Google Scholar] [CrossRef]
- Han, Z.; Sagum, C.A.; Takizawa, F.; Ruthel, G.; Berry, C.T.; Kong, J.; Sunyer, J.O.; Freedman, B.D.; Bedford, M.T.; Sidhu, S.S.; et al. Ubiquitin Ligase WWP1 Interacts with Ebola Virus VP40 To Regulate Egress. J. Virol. 2017, 91, e00812-17. [Google Scholar] [CrossRef]
- Okumura, A.; Rasmussen, A.L.; Halfmann, P.; Feldmann, F.; Yoshimura, A.; Feldmann, H.; Kawaoka, Y.; Harty, R.N.; Katze, M.G. Suppressor of Cytokine Signaling 3 Is an Inducible Host Factor That Regulates Virus Egress during Ebola Virus Infection. J. Virol. 2015, 89, 10399–10406. [Google Scholar] [CrossRef]
- Nascimento, R.; Costa, H.; Parkhouse, R.M.E. Virus manipulation of cell cycle. Protoplasma 2012, 249, 519–528. [Google Scholar] [CrossRef]
- Fehr, A.R.; Yu, D. Control the Host Cell Cycle: Viral Regulation of the Anaphase-Promoting Complex. J. Virol. 2013, 87, 8818–8825. [Google Scholar] [CrossRef]
- Dove, B.; Brooks, G.; Bicknell, K.; Wurm, T.; Hiscox, J.A. Cell Cycle Perturbations Induced by Infection with the Coronavirus Infectious Bronchitis Virus and Their Effect on Virus Replication. J. Virol. 2006, 80, 4147–4156. [Google Scholar] [CrossRef]
- Chaurushiya, M.S.; Weitzman, M.D. Viral manipulation of DNA repair and cell cycle checkpoints. DNA Repair 2009, 8, 1166–1176. [Google Scholar] [CrossRef]
- Santiago, F.; Clark, E.; Chong, S.; Molina, C.; Mozafari, F.; Mahieux, R.; Fujii, M.; Azimi, N.; Kashanchi, F. Transcriptional Up-Regulation of the Cyclin D2 Gene and Acquisition of New Cyclin-Dependent Kinase Partners in Human T-Cell Leukemia Virus Type 1-Infected Cells. J. Virol. 1999, 73, 9917–9927. [Google Scholar]
- Kehn, K.; Deng, L.; de la Fuente, C.; Strouss, K.; Wu, K.; Maddukuri, A.; Baylor, S.; Rufner, R.; Pumfery, A.; Bottazzi, M.E.; et al. The role of cyclin D2 and p21/waf1 in human T-cell leukemia virus type 1 infected cells. Retrovirology 2004, 1, 6. [Google Scholar] [CrossRef][Green Version]
- Zhou, L.; Kang, D.; Xu, C.; Zhao, W.; Tian, B.; Chen, L. Expression of cyclin D1 and cyclin E significantly associates with human papillomavirus subtypes in Bowenoid papulosis. Acta Histochem. 2013, 115, 339–343. [Google Scholar] [CrossRef]
- Ripple, M.J.; Parker Struckhoff, A.; Trillo-Tinoco, J.; Li, L.; Margolin, D.A.; McGoey, R.; Valle, L.D. Activation of c-Myc and Cyclin D1 by JCV T-Antigen and β-Catenin in Colon Cancer. PLoS ONE 2014, 9, e106257. [Google Scholar] [CrossRef]
- Xiang, Z.; Liang, Z.; Yanfeng, H.; Leitao, K. Persistence of RSV promotes proliferation and epithelial-mesenchymal transition of bronchial epithelial cells through Nodal signaling. J. Med. Microbiol. 2017, 66, 1499–1505. [Google Scholar] [CrossRef]
- Xu, Y.; Shi, Y.; Yuan, Q.; Liu, X.; Yan, B.; Chen, L.; Tao, Y.; Cao, Y. Epstein-Barr Virus encoded LMP1 regulates cyclin D1 promoter activity by nuclear EGFR and STAT3 in CNE1 cells. J. Exp. Clin. Cancer Res. CR 2013, 32, 90. [Google Scholar] [CrossRef]
- Casimiro, M.C.; Velasco-Velázquez, M.; Aguirre-Alvarado, C.; Pestell, R.G. Overview of cyclins D1 function in cancer and the CDK inhibitor landscape: Past and present. Expert Opin. Investig. Drugs 2014, 23, 295–304. [Google Scholar] [CrossRef]
- Pestell, R.G. New Roles of Cyclin D1. Am. J. Pathol. 2013, 183, 3–9. [Google Scholar] [CrossRef]
- Klein, E.A.; Assoian, R.K. Transcriptional regulation of the cyclin D1 gene at a glance. J. Cell Sci. 2008, 121, 3853–3857. [Google Scholar] [CrossRef]
- Björndal, A.S.; Szekely, L.; Elgh, F. Ebola virus infection inversely correlates with the overall expression levels of promyelocytic leukaemia (PML) protein in cultured cells. BMC Microbiol. 2003, 3, 6. [Google Scholar] [CrossRef]
- Nanbo, A.; Watanabe, S.; Halfmann, P.; Kawaoka, Y. The spatio-temporal distribution dynamics of Ebola virus proteins and RNA in infected cells. Sci. Rep. 2013, 3, srep01206. [Google Scholar] [CrossRef]
- Vecchio, K.D.; Frick, C.T.; Gc, J.B.; Oda, S.; Gerstman, B.S.; Saphire, E.O.; Chapagain, P.P.; Stahelin, R.V. A cationic, C-terminal patch and structural rearrangements in Ebola virus matrix VP40 protein control its interactions with phosphatidylserine. J. Biol. Chem. 2018, 293, 3335–3349. [Google Scholar] [CrossRef]
- Kota, K.P.; Benko, J.G.; Mudhasani, R.; Retterer, C.; Tran, J.P.; Bavari, S.; Panchal, R.G. High Content Image Based Analysis Identifies Cell Cycle Inhibitors as Regulators of Ebola Virus Infection. Viruses 2012, 4, 1865–1877. [Google Scholar] [CrossRef]
- Wauquier, N.; Becquart, P.; Padilla, C.; Baize, S.; Leroy, E.M. Human Fatal Zaire Ebola Virus Infection Is Associated with an Aberrant Innate Immunity and with Massive Lymphocyte Apoptosis. PLoS Negl. Trop. Dis. 2010, 4, pii: e837. [Google Scholar] [CrossRef]
- Gupta, M.; Spiropoulou, C.; Rollin, P.E. Ebola virus infection of human PBMCs causes massive death of macrophages, CD4 and CD8 T cell sub-populations in vitro. Virology 2007, 364, 45–54. [Google Scholar] [CrossRef]
- Yaddanapudi, K.; Palacios, G.; Towner, J.S.; Chen, I.; Sariol, C.A.; Nichol, S.T.; Lipkin, W.I. Implication of a retrovirus-like glycoprotein peptide in the immunopathogenesis of Ebola and Marburg viruses. FASEB J. 2006, 20, 2519–2530. [Google Scholar] [CrossRef]
- Martin, B.; Reynard, O.; Volchkov, V.; Decroly, E. Filovirus proteins for antiviral drug discovery: Structure/function of proteins involved in assembly and budding. Antiviral Res. 2018, 150, 183–192. [Google Scholar] [CrossRef]
- Watanabe, S.; Noda, T.; Kawaoka, Y. Functional Mapping of the Nucleoprotein of Ebola Virus. J. Virol. 2006, 80, 3743–3751. [Google Scholar] [CrossRef]
- Huang, Y.; Xu, L.; Sun, Y.; Nabel, G.J. The Assembly of Ebola Virus Nucleocapsid Requires Virion-Associated Proteins 35 and 24 and Posttranslational Modification of Nucleoprotein. Mol. Cell 2002, 10, 307–316. [Google Scholar] [CrossRef]
- Kirchdoerfer, R.N.; Abelson, D.M.; Li, S.; Wood, M.R.; Saphire, E.O. Assembly of the Ebola Virus Nucleoprotein from a Chaperoned VP35 Complex. Cell Rep. 2015, 12, 140–149. [Google Scholar] [CrossRef]
- Bharat, T.A.M.; Noda, T.; Riches, J.D.; Kraehling, V.; Kolesnikova, L.; Becker, S.; Kawaoka, Y.; Briggs, J.A.G. Structural dissection of Ebola virus and its assembly determinants using cryo-electron tomography. Proc. Natl. Acad. Sci. USA 2012, 109, 4275–4280. [Google Scholar] [CrossRef]
- Martin, B.; Canard, B.; Decroly, E. Filovirus proteins for antiviral drug discovery: Structure/function bases of the replication cycle. Antiviral Res. 2017, 141, 48–61. [Google Scholar] [CrossRef]
- Dong, S.; Yang, P.; Li, G.; Liu, B.; Wang, W.; Liu, X.; Xia, B.; Yang, C.; Lou, Z.; Guo, Y.; et al. Insight into the Ebola virus nucleocapsid assembly mechanism: Crystal structure of Ebola virus nucleoprotein core domain at 1.8 Å resolution. Protein Cell 2015, 6, 351–362. [Google Scholar] [CrossRef]
- Dziubańska, P.J.; Derewenda, U.; Ellena, J.F.; Engel, D.A.; Derewenda, Z.S. The structure of the C-terminal domain of the Zaire ebolavirus nucleoprotein. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 2420–2429. [Google Scholar] [CrossRef]
- Noda, T.; Hagiwara, K.; Sagara, H.; Kawaoka, Y. Characterization of the Ebola virus nucleoprotein–RNA complex. J. Gen. Virol. 2010, 91, 1478–1483. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, W.; Ji, W.; Deng, M.; Sun, Y.; Zhou, H.; Yang, C.; Deng, F.; Wang, H.; Hu, Z.; et al. Crimean–Congo hemorrhagic fever virus nucleoprotein reveals endonuclease activity in bunyaviruses. Proc. Natl. Acad. Sci. USA 2012, 109, 5046–5051. [Google Scholar] [CrossRef]
- Hastie, K.M.; Kimberlin, C.R.; Zandonatti, M.A.; MacRae, I.J.; Saphire, E.O. Structure of the Lassa virus nucleoprotein reveals a dsRNA-specific 3′ to 5′ exonuclease activity essential for immune suppression. Proc. Natl. Acad. Sci. USA 2011, 108, 2396–2401. [Google Scholar] [CrossRef]
- Qi, X.; Lan, S.; Wang, W.; Schelde, L.M.; Dong, H.; Wallat, G.D.; Ly, H.; Liang, Y.; Dong, C. Cap binding and immune evasion revealed by Lassa nucleoprotein structure. Nature 2010, 468, 779–783. [Google Scholar] [CrossRef]
- Hatakeyama, D.; Ohmi, N.; Saitoh, A.; Makiyama, K.; Morioka, M.; Okazaki, H.; Kuzuhara, T. Acetylation of lysine residues in the recombinant nucleoprotein and VP40 matrix protein of Zaire Ebolavirus by eukaryotic histone acetyltransferases. Biochem. Biophys. Res. Commun. 2018, 504, 635–640. [Google Scholar] [CrossRef]
- Fujisawa, T.; Filippakopoulos, P. Functions of bromodomain-containing proteins and their roles in homeostasis and cancer. Nat. Rev. Mol. Cell Biol. 2017, 18, 246–262. [Google Scholar] [CrossRef]
- Ren, W.; Wang, C.; Wang, Q.; Zhao, D.; Zhao, K.; Sun, D.; Liu, X.; Han, C.; Hou, J.; Li, X.; et al. Bromodomain protein Brd3 promotes Ifnb1 transcription via enhancing IRF3/p300 complex formation and recruitment to Ifnb1 promoter in macrophages. Sci. Rep. 2017, 7, 39986. [Google Scholar] [CrossRef]
- García-Dorival, I.; Wu, W.; Armstrong, S.D.; Barr, J.N.; Carroll, M.W.; Hewson, R.; Hiscox, J.A. Elucidation of the Cellular Interactome of Ebola Virus Nucleoprotein and Identification of Therapeutic Targets. J. Proteome Res. 2016, 15, 4290–4303. [Google Scholar] [CrossRef]
- Proserpio, V.; Fittipaldi, R.; Ryall, J.G.; Sartorelli, V.; Caretti, G. The methyltransferase SMYD3 mediates the recruitment of transcriptional cofactors at the myostatin and c-Met genes and regulates skeletal muscle atrophy. Genes Dev. 2013, 27, 1299–1312. [Google Scholar] [CrossRef]
- Ebihara, H.; Takada, A.; Kobasa, D.; Jones, S.; Neumann, G.; Theriault, S.; Bray, M.; Feldmann, H.; Kawaoka, Y. Molecular Determinants of Ebola Virus Virulence in Mice. PLoS Pathog. 2006, 2, e73. [Google Scholar] [CrossRef]
- Hoenen, T.; Shabman, R.S.; Groseth, A.; Herwig, A.; Weber, M.; Schudt, G.; Dolnik, O.; Basler, C.F.; Becker, S.; Feldmann, H. Inclusion Bodies Are a Site of Ebolavirus Replication. J. Virol. 2012, 86, 11779–11788. [Google Scholar] [CrossRef]
- Martin, B.; Hoenen, T.; Canard, B.; Decroly, E. Filovirus proteins for antiviral drug discovery: A structure/function analysis of surface glycoproteins and virus entry. Antiviral Res. 2016, 135, 1–14. [Google Scholar] [CrossRef]
- Miller, E.H.; Chandran, K. Filovirus entry into cells – new insights. Curr. Opin. Virol. 2012, 2, 206–214. [Google Scholar] [CrossRef]
- Simmons, G. Filovirus Entry. In Viral Entry into Host Cells; Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2013; pp. 83–94. ISBN 978-1-4614-7650-4. [Google Scholar]
- Saeed, M.F.; Kolokoltsov, A.A.; Albrecht, T.; Davey, R.A. Cellular Entry of Ebola Virus Involves Uptake by a Macropinocytosis-Like Mechanism and Subsequent Trafficking through Early and Late Endosomes. PLoS Pathog. 2010, 6, e1001110. [Google Scholar] [CrossRef]
- Nanbo, A.; Imai, M.; Watanabe, S.; Noda, T.; Takahashi, K.; Neumann, G.; Halfmann, P.; Kawaoka, Y. Ebolavirus Is Internalized into Host Cells via Macropinocytosis in a Viral Glycoprotein-Dependent Manner. PLoS Pathog. 2010, 6, e1001121. [Google Scholar] [CrossRef]
- White, J.M.; Schornberg, K.L. A new player in the puzzle of filovirus entry. Nat. Rev. Microbiol. 2012, 10, 317–322. [Google Scholar] [CrossRef]
- Markosyan, R.M.; Miao, C.; Zheng, Y.-M.; Melikyan, G.B.; Liu, S.-L.; Cohen, F.S. Induction of Cell-Cell Fusion by Ebola Virus Glycoprotein: Low pH Is Not a Trigger. PLoS Pathog. 2016, 12, e1005373. [Google Scholar] [CrossRef]
- Volchkov, V.E.; Feldmann, H.; Volchkova, V.A.; Klenk, H.-D. Processing of the Ebola virus glycoprotein by the proprotein convertase furin. Proc. Natl. Acad. Sci. USA 1998, 95, 5762–5767. [Google Scholar] [CrossRef]
- Jeffers, S.A.; Sanders, D.A.; Sanchez, A. Covalent Modifications of the Ebola Virus Glycoprotein. J. Virol. 2002, 76, 12463–12472. [Google Scholar] [CrossRef]
- Gruenberg, J.; Stenmark, H. The biogenesis of multivesicular endosomes. Nat. Rev. Mol. Cell Biol. 2004, 5, 317–323. [Google Scholar] [CrossRef]
- Gadila, S.K.G.; Kim, K. Cargo trafficking from the trans-Golgi network towards the endosome. Biol. Cell 2016, 108, 205–218. [Google Scholar] [CrossRef]
- Pallesen, J.; Murin, C.D.; de Val, N.; Cottrell, C.A.; Hastie, K.M.; Turner, H.L.; Fusco, M.L.; Flyak, A.I.; Zeitlin, L.; Crowe, J.E., Jr.; et al. Structures of Ebola virus GP and sGP in complex with therapeutic antibodies. Nat. Microbiol. 2016, 1, 16128. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Hope, T.J. Full-length Ebola glycoprotein accumulates in the endoplasmic reticulum. Virol. J. 2011, 8, 11. [Google Scholar] [CrossRef]
- Escudero-Pérez, B.; Volchkova, V.A.; Dolnik, O.; Lawrence, P.; Volchkov, V.E. Shed GP of Ebola Virus Triggers Immune Activation and Increased Vascular Permeability. PLoS Pathog. 2014, 10, e1004509. [Google Scholar] [CrossRef]
- Lai, C.-Y.; Strange, D.P.; Wong, T.A.S.; Lehrer, A.T.; Verma, S. Ebola Virus Glycoprotein Induces an Innate Immune Response In vivo via TLR4. Front. Microbiol. 2017, 8, 1571. [Google Scholar] [CrossRef]
- Okumura, A.; Pitha, P.M.; Yoshimura, A.; Harty, R.N. Interaction between Ebola Virus Glycoprotein and Host Toll-Like Receptor 4 Leads to Induction of Proinflammatory Cytokines and SOCS1. J. Virol. 2010, 84, 27–33. [Google Scholar] [CrossRef]
- Iampietro, M.; Santos, R.I.; Lubaki, N.M.; Bukreyev, A. Ebola Virus Shed Glycoprotein Triggers Differentiation, Infection, and Death of Monocytes Through Toll-Like Receptor 4 Activation. J. Infect. Dis. 2018, 218, S327–S334. [Google Scholar] [CrossRef]
- Martinez, O.; Johnson, J.C.; Honko, A.; Yen, B.; Shabman, R.S.; Hensley, L.E.; Olinger, G.G.; Basler, C.F. Ebola Virus Exploits a Monocyte Differentiation Program To Promote Its Entry. J. Virol. 2013, 87, 3801–3814. [Google Scholar] [CrossRef]
- Edri, A.; Shemesh, A.; Iraqi, M.; Matalon, O.; Brusilovsky, M.; Hadad, U.; Radinsky, O.; Gershoni-Yahalom, O.; Dye, J.M.; Mandelboim, O.; et al. The Ebola-Glycoprotein Modulates the Function of Natural Killer Cells. Front. Immunol. 2018, 9, 1428. [Google Scholar] [CrossRef]
- Iampietro, M.; Younan, P.; Nishida, A.; Dutta, M.; Lubaki, N.M.; Santos, R.I.; Koup, R.A.; Katze, M.G.; Bukreyev, A. Ebola virus glycoprotein directly triggers T lymphocyte death despite of the lack of infection. PLoS Pathog. 2017, 13, e1006397. [Google Scholar] [CrossRef]
- Maruyama, T.; Parren, P.W.H.I.; Sanchez, A.; Rensink, I.; Rodriguez, L.L.; Khan, A.S.; Peters, C.J.; Burton, D.R. Recombinant Human Monoclonal Antibodies to Ebola Virus. J. Infect. Dis. 1999, 179, S235–S239. [Google Scholar] [CrossRef]
- Mohan, G.S.; Li, W.; Ye, L.; Compans, R.W.; Yang, C. Antigenic Subversion: A Novel Mechanism of Host Immune Evasion by Ebola Virus. PLoS Pathog. 2012, 8, e1003065. [Google Scholar] [CrossRef]
- Bixler, S.L.; Goff, A.J. The Role of Cytokines and Chemokines in Filovirus Infection. Viruses 2015, 7, 5489–5507. [Google Scholar] [CrossRef]
- Villinger, F.; Rollin, P.E.; Brar, S.S.; Chikkala, N.F.; Winter, J.; Sundstrom, J.B.; Zaki, S.R.; Swanepoel, R.; Ansari, A.A.; Peters, C.J. Markedly Elevated Levels of Interferon (IFN)-γ, IFN-α, Interleukin (IL)-2, IL-10, and Tumor Necrosis Factor-α Associated with Fatal Ebola Virus Infection. J. Infect. Dis. 1999, 179, S188–S191. [Google Scholar] [CrossRef]
- Baize, S.; Leroy, E.M.; Georges, A.J.; Georges-Courbot, M.C.; Capron, M.; Bedjabaga, I.; Lansoud-Soukate, J.; Mavoungou, E. Inflammatory responses in Ebola virus-infected patients. Clin. Exp. Immunol. 2002, 128, 163–168. [Google Scholar] [CrossRef]
- Trinchieri, G. Interleukin-12 and interferon-gamma. Do they always go together? Am. J. Pathol. 1995, 147, 1534–1538. [Google Scholar]
- Szabo, S.J.; Dighe, A.S.; Gubler, U.; Murphy, K.M. Regulation of the Interleukin (IL)-12R β2 Subunit Expression in Developing T Helper 1 (Th1) and Th2 Cells. J. Exp. Med. 1997, 185, 817–824. [Google Scholar] [CrossRef]
- Otani, T.; Nakamura, S.; Toki, M.; Motoda, R.; Kurimoto, M.; Orita, K. Identification of IFN-gamma-producing cells in IL-12/IL-18-treated mice. Cell. Immunol. 1999, 198, 111–119. [Google Scholar] [CrossRef]
- Wei, Y.P.; Kita, M.; Shinmura, K.; Yan, X.Q.; Fukuyama, R.; Fushiki, S.; Imanishi, J. Expression of IFN-gamma in cerebrovascular endothelial cells from aged mice. J. Interferon Cytokine Res. Off. J. Int. Soc. Interferon Cytokine Res. 2000, 20, 403–409. [Google Scholar] [CrossRef]
- Suo, Z.; Tan, J.; Placzek, A.; Crawford, F.; Fang, C.; Mullan, M. Alzheimer’s beta-amyloid peptides induce inflammatory cascade in human vascular cells: The roles of cytokines and CD40. Brain Res. 1998, 807, 110–117. [Google Scholar] [CrossRef]
- Hakonarson, H.; Maskeri, N.; Carter, C.; Grunstein, M.M. Regulation of TH1- and TH2-type cytokine expression and action in atopic asthmatic sensitized airway smooth muscle. J. Clin. Invest. 1999, 103, 1077–1087. [Google Scholar] [CrossRef]
- Timoshanko, J.R.; Holdsworth, S.R.; Kitching, A.R.; Tipping, P.G. IFN-γ Production by Intrinsic Renal Cells and Bone Marrow-Derived Cells Is Required for Full Expression of Crescentic Glomerulonephritis in Mice. J. Immunol. 2002, 168, 4135–4141. [Google Scholar] [CrossRef]
- Baize, S.; Leroy, E.M.; Georges-Courbot, M.-C.; Capron, M.; Lansoud-Soukate, J.; Debré, P.; Fisher-Hoch, S.P.; McCormick, J.B.; Georges, A.J. Defective humoral responses and extensive intravascular apoptosis are associated with fatal outcome in Ebola virus-infected patients. Nat. Med. 1999, 5, 423–426. [Google Scholar] [CrossRef]
- Steel, J.C.; Waldmann, T.A.; Morris, J.C. Interleukin-15 biology and its therapeutic implications in cancer. Trends Pharmacol. Sci. 2012, 33, 35–41. [Google Scholar] [CrossRef]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte Chemoattractant Protein-1 (MCP-1): An Overview. J. Interferon Cytokine Res. 2009, 29, 313–326. [Google Scholar] [CrossRef]
- Kindrachuk, J.; Wahl-Jensen, V.; Safronetz, D.; Trost, B.; Hoenen, T.; Arsenault, R.; Feldmann, F.; Traynor, D.; Postnikova, E.; Kusalik, A.; et al. Ebola Virus Modulates Transforming Growth Factor β Signaling and Cellular Markers of Mesenchyme-Like Transition in Hepatocytes. J. Virol. 2014, 88, 9877–9892. [Google Scholar] [CrossRef]
- Zhang, H.-G.; Liu, C.; Su, K.; Yu, S.; Zhang, L.; Zhang, S.; Wang, J.; Cao, X.; Grizzle, W.; Kimberly, R.P. A Membrane Form of TNF-α Presented by Exosomes Delays T Cell Activation-Induced Cell Death. J. Immunol. 2006, 176, 7385–7393. [Google Scholar] [CrossRef]
- Chen, T.; Guo, J.; Yang, M.; Zhu, X.; Cao, X. Chemokine-Containing Exosomes Are Released from Heat-Stressed Tumor Cells via Lipid Raft-Dependent Pathway and Act as Efficient Tumor Vaccine. J. Immunol. 2011, 186, 2219–2228. [Google Scholar] [CrossRef]
- Zhang, B.; Shen, L.; Shi, H.; Pan, Z.; Wu, L.; Yan, Y.; Zhang, X.; Mao, F.; Qian, H.; Xu, W. Exosomes from Human Umbilical Cord Mesenchymal Stem Cells: Identification, Purification, and Biological Characteristics. Stem Cells Int. 2016, 2016, 1929536. [Google Scholar] [CrossRef]
- Nickel, W.; Rabouille, C. Mechanisms of regulated unconventional protein secretion. Nat. Rev. Mol. Cell Biol. 2009, 10, 148–155. [Google Scholar] [CrossRef]
- Konadu, K.A.; Chu, J.; Huang, M.B.; Amancha, P.K.; Armstrong, W.; Powell, M.D.; Villinger, F.; Bond, V.C. Association of Cytokines With Exosomes in the Plasma of HIV-1–Seropositive Individuals. J. Infect. Dis. 2015, 211, 1712–1716. [Google Scholar] [CrossRef]
- Fitzgerald, W.; Freeman, M.L.; Lederman, M.M.; Vasilieva, E.; Romero, R.; Margolis, L. A System of Cytokines Encapsulated in ExtraCellular Vesicles. Sci. Rep. 2018, 8, 8973. [Google Scholar] [CrossRef]
- Smith, J.R.; Todd, S.; Ashander, L.M.; Charitou, T.; Ma, Y.; Yeh, S.; Crozier, I.; Michael, M.Z.; Appukuttan, B.; Williams, K.A.; et al. Retinal Pigment Epithelial Cells are a Potential Reservoir for Ebola Virus in the Human Eye. Transl. Vis. Sci. Technol. 2017, 6, 12. [Google Scholar] [CrossRef]
- Zeng, X.; Blancett, C.D.; Koistinen, K.A.; Schellhase, C.W.; Bearss, J.J.; Radoshitzky, S.R.; Honnold, S.P.; Chance, T.B.; Warren, T.K.; Froude, J.W.; et al. Identification and pathological characterization of persistent asymptomatic Ebola virus infection in rhesus monkeys. Nat. Microbiol. 2017, 2, 17113. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pleet, M.L.; DeMarino, C.; Stonier, S.W.; Dye, J.M.; Jacobson, S.; Aman, M.J.; Kashanchi, F. Extracellular Vesicles and Ebola Virus: A New Mechanism of Immune Evasion. Viruses 2019, 11, 410. https://doi.org/10.3390/v11050410
Pleet ML, DeMarino C, Stonier SW, Dye JM, Jacobson S, Aman MJ, Kashanchi F. Extracellular Vesicles and Ebola Virus: A New Mechanism of Immune Evasion. Viruses. 2019; 11(5):410. https://doi.org/10.3390/v11050410
Chicago/Turabian StylePleet, Michelle L., Catherine DeMarino, Spencer W. Stonier, John M. Dye, Steven Jacobson, M. Javad Aman, and Fatah Kashanchi. 2019. "Extracellular Vesicles and Ebola Virus: A New Mechanism of Immune Evasion" Viruses 11, no. 5: 410. https://doi.org/10.3390/v11050410
APA StylePleet, M. L., DeMarino, C., Stonier, S. W., Dye, J. M., Jacobson, S., Aman, M. J., & Kashanchi, F. (2019). Extracellular Vesicles and Ebola Virus: A New Mechanism of Immune Evasion. Viruses, 11(5), 410. https://doi.org/10.3390/v11050410