Inflammatory and Humoral Immune Response during Ebola Virus Infection in Survivor and Fatal Cases Occurred in Sierra Leone during the 2014–2016 Outbreak in West Africa

 ,
,  ,
,  , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Group
2.2. Cytokines Gene Expression
2.3. Humoral Response Analysis
2.4. Statistical Analysis
3. Results
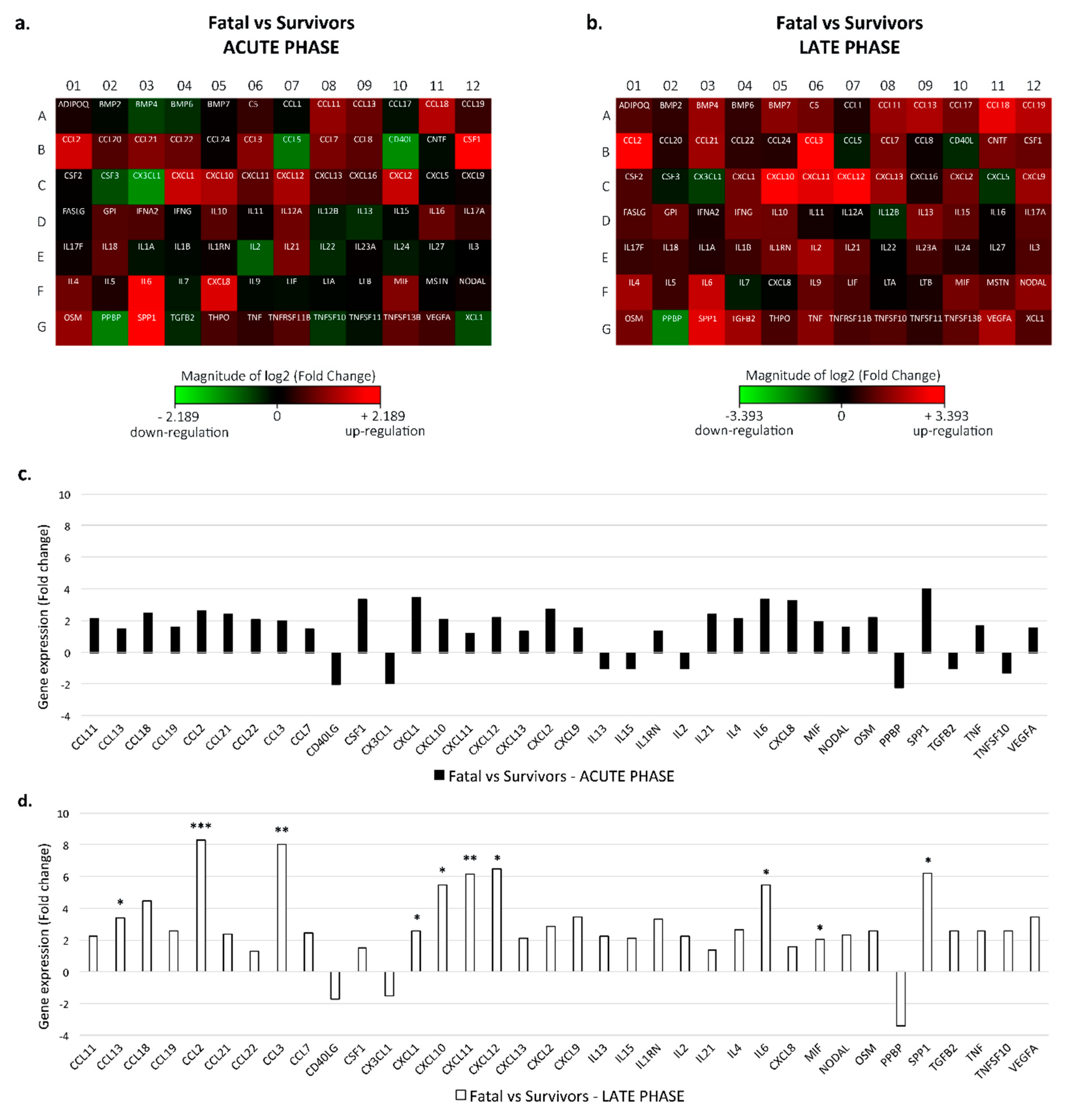
3.1. Cytokine Expression
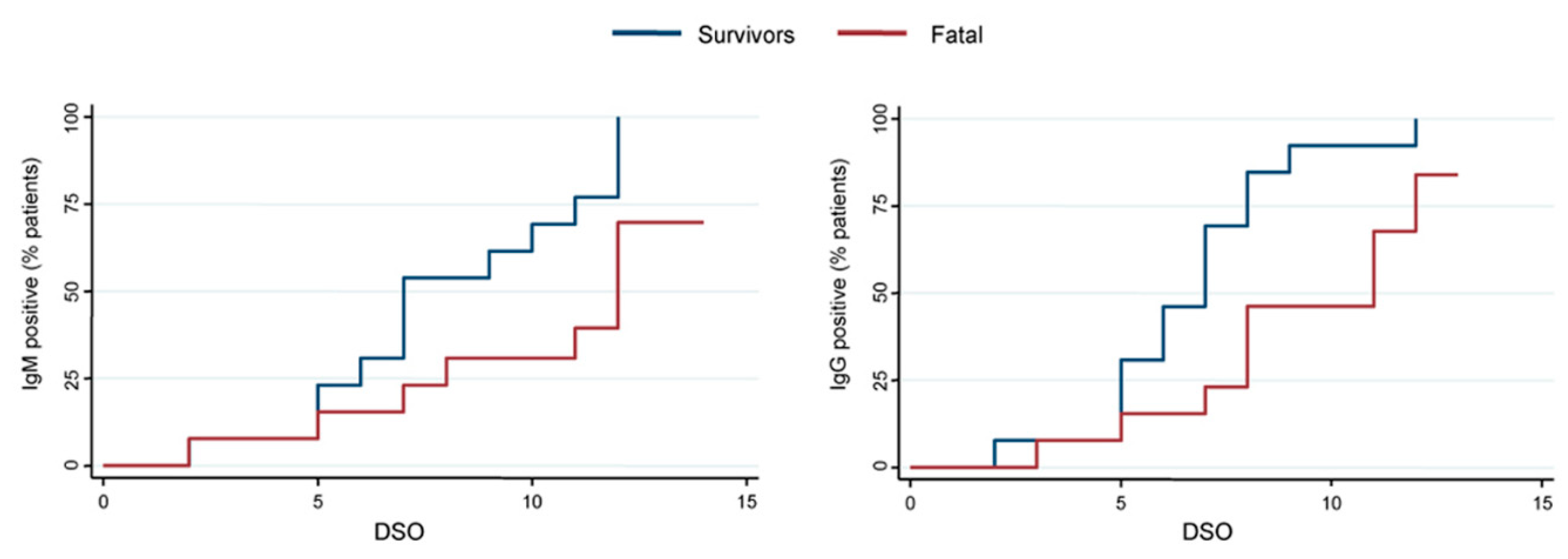
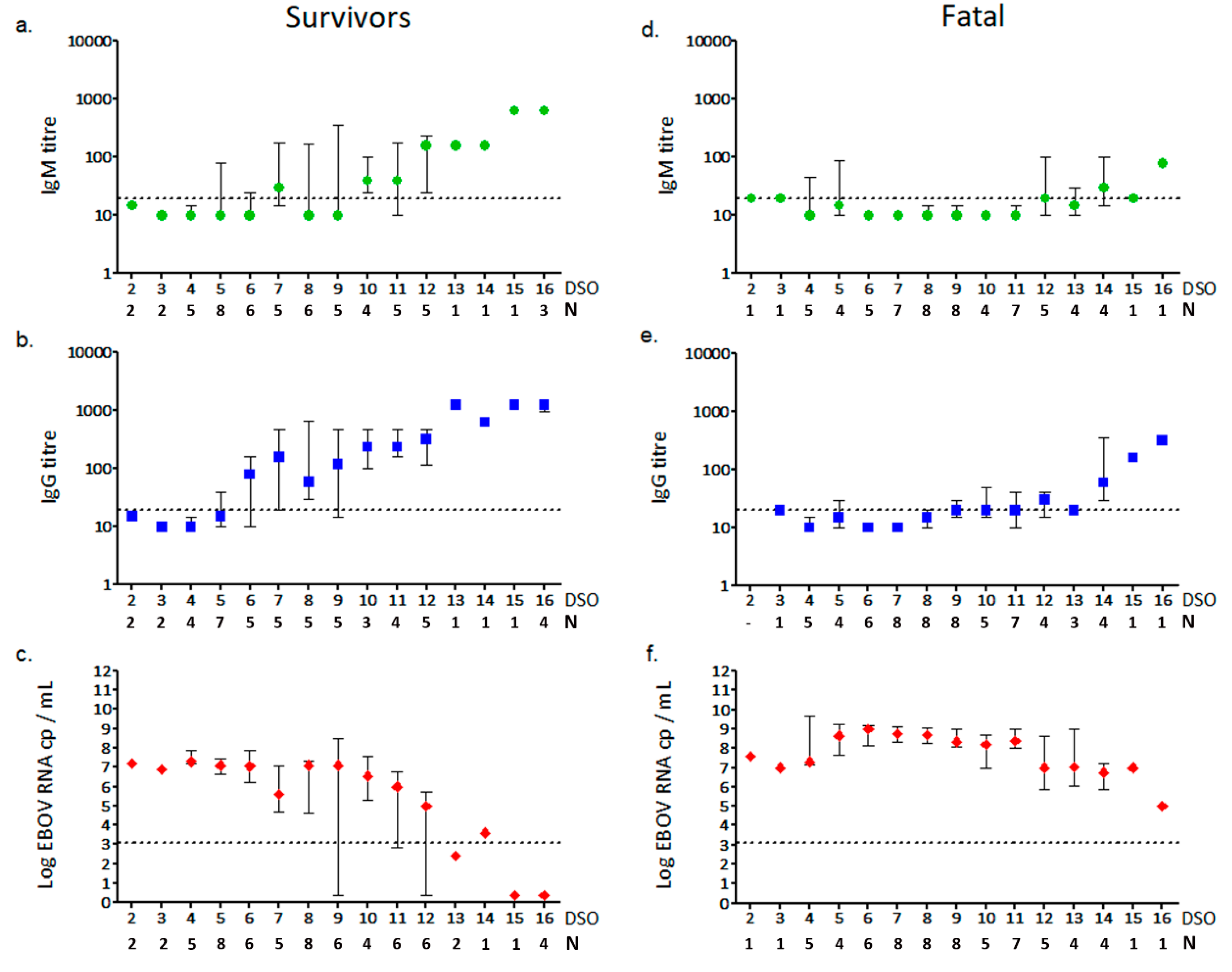
3.2. Anti-EBOV IgM and IgG Response
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Feldmann, H.; Sanchez, A.; Thomas, W.G. Filoviridae: Marburg and Ebola Viruses. In Fields Virology, 6th ed.; Wolters Kluwer N.V.: South Holland, The Netherlands, 2013; pp. 923–956. [Google Scholar]
- Goldstein, T.; Anthony, S.J.; Gbakima, A.; Bird, B.H.; Bangura, J.; Tremeau-Bravard, A.; Belaganahalli, M.N.; Wells, H.L.; Dhanota, J.K.; Liang, E.; et al. The discovery of Bombali virus adds further support for bats as hosts of ebolaviruses. Nat. Microbiol. 2018, 3, 1084–1089. [Google Scholar] [CrossRef]
- WHO. Ebola Situation Reports. Available online: https://www.who.int/ebola/situation-reports/drc-2018/en/ (accessed on 5 April 2019).
- CDC. Ebola History. Available online: https://www.cdc.gov/vhf/ebola/history/2014-2016-outbreak/index.html (accessed on 26 September 2018).
- Falasca, L.; Agrati, C.; Petrosillo, N.; Di Caro, A.; Capobianchi, M.R.; Ippolito, G.; Piacentini, M. Molecular mechanisms of Ebola virus pathogenesis: Focus on cell death. Cell Death Differ. 2015, 22, 1250–1259. [Google Scholar] [CrossRef] [PubMed]
- Cimini, E.; Viola, D.; Cabeza-Cabrerizo, M.; Romanelli, A.; Tumino, N.; Sacchi, A.; Bordoni, V.; Casetti, R.; Turchi, F.; Martini, F.; et al. Different features of Vδ2 T and NK cells in fatal and non-fatal human Ebola infections. PLoS Negl. Trop. Dis. 2017, 11, 1–11. [Google Scholar] [CrossRef]
- Menicucci, A.R.; Versteeg, K.; Woolsey, C.; Mire, C.E.; Geisbert, J.B.; Cross, R.W.; Agans, K.N.; Jankeel, A.; Geisbert, T.W.; Messaoudi, I. Transcriptome Analysis of Circulating Immune Cell Subsets Highlight the Role of Monocytes in Zaire Ebola Virus Makona Pathogenesis. Front. Immunol. 2017, 8, 1372. [Google Scholar] [CrossRef]
- Kumar, A. Ebola Virus Altered Innate and Adaptive Immune Response Signalling Pathways: Implications for Novel Therapeutic Approaches. Infect. Disord. Drug Targets 2016, 16, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Bixler, S.L.; Goff, A.J. The role of cytokines and chemokines in filovirus infection. Viruses 2015, 7, 5489–5507. [Google Scholar] [CrossRef] [PubMed]
- Eisfeld, A.J.; Halfmann, P.J.; Wendler, J.P.; Kyle, J.E.; Burnum-Johnson, K.E.; Peralta, Z.; Maemura, T.; Walters, K.B.; Watanabe, T.; Fukuyama, S.; et al. Multi-platform ’Omics Analysis of Human Ebola Virus Disease Pathogenesis. Cell Host Microbe. 2017, 22, 817–829.e8. [Google Scholar] [CrossRef] [PubMed]
- McElroy, A.K.; Akondy, R.S.; Davis, C.W.; Ellebedy, A.H.; Mehta, A.K.; Kraft, C.S.; Lyon, G.M.; Ribner, B.S.; Varkey, J.; Sidney, J.; et al. Human Ebola virus infection results in substantial immune activation. Proc. Natl. Acad. Sci. USA 2015, 112, 4719–4724. [Google Scholar] [CrossRef]
- Kerber, R.; Krumkamp, R.; Korva, M.; Rieger, T.; Wurr, S.; Duraffour, S.; Oestereich, L.; Gabriel, M.; Sissoko, D.; Anglaret, X.; et al. Kinetics of Soluble Mediators of the Host Response in Ebola Virus Disease. J. Infect. Dis. 2018, 218, S496–S503. [Google Scholar] [CrossRef] [PubMed]
- Wauquier, N.; Becquart, P.; Padilla, C.; Baize, S.; Leroy, E.M. Human fatal zaire ebola virus infection is associated with an aberrant innate immunity and with massive lymphocyte apoptosis. PLoS Negl. Trop. Dis. 2010, 4, e837. [Google Scholar] [CrossRef]
- Ruibal, P.; Oestereich, L.; Lüdtke, A.; Becker-Ziaja, B.; Wozniak, D.M.; Kerber, R.; Korva, M.; Cabeza-Cabrerizo, M.; Bore, J.A.; Koundouno, F.R.; et al. Unique human immune signature of Ebola virus disease in Guinea. Nature 2016, 533, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Agrati, C.; Castilletti, C.; Casetti, R.; Sacchi, A.; Falasca, L.; Turchi, F.; Tumino, N.; Bordoni, V.; Cimini, E.; Viola, D.; et al. Longitudinal characterization of dysfunctional T cell-activation during human acute Ebola infection. Cell Death Dis. 2016, 7, e2164. [Google Scholar] [CrossRef]
- Baize, S.; Leroy, E.M.; Georges-Courbot, M.-C.; Capron, M.; Lansoud-Soukate, J.; Debré, P.; Fisher-Hoch, S.P.; McCormick, J.B.; Georges, A.J. Defective humoral responses and extensive intravascular apoptosis are associated with fatal outcome in Ebola virus-infected patients. Nat. Med. 1999, 5, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Rowe, A.K.; Bertolli, J.; Khan, A.S.; Mukunu, R.; Muyembe-Tamfum, J.J.; Bressler, D.; Williams, A.J.; Peters, C.J.; Rodriguez, L.; Feldmann, H.; et al. Clinical, Virologic, and Immunologic Follow-Up of Convalescent Ebola Hemorrhagic Fever Patients and Their Household Contacts, Kikwit, Democratic Republic of the Congo. J. Infect. Dis. 1999, 179, S28–S35. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.-Y.; Strange, D.P.; Wong, T.A.S.; Lehrer, A.T.; Verma, S. Ebola Virus Glycoprotein Induces an Innate Immune Response In vivo via TLR4. Front. Microbiol. 2017, 8, 1571. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Jiang, J.F.; Deng, Y.Q.; Jiang, B.G.; Fan, H.; Han, J.F.; Hu, Y.; Zhuang, D.M.; Kargbo, D.; An, X.P.; et al. Features of ebola virus disease at the late outbreak stage in Sierra Leone: Clinical, virological, immunological, and evolutionary analyses. J. Infect. Dis. 2017, 215, 1107–1110. [Google Scholar] [CrossRef] [PubMed]
- Radinsky, O.; Edri, A.; Brusilovsky, M.; Fedida-Metula, S.; Sobarzo, A.; Gershoni-Yahalom, O.; Lutwama, J.; Dye, J.; Lobel, L.; Porgador, A. Sudan ebolavirus long recovered survivors produce GP-specific Abs that are of the IgG1 subclass and preferentially bind FcγRI. Sci. Rep. 2017, 7, 6054. [Google Scholar] [CrossRef]
- Colavita, F.; Venditti, C.; Vulcano, A.; Castilletti, C.; Zaccaro, P.; Mazzarelli, A.; Quartu, S.; Coen, S.; Cannas, A.; Chiappini, R.; et al. INMI/Emergency NGO Italian laboratory established in Sierra Leone during Ebola virus disease outbreak in West Africa. Clin. Microbiol. Infect. Dis. 2016, 1, 1–9. [Google Scholar] [CrossRef][Green Version]
- Vernet, M.-A.; Reynard, S.; Fizet, A.; Schaeffer, J.; Pannetier, D.; Guedj, J.; Rives, M.; Georges, N.; Garcia-Bonnet, N.; Sylla, A.I.; et al. Clinical, virological, and biological parameters associated with outcomes of Ebola virus infection in Macenta, Guinea. JCI Insight 2017, 2, e88864. [Google Scholar] [CrossRef]
- Lanini, S.; Portella, G.; Vairo, F.; Kobinger, G.P.; Pesenti, A.; Langer, M.; Kabia, S.; Brogiato, G.; Amone, J.; Castilletti, C.; et al. Blood kinetics of Ebola virus in survivors and nonsurvivors. J. Clin. Investig. 2015, 125, 4692–4698. [Google Scholar] [CrossRef]
- Murdoch, C.; Finn, A. Chemokine receptors and their role in inflammation and infectious diseases. Blood 2000, 95, 3032–3043. [Google Scholar]
- Davenport, R.D. An introduction to chemokines and their roles in transfusion medicine. Vox Sang. 2009, 96, 183–198. [Google Scholar] [CrossRef]
- Yeaman, M.R. Platelets: At the nexus of antimicrobial defence. Nat. Rev. Microbiol. 2014, 12, 426–437. [Google Scholar] [CrossRef]
- Kash, J.C.; Walters, K.-A.; Kindrachuk, J.; Baxter, D.; Scherler, K.; Janosko, K.B.; Adams, R.D.; Herbert, A.S.; James, R.M.; Stonier, S.W.; et al. Longitudinal peripheral blood transcriptional analysis of a patient with severe Ebola virus disease. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Misasi, J.; Sullivan, N.J. Camouflage and Misdirection: The Full-On Assault of Ebola Virus Disease. Cell 2014, 159, 477–486. [Google Scholar] [CrossRef]
- Ksiazek, T.G.; Rollin, P.E.; Williams, A.J.; Bressler, D.S.; Martin, M.L.; Swanepoel, R.; Burt, F.J.; Leman, P.A.; Khan, A.S.; Rowe, A.K.; et al. Clinical Virology of Ebola Hemorrhagic Fever (EHF): Virus, Virus Antigen, and IgG and IgM Antibody Findings among EHF Patients in Kikwit, Democratic Republic of the Congo, 1995. J. Infect. Dis. 1999, 179, S177–S187. [Google Scholar] [CrossRef]
- Baize, S.; Leroy, E.M.; Georges, A.J.; Capron, M.; Bedjabaga, I.; Lansoud-Soukate, J.; Mavoungou, E. Inflammatory responses in Ebola virus-infected patients. Clin. Exp. Immunol. 2002, 128, 163–168. [Google Scholar] [CrossRef]
- Liu, X.; Speranza, E.; Muñoz-Fontela, C.; Haldenby, S.; Rickett, N.Y.; Garcia-Dorival, I.; Fang, Y.; Hall, Y.; Zekeng, E.-G.; Lüdtke, A.; et al. Transcriptomic signatures differentiate survival from fatal outcomes in humans infected with Ebola virus. Genome Biol. 2017, 18, 4. [Google Scholar] [CrossRef]
- Balzano, F.; Deiana, M.; Dei Giudici, S.; Oggiano, A.; Baralla, A.; Pasella, S.; Mannu, A.; Pescatori, M.; Porcu, B.; Fanciulli, G.; et al. miRNA Stability in Frozen Plasma Samples. Molecules 2015, 20, 19030–19040. [Google Scholar] [CrossRef]
- Huang, X.; Yuan, T.; Tschannen, M.; Sun, Z.; Jacob, H.; Du, M.; Liang, M.; Dittmar, R.L.; Liu, Y.; Liang, M.; et al. Characterization of human plasma-derived exosomal RNAs by deep sequencing. BMC Genom. 2013, 14, 319. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Gene | Correlation Coefficient (r) | 95% Confidence Interval | p-Value |
|---|---|---|---|
| BMP7 | 0.694 | 0.288 to 0.889 | 0.003 |
| CCL11 | 0.753 | 0.397 to 0.912 | 0.0008 |
| CCL13 | 0.718 | 0.330 to 0.898 | 0.0017 |
| CCL18 | 0.677 | 0.257 to 0.881 | 0.004 |
| CCL2 | 0.859 | 0.622 to 0.952 | <0.0001 |
| CCL21 | 0.771 | 0.432 to 0.919 | 0.0005 |
| CCL3 | 0.612 | 0.005 to 0.809 | 0.043 |
| CSF1 | 0.700 | 0.298 to 0.891 | 0.0025 |
| CXCL12 | 0.821 | 0.536 to 0.938 | <0.0001 |
| CXCL13 | 0.644 | 0.202 to 0.868 | 0.0071 |
| GPI | 0.700 | 0.298 to 0.891 | 0.0025 |
| IL10 | 0.612 | 0.151 to 0.854 | 0.012 |
| IL12A | 0.641 | 0.191 to 0.867 | 0.007 |
| IL15 | 0.688 | 0.277 to 0.886 | 0.0032 |
| IL18 | 0.685 | 0.272 to 0.885 | 0.0034 |
| IL23A | 0.527 | 0.025 to 0.816 | 0.036 |
| IL3 | 0.635 | 0.188 to 0.864 | 0.008 |
| MIF | 0.724 | 0.341 to 0.900 | 0.0015 |
| PPBP | −0.621 | −0.858 to −0.165 | 0.0103 |
| SPP1 | 0.668 | 0.242 to 0.878 | 0.005 |
| TNF | 0.709 | 0.314 to 0.895 | 0.0021 |
| TNFRSF11B | 0.624 | 0.169 to 0.859 | 0.0099 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colavita, F.; Biava, M.; Castilletti, C.; Lanini, S.; Miccio, R.; Portella, G.; Vairo, F.; Ippolito, G.; Capobianchi, M.R.; Di Caro, A.; et al. Inflammatory and Humoral Immune Response during Ebola Virus Infection in Survivor and Fatal Cases Occurred in Sierra Leone during the 2014–2016 Outbreak in West Africa. Viruses 2019, 11, 373. https://doi.org/10.3390/v11040373
Colavita F, Biava M, Castilletti C, Lanini S, Miccio R, Portella G, Vairo F, Ippolito G, Capobianchi MR, Di Caro A, et al. Inflammatory and Humoral Immune Response during Ebola Virus Infection in Survivor and Fatal Cases Occurred in Sierra Leone during the 2014–2016 Outbreak in West Africa. Viruses. 2019; 11(4):373. https://doi.org/10.3390/v11040373
Chicago/Turabian StyleColavita, Francesca, Mirella Biava, Concetta Castilletti, Simone Lanini, Rossella Miccio, Gina Portella, Francesco Vairo, Giuseppe Ippolito, Maria Rosaria Capobianchi, Antonino Di Caro, and et al. 2019. "Inflammatory and Humoral Immune Response during Ebola Virus Infection in Survivor and Fatal Cases Occurred in Sierra Leone during the 2014–2016 Outbreak in West Africa" Viruses 11, no. 4: 373. https://doi.org/10.3390/v11040373
APA StyleColavita, F., Biava, M., Castilletti, C., Lanini, S., Miccio, R., Portella, G., Vairo, F., Ippolito, G., Capobianchi, M. R., Di Caro, A., & Lalle, E. (2019). Inflammatory and Humoral Immune Response during Ebola Virus Infection in Survivor and Fatal Cases Occurred in Sierra Leone during the 2014–2016 Outbreak in West Africa. Viruses, 11(4), 373. https://doi.org/10.3390/v11040373