HIV-1 Envelope Glycoprotein at the Interface of Host Restriction and Virus Evasion



Abstract
1. Introduction
2. HIV-1 Env Is Attacked by Host Innate Immunity
3. A Long and Challenging Journey for Env Protein to Reach Virus Particles
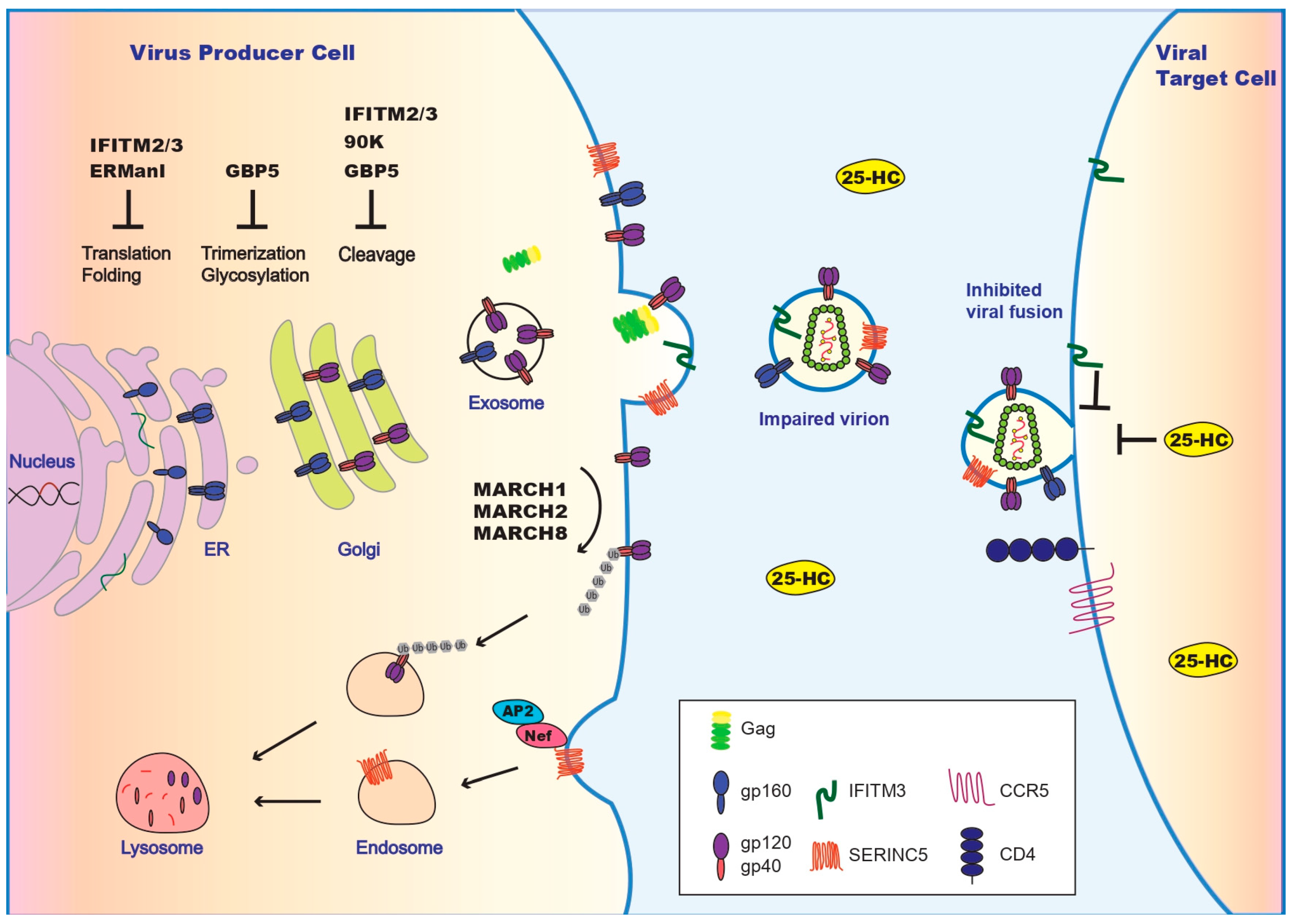
3.1. ER-Associated Degradation: Traps at Its Place of Birth
3.2. GBP5, 90K, and IFITM3: Blocks Along Env’s Route to the Virus Assembly Site
3.3. MARCH1, MARCH2, and MARCH8: Removing HIV-1 Env from the Cell Surface
4. Cellular Antagonists of Env Protein in HIV-1 Particles
5. Env Antagonists in the Membrane of Target Cells: The Other Half of the Fusion Story
6. Env Protein Fights Back: Evasion of Host Restriction on Virus Entry and Beyond
7. Viral Envelope Protein under the Suppressive Pressure of Both Adaptive and Innate Immunity
8. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Checkley, M.A.; Luttge, B.G.; Freed, E.O. HIV-1 Envelope Glycoprotein Biosynthesis, Trafficking, and Incorporation. J. Mol. Boil. 2011, 410, 582–608. [Google Scholar] [CrossRef] [PubMed]
- Quiñones-Kochs, M.I.; Buonocore, L.; Rose, J.K. Role of N-Linked Glycans in a Human Immunodeficiency Virus Envelope Glycoprotein: Effects on Protein Function and the Neutralizing Antibody Response. J. Virol. 2002, 76, 4199–4211. [Google Scholar] [CrossRef] [PubMed]
- Burton, D.R.; Mascola, J.R. Antibody responses to envelope glycoproteins in HIV-1 infection. Nat. Immunol. 2015, 16, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Jardine, J.G.; Ota, T.; Sok, D.; Pauthner, M.; Kulp, D.W.; Kalyuzhniy, O.; Skog, P.D.; Thinnes, T.C.; Bhullar, D.; Briney, B.; et al. HIV-1 VACCINES. Priming a broadly neutralizing antibody response to HIV-1 using a germline-targeting immunogen. Science 2015, 349, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Chung, A.; Rollman, E.; Johansson, S.; Kent, S.; Stratov, I. The Utility of ADCC Responses in HIV Infection. Curr. HIV Res. 2008, 6, 515–519. [Google Scholar] [CrossRef] [PubMed]
- Rerks-Ngarm, S.; Pitisuttithum, P.; Nitayaphan, S.; Kaewkungwal, J.; Chiu, J.; Paris, R.; Premsri, N.; Namwat, C.; De Souza, M.; Adams, E.; et al. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N. Engl. J. Med. 2009, 361, 2209–2220. [Google Scholar] [CrossRef] [PubMed]
- Richard, J.; Prévost, J.; Alsahafi, N.; Ding, S.; Finzi, A. Impact of HIV-1 Envelope Conformation on ADCC Responses. Trends Microbiol. 2018, 26, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Sparrer, K.M.; Gack, M.U. Intracellular detection of viral nucleic acids. Curr. Opin. Microbiol. 2015, 26, 1–9. [Google Scholar] [CrossRef]
- Drappier, M.; Michiels, T. Inhibition of the OAS/RNase L pathway by viruses. Curr. Opin. Virol. 2015, 15, 19–26. [Google Scholar] [CrossRef]
- Degols, G.; Eldin, P.; Mechti, N. ISG20, an actor of the innate immune response. Biochimie 2007, 89, 831–835. [Google Scholar] [CrossRef]
- Refsland, E.W.; Harris, R.S. The APOBEC3 Family of Retroelement Restriction Factors. Curr. Top. Microbiol. Immunol. 2013, 371, 1–27. [Google Scholar]
- Lahouassa, H.; Daddacha, W.; Hofmann, H.; Ayinde, D.; Logue, E.C.; Dragin, L.; Bloch, N.; Maudet, C.; Bertrand, M.; Gramberg, T.; et al. Erratum: SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat. Immunol. 2012, 13, 223–228. [Google Scholar] [CrossRef]
- MacMicking, J.D. Interferon-inducible effector mechanisms in cell-autonomous immunity. Nat. Rev. Immunol. 2012, 12, 367–382. [Google Scholar] [CrossRef]
- Lu, J.; Pan, Q.; Rong, L.; He, W.; Liu, S.-L.; Liang, C. The IFITM Proteins Inhibit HIV-1 Infection. J. Virol. 2011, 85, 2126–2137. [Google Scholar] [CrossRef]
- Li, M.; Kao, E.; Gao, X.; Sandig, H.; Limmer, K.; Pavon-Eternod, M.; Jones, T.E.; Landry, S.; Pan, T.; Weitzman, M.D.; et al. Codon-usage-based inhibition of HIV protein synthesis by human schlafen 11. Nature 2012, 491, 125–128. [Google Scholar] [CrossRef] [PubMed]
- Clerzius, G.; Gelinas, J.F.; Gatignol, A. Multiple levels of PKR inhibition during HIV-1 replication. Rev. Med. Virol. 2011, 21, 42–53. [Google Scholar] [CrossRef]
- Qi, M.; Williams, J.A.; Chu, H.; Chen, X.; Wang, J.-J.; Ding, L.; Akhirome, E.; Wen, X.; Lapierre, L.A.; Goldenring, J.R.; et al. Rab11-FIP1C and Rab14 Direct Plasma Membrane Sorting and Particle Incorporation of the HIV-1 Envelope Glycoprotein Complex. PLoS Pathog. 2013, 9, e1003278. [Google Scholar] [CrossRef]
- Kirschman, J.; Qi, M.; Ding, L.; Hammonds, J.; Dienger-Stambaugh, K.; Wang, J.-J.; Lapierre, L.A.; Goldenring, J.R.; Spearman, P. HIV-1 Envelope Glycoprotein Trafficking through the Endosomal Recycling Compartment Is Required for Particle Incorporation. J. Virol. 2018, 92, e01893-17. [Google Scholar] [CrossRef]
- Kluge, S.F.; Sauter, D.; Kirchhoff, F. SnapShot: Antiviral Restriction Factors. Cell 2015, 163, 774. [Google Scholar] [CrossRef]
- Land, A.; Zonneveld, D.; Braakman, I. Folding of HIV-1 Envelope glycoprotein involves extensive isomerization of disulfide bonds and conformation-dependent leader peptide cleavage. FASEB J. 2003, 17, 1058–1067. [Google Scholar] [CrossRef] [PubMed]
- Meusser, B.; Hirsch, C.; Jarosch, E.; Sommer, T. ERAD: The long road to destruction. Nat. Cell Biol. 2005, 7, 766–772. [Google Scholar] [CrossRef] [PubMed]
- Henrissat, B.; Davies, G. Structural and sequence-based classification of glycoside hydrolases. Curr. Opin. Struct. Boil. 1997, 7, 637–644. [Google Scholar] [CrossRef]
- Gonzalez, D.S.; Karaveg, K.; Vandersall-Nairn, A.S.; Lal, A.; Moremen, K.W. Identification, Expression, and Characterization of a cDNA Encoding Human Endoplasmic Reticulum Mannosidase I, the Enzyme That Catalyzes the First Mannose Trimming Step in Mammalian Asn-linked Oligosaccharide Biosynthesis. J. Boil. Chem. 1999, 274, 21375–21386. [Google Scholar] [CrossRef]
- Zhou, T.; Frabutt, D.A.; Moremen, K.W.; Zheng, Y.H. ER ManI (Endoplasmic Reticulum Class I alpha-Mannosidase) Is Required for HIV-1 Envelope Glycoprotein Degradation via Endoplasmic Reticulum-associated Protein Degradation Pathway. J. Biol. Chem. 2015, 290, 22184–22192. [Google Scholar] [CrossRef]
- Zhou, T.; Dang, Y.; Zheng, Y.-H.; Sundquist, W.I. The Mitochondrial Translocator Protein, TSPO, Inhibits HIV-1 Envelope Glycoprotein Biosynthesis via the Endoplasmic Reticulum-Associated Protein Degradation Pathway. J. Virol. 2014, 88, 3474–3484. [Google Scholar] [CrossRef]
- Frabutt, D.A.; Wang, B.; Riaz, S.; Schwartz, R.C.; Zheng, Y.H. Innate Sensing of Influenza A Virus Hemagglutinin Glycoproteins by the Host Endoplasmic Reticulum (ER) Stress Pathway Triggers a Potent Antiviral Response via ER-Associated Protein Degradation. J. Virol. 2018, 92, e01690-17. [Google Scholar] [CrossRef]
- Zhang, X.; Zhou, T.; Frabutt, D.A.; Zheng, Y.-H. HIV-1 Vpr increases Env expression by preventing Env from endoplasmic reticulum-associated protein degradation (ERAD). Virology 2016, 496, 194–202. [Google Scholar] [CrossRef]
- Casini, A.; Olivieri, M.; Vecchi, L.; Burrone, O.R.; Cereseto, A. Reduction of HIV-1 infectivity through endoplasmic reticulum-associated degradation-mediated Env depletion. J. Virol. 2015, 89, 2966–2971. [Google Scholar] [CrossRef] [PubMed]
- Jejcic, A.; Daniels, R.; Goobar-Larsson, L.; Hebert, D.N.; Vahlne, A. Small Molecule Targets Env for Endoplasmic Reticulum-Associated Protein Degradation and Inhibits Human Immunodeficiency Virus Type 1 Propagation. J. Virol. 2009, 83, 10075–10084. [Google Scholar] [CrossRef] [PubMed]
- Margottin, F.; Bour, S.P.; Durand, H.; Selig, L.; Benichou, S.; Richard, V.; Thomas, D.; Strebel, K.; Benarous, R. A novel human WD protein, h-βTrCp, that interacts with HIV-1 Vpu connects CD4 to the ER degradation pathway through an F-box motif. Mol. Cell 1998, 1, 565–574. [Google Scholar] [CrossRef]
- Krapp, C.; Hotter, D.; Gawanbacht, A.; McLaren, P.J.; Kluge, S.F.; Stürzel, C.M.; Mack, K.; Reith, E.; Engelhart, S.; Ciuffi, A.; et al. Guanylate Binding Protein (GBP) 5 Is an Interferon-Inducible Inhibitor of HIV-1 Infectivity. Cell Host Microbe 2016, 19, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Lodermeyer, V.; Suhr, K.; Schrott, N.; Kolbe, C.; Stürzel, C.M.; Krnavek, D.; Münch, J.; Dietz, C.; Waldmann, T.; Kirchhoff, F.; et al. 90K, an interferon-stimulated gene product, reduces the infectivity of HIV-1. Retrovirology 2013, 10, 111. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Li, M.; Wilkins, J.; Ding, S.; Swartz, T.H.; Esposito, A.M.; Zheng, Y.-M.; Freed, E.O.; Liang, C.; Chen, B.K.; et al. IFITM Proteins Restrict HIV-1 Infection by Antagonizing the Envelope Glycoprotein. Cell Rep. 2015, 13, 145–156. [Google Scholar] [CrossRef]
- Vestal, D.J.; Jeyaratnam, J.A. The Guanylate-Binding Proteins: Emerging Insights into the Biochemical Properties and Functions of This Family of Large Interferon-Induced Guanosine Triphosphatase. J. Interface Cytokine Res. 2011, 31, 89–97. [Google Scholar] [CrossRef]
- Kim, B.H.; Shenoy, A.R.; Kumar, P.; Bradfield, C.J.; MacMicking, J.D. IFN-inducible GTPases in host cell defense. Cell Host Microbe 2012, 12, 432–444. [Google Scholar] [CrossRef]
- Pan, W.; Zuo, X.; Feng, T.; Shi, X.; Dai, J. Guanylate-binding protein 1 participates in cellular antiviral response to dengue virus. Virol. J. 2012, 9, 292. [Google Scholar] [CrossRef]
- Anderson, S.L.; Carton, J.M.; Lou, J.; Xing, L.; Rubin, B.Y. Interferon-Induced Guanylate Binding Protein-1 (GBP-1) Mediates an Antiviral Effect against Vesicular Stomatitis Virus and Encephalomyocarditis Virus. Virology 1999, 256, 8–14. [Google Scholar] [CrossRef] [PubMed]
- McLaren, P.J.; Gawanbacht, A.; Pyndiah, N.; Krapp, C.; Hotter, D.; Kluge, S.F.; Götz, N.; Heilmann, J.; Mack, K.; Sauter, D.; et al. Identification of potential HIV restriction factors by combining evolutionary genomic signatures with functional analyses. Retrovirology 2015, 12, 41. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Hizukuri, Y.; Yamashiro, K.; Makita, N.; Ohnishi, K.; Takeya, M.; Komohara, Y.; Hayashi, Y. Guanylate-binding protein 5 is a marker of interferon-gamma-induced classically activated macrophages. Clin. Transl. Immunol. 2016, 5, e111. [Google Scholar] [CrossRef]
- Guerrero, S.; Batisse, J.; Libre, C.; Bernacchi, S.; Marquet, R.; Paillart, J.-C.; Boehr, D. HIV-1 Replication and the Cellular Eukaryotic Translation Apparatus. Viruses 2015, 7, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E.R.; Dunfee, R.L.; Stanton, J.; Bogdan, D.; Kunstman, K.; Wolinsky, S.M.; Gabuzda, D. High Frequency of Defective vpu Compared with tat and rev Genes in Brain from Patients with HIV Type 1-Associated Dementia. AIDS Res. Hum. Retrovir. 2007, 23, 575–580. [Google Scholar] [CrossRef]
- Brakebusch, C.; Sures, I.; Jallal, B.; Mossie, K.; Fusco, O.; Iacobelli, S.; Ullrich, A. Isolation and Functional Characterization of the Human 90K Promoter. Genomics 1999, 57, 268–278. [Google Scholar] [CrossRef]
- Briggs, N.C.; Natoli, C.; Tinari, N.; D’egidio, M.A.; Goedert, J.J.; Iacobelli, S. A 90-kDa protein serum marker for the prediction of progression to AIDS in a cohort of HIV-1+ homosexual men. AIDS Res. Hum. Retrovir. 1993, 9, 811–816. [Google Scholar] [CrossRef]
- Darcissac, E.C.A.; Vidal, V.; De La Tribonnière, X.; Mouton, Y.; Bahr, G.M. Variations in serum IL-7 and 90K/Mac-2 binding protein (Mac−2 BP) levels analysed in cohorts of HIV-1 patients and correlated with clinical changes following antiretroviral therapy. Clin. Exp. Immunol. 2001, 126, 287–294. [Google Scholar] [CrossRef]
- Loh, L.C.; Keeler, V.; Laferte, S. Monoclonal antibodies specific for human tumor-associated antigen 90K/Mac-2 binding protein: Tools to examine protein conformation and function. J. Cell. Biochem. 2000, 77, 540–559. [Google Scholar]
- Wang, Q.; Zhang, X.; Han, Y.; Wang, X.; Gao, G. M2BP inhibits HIV-1 virion production in a vimentin filaments-dependent manner. Sci. Rep. 2016, 6, 32736. [Google Scholar] [CrossRef] [PubMed]
- Lodermeyer, V.; Ssebyatika, G.; Passos, V.; Ponnurangam, A.; Malassa, A.; Ewald, E.; Stürzel, C.M.; Kirchhoff, F.; Rotger, M.; Falk, C.S.; et al. The Antiviral Activity of the Cellular Glycoprotein LGALS3BP/90K Is Species Specific. J. Virol. 2018, 92, e00226-18. [Google Scholar] [CrossRef]
- Bailey, C.C.; Zhong, G.; Huang, I.-C.; Farzan, M. IFITM-Family Proteins: The Cell’s First Line of Antiviral Defense. Annu. Rev. Virol. 2014, 1, 261–283. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Pan, Q.; Ding, S.; Wang, Z.; Yu, J.; Finzi, A.; Liu, S.-L.; Liang, C. The V3 Loop of HIV-1 Env Determines Viral Susceptibility to IFITM3 Impairment of Viral Infectivity. J. Virol. 2017, 91, e02441-16. [Google Scholar] [CrossRef]
- Bartee, E.; Mansouri, M.; Nerenberg, B.T.H.; Gouveia, K.; Früh, K. Downregulation of major histocompatibility complex class I by human ubiquitin ligases related to viral immune evasion proteins. J. Virol. 2004, 78, 1109–1120. [Google Scholar] [CrossRef] [PubMed]
- Goto, E.; Ishido, S.; Sato, Y.; Ohgimoto, S.; Ohgimoto, K.; Nagano-Fujii, M.; Hotta, H. c-MIR, a Human E3 Ubiquitin Ligase, Is a Functional Homolog of Herpesvirus Proteins MIR1 and MIR2 and Has Similar Activity. J. Boil. Chem. 2003, 278, 14657–14668. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Shin, J.-S. Molecular mechanism and cellular function of MHCII ubiquitination. Immunol. Rev. 2015, 266, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Fujita, H.; Iwabu, Y.; Tokunaga, K.; Tanaka, Y. Membrane-associated RING-CH (MARCH) 8 mediates the ubiquitination and lysosomal degradation of the transferrin receptor. J. Cell Sci. 2013, 126, 2798–2809. [Google Scholar] [CrossRef]
- Roy, N.; Pacini, G.; Berlioz-Torrent, C.; Janvier, K. Characterization of E3 ligases involved in lysosomal sorting of the HIV-1 restriction factor BST2. J. Cell Sci. 2017, 130, 1596–1611. [Google Scholar] [CrossRef] [PubMed]
- Tada, T.; Zhang, Y.; Koyama, T.; Tobiume, M.; Tsunetsugu-Yokota, Y.; Yamaoka, S.; Fujita, H.; Tokunaga, K. MARCH8 inhibits HIV-1 infection by reducing virion incorporation of envelope glycoproteins. Nat. Med. 2015, 21, 1502–1507. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tada, T.; Ozono, S.; Yao, W.; Tanaka, M.; Yamaoka, S.; Kishigami, S.; Fujita, H.; Tokunaga, K. Membrane-associated RING-CH (MARCH) 1 and 2 are MARCH family members that inhibit HIV-1 infection. J. Boil. Chem. 2019, 294, 3397–3405. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lu, J.; Liu, X. MARCH2 is upregulated in HIV-1 infection and inhibits HIV-1 production through envelope protein translocation or degradation. Virology 2018, 518, 293–300. [Google Scholar] [CrossRef]
- Tartour, K.; Appourchaux, R.; Gaillard, J.; Nguyen, X.-N.; Durand, S.; Turpin, J.; Beaumont, E.; Roch, E.; Berger, G.; Mahieux, R.; et al. IFITM proteins are incorporated onto HIV-1 virion particles and negatively imprint their infectivity. Retrovirology 2014, 11, 103. [Google Scholar] [CrossRef] [PubMed]
- Compton, A.A.; Bruel, T.; Porrot, F.; Mallet, A.; Sachse, M.; Euvrard, M.; Liang, C.; Casartelli, N.; Schwartz, O. IFITM Proteins Incorporated into HIV-1 Virions Impair Viral Fusion and Spread. Cell Host Microbe 2014, 16, 736–747. [Google Scholar] [CrossRef]
- Foster, T.L.; Wilson, H.; Iyer, S.S.; Coss, K.; Doores, K.; Smith, S.; Kellam, P.; Finzi, A.; Borrow, P.; Hahn, B.H.; et al. Resistance of Transmitted Founder HIV-1 to IFITM-Mediated Restriction. Cell Host Microbe 2016, 20, 429–442. [Google Scholar] [CrossRef]
- Tartour, K.; Nguyen, X.-N.; Appourchaux, R.; Assil, S.; Barateau, V.; Bloyet, L.-M.; Gaillard, J.B.; Confort, M.-P.; Escudero-Perez, B.; Gruffat, H.; et al. Interference with the production of infectious viral particles and bimodal inhibition of replication are broadly conserved antiviral properties of IFITMs. PLoS Pathog. 2017, 13, e1006610. [Google Scholar] [CrossRef]
- Inuzuka, M.; Hayakawa, M.; Ingi, T. Serinc, an Activity-regulated Protein Family, Incorporates Serine into Membrane Lipid Synthesis. J. Boil. Chem. 2005, 280, 35776–35783. [Google Scholar] [CrossRef] [PubMed]
- Rosa, A.; Chande, A.; Ziglio, S.; De Sanctis, V.; Bertorelli, R.; Goh, S.L.; McCauley, S.M.; Nowosielska, A.; Antonarakis, S.E.; Luban, J.; et al. HIV-1 Nef promotes infection by excluding SERINC5 from virion incorporation. Nature 2015, 526, 212–217. [Google Scholar] [CrossRef]
- Usami, Y.; Wu, Y.; Göttlinger, H.G. SERINC3 and SERINC5 restrict HIV-1 infectivity and are counteracted by Nef. Nature 2015, 526, 218–223. [Google Scholar] [CrossRef]
- Sood, C.; Marin, M.; Chande, A.; Pizzato, M.; Melikyan, G.B. SERINC5 protein inhibits HIV-1 fusion pore formation by promoting functional inactivation of envelope glycoproteins. J. Boil. Chem. 2017, 292, 6014–6026. [Google Scholar] [CrossRef]
- Trautz, B.; Wiedemann, H.; Lüchtenborg, C.; Pierini, V.; Kranich, J.; Glass, B.; Kräusslich, H.-G.; Brocker, T.; Pizzato, M.; Ruggieri, A.; et al. The host-cell restriction factor SERINC5 restricts HIV-1 infectivity without altering the lipid composition and organization of viral particles. J. Boil. Chem. 2017, 292, 13702–13713. [Google Scholar] [CrossRef] [PubMed]
- Beitari, S.; Ding, S.; Pan, Q.; Finzi, A.; Liang, C. Effect of HIV-1 Env on SERINC5 Antagonism. J. Virol. 2017, 91, e02214-16. [Google Scholar] [CrossRef]
- Shi, J.; Xiong, R.; Zhou, T.; Su, P.; Zhang, X.; Qiu, X.; Li, H.; Li, S.; Yu, C.; Wang, B.; et al. HIV-1 Nef Antagonizes SERINC5 Restriction by Downregulation of SERINC5 via the Endosome/Lysosome System. J. Virol. 2018, 92, e00196-18. [Google Scholar] [CrossRef]
- Ahi, Y.S.; Zhang, S.; Thappeta, Y.; Denman, A.; Feizpour, A.; Gummuluru, S.; Reinhard, B.; Muriaux, D.; Fivash, M.J.; Rein, A. Functional Interplay Between Murine Leukemia Virus Glycogag, Serinc5, and Surface Glycoprotein Governs Virus Entry, with Opposite Effects on Gammaretroviral and Ebolavirus Glycoproteins. mBio 2016, 7, e01985-16. [Google Scholar] [CrossRef] [PubMed]
- Chande, A.; Cuccurullo, E.C.; Rosa, A.; Ziglio, S.; Carpenter, S.; Pizzato, M. S2 from equine infectious anemia virus is an infectivity factor which counteracts the retroviral inhibitors SERINC5 and SERINC3. Proc. Natl. Acad. Sci. USA 2016, 113, 13197–13202. [Google Scholar] [CrossRef] [PubMed]
- Brass, A.L.; Huang, I.C.; Benita, Y.; John, S.P.; Krishnan, M.N.; Feeley, E.M.; Ryan, B.J.; Weyer, J.L.; Van Der Weyden, L.; Fikrig, E.; et al. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 2009, 139, 1243–1254. [Google Scholar] [CrossRef]
- Li, K.; Markosyan, R.M.; Zheng, Y.-M.; Golfetto, O.; Bungart, B.; Li, M.; Ding, S.; He, Y.; Liang, C.; Lee, J.C.; et al. IFITM Proteins Restrict Viral Membrane Hemifusion. PLoS Pathog. 2013, 9, e1003124. [Google Scholar] [CrossRef]
- Desai, T.M.; Marin, M.; Chin, C.R.; Savidis, G.; Brass, A.L.; Melikyan, G.B. IFITM3 Restricts Influenza A Virus Entry by Blocking the Formation of Fusion Pores following Virus-Endosome Hemifusion. PLoS Pathog. 2014, 10, e1004048. [Google Scholar] [CrossRef] [PubMed]
- Amini-Bavil-Olyaee, S.; Choi, Y.J.; Lee, J.H.; Shi, M.; Huang, I.-C.; Farzan, M.; Jung, J.U. The antiviral effector IFITM3 disrupts intracellular cholesterol homeostasis to block viral entry. Cell Host Microbe 2013, 13, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Wrensch, F.; Winkler, M.; Pöhlmann, S. IFITM Proteins Inhibit Entry Driven by the MERS-Coronavirus Spike Protein: Evidence for Cholesterol-Independent Mechanisms. Viruses 2014, 6, 3683–3698. [Google Scholar] [CrossRef] [PubMed]
- Lewin, A.R.; Reid, L.E.; McMahon, M.; Stark, G.R.; Kerr, I.M. Molecular analysis of a human interferon-inducible gene family. Eur. J. Biochem. 1991, 199, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Jia, R.; Pan, Q.; Ding, S.; Rong, L.; Liu, S.-L.; Geng, Y.; Qiao, W.; Liang, C. The N-Terminal Region of IFITM3 Modulates Its Antiviral Activity by Regulating IFITM3 Cellular Localization. J. Virol. 2012, 86, 13697–13707. [Google Scholar] [CrossRef] [PubMed]
- Jia, R.; Xu, F.; Qian, J.; Yao, Y.; Miao, C.; Zheng, Y.-M.; Liu, S.-L.; Guo, F.; Geng, Y.; Qiao, W.; et al. Identification of an endocytic signal essential for the antiviral action of IFITM3. Cell. Microbiol. 2014, 16, 1080–1093. [Google Scholar] [CrossRef]
- Bailey, C.C.; Kondur, H.R.; Huang, I.-C.; Farzan, M. Interferon-induced Transmembrane Protein 3 Is a Type II Transmembrane Protein. J. Boil. Chem. 2013, 288, 32184–32193. [Google Scholar] [CrossRef]
- Li, K.; Jia, R.; Li, M.; Zheng, Y.M.; Miao, C.; Yao, Y.; Ji, H.L.; Geng, Y.; Qiao, W.; Albritton, L.M.; et al. A sorting signal suppresses IFITM1 restriction of viral entry. J. Biol. Chem. 2015, 290, 4248–4259. [Google Scholar] [CrossRef]
- Park, K.; Scott, A.L. Cholesterol 25-hydroxylase production by dendritic cells and macrophages is regulated by type I interferons. J. Leukoc. Boil. 2010, 88, 1081–1087. [Google Scholar] [CrossRef]
- Lund, E.G.; Kerr, T.A.; Sakai, J.; Li, W.P.; Russell, D.W. cDNA cloning of mouse and human cholesterol 25-hydroxylases, polytopic membrane proteins that synthesize a potent oxysterol regulator of lipid metabolism. J. Biol. Chem. 1998, 273, 34316–34327. [Google Scholar] [CrossRef]
- Liu, S.Y.; Aliyari, R.; Chikere, K.; Li, G.; Marsden, M.D.; Smith, J.K.; Pernet, O.; Guo, H.; Nusbaum, R.; Zack, J.A.; et al. Interferon-inducible cholesterol-25-hydroxylase broadly inhibits viral entry by production of 25-hydroxycholesterol. Immunity 2013, 38, 92–105. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, S.; Yi, Z.; Tian, H.; Aliyari, R.; Li, Y.; Chen, G.; Liu, P.; Zhong, J.; Chen, X.; et al. Interferon-Inducible Cholesterol-25-Hydroxylase Inhibits Hepatitis C Virus Replication via Distinct Mechanisms. Sci. Rep. 2014, 4, 7242. [Google Scholar] [CrossRef]
- Li, C.; Deng, Y.-Q.; Wang, S.; Ma, F.; Aliyari, R.; Huang, X.-Y.; Zhang, N.-N.; Watanabe, M.; Dong, H.-L.; Liu, P.; et al. 25-Hydroxycholesterol Protects Host against Zika Virus Infection and Its Associated Microcephaly in a Mouse Model. Immunity 2017, 46, 446–456. [Google Scholar] [CrossRef]
- Kandutsch, A.A.; Chen, H.W. Inhibition of sterol synthesis in cultured mouse cells by cholesterol derivatives oxygenated in the side chain. J. Boil. Chem. 1974, 249, 6057–6061. [Google Scholar]
- Goldstein, J.L.; Brunschede, G.Y.; Brown, M.S. Inhibition of proteolytic degradation of low density lipoprotein in human fibroblasts by chloroquine, concanavalin A, and Triton WR 1339. J. Boil. Chem. 1975, 250, 7854–7862. [Google Scholar]
- McDonald, J.G.; Russell, D.W. Editorial: 25-Hydroxycholesterol: A new life in immunology. J. Leukoc. Boil. 2010, 88, 1071–1072. [Google Scholar] [CrossRef]
- Diczfalusy, U.; Olofsson, K.E.; Carlsson, A.-M.; Gong, M.; Golenbock, D.T.; Rooyackers, O.; Fläring, U.; Björkbacka, H. Marked upregulation of cholesterol 25-hydroxylase expression by lipopolysaccharide. J. Res. 2009, 50, 2258–2264. [Google Scholar] [CrossRef]
- Bauman, D.R.; Bitmansour, A.D.; McDonald, J.G.; Thompson, B.M.; Liang, G.; Russell, D.W. 25-Hydroxycholesterol secreted by macrophages in response to Toll-like receptor activation suppresses immunoglobulin A production. Proc. Natl. Acad. Sci. USA 2009, 106, 16764–16769. [Google Scholar] [CrossRef]
- Gomes, B.; Gonçalves, S.; Disalvo, A.; Hollmann, A.; Santos, N.C. Effect of 25-hydroxycholesterol in viral membrane fusion: Insights on HIV inhibition. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1171–1178. [Google Scholar] [CrossRef]
- Shrivastava-Ranjan, P.; Bergeron, É.; Chakrabarti, A.K.; Albariño, C.G.; Flint, M.; Nichol, S.T.; Spiropoulou, C.F. 25-Hydroxycholesterol Inhibition of Lassa Virus Infection through Aberrant GP1 Glycosylation. mBio 2016, 7, e01808-16. [Google Scholar] [CrossRef]
- Doms, A.; Sanabria, T.; Hansen, J.N.; Altan-Bonnet, N.; Holm, G.H. 25-Hydroxycholesterol Production by the Cholesterol-25-Hydroxylase Interferon-Stimulated Gene Restricts Mammalian Reovirus Infection. J. Virol. 2018, 92, e01047-18. [Google Scholar] [CrossRef]
- Anafu, A.A.; Bowen, C.H.; Chin, C.R.; Brass, A.L.; Holm, G.H. Interferon-inducible Transmembrane Protein 3 (IFITM3) Restricts Reovirus Cell Entry. J. Boil. Chem. 2013, 288, 17261–17271. [Google Scholar] [CrossRef]
- Ikeda, T.; Symeonides, M.; Albin, J.S.; Li, M.; Thali, M.; Harris, R.S. HIV-1 adaptation studies reveal a novel Env-mediated homeostasis mechanism for evading lethal hypermutation by APOBEC3G. PLoS Pathog. 2018, 14, e1007010. [Google Scholar] [CrossRef]
- Ding, S.; Pan, Q.; Liu, S.-L.; Liang, C. HIV-1 mutates to evade IFITM1 restriction. Virology 2014, 454, 11–24. [Google Scholar] [CrossRef]
- Gerlach, T.; Hensen, L.; Matrosovich, T.; Bergmann, J.; Winkler, M.; Peteranderl, C.; Klenk, H.-D.; Weber, F.; Herold, S.; Pöhlmann, S.; et al. pH Optimum of Hemagglutinin-Mediated Membrane Fusion Determines Sensitivity of Influenza A Viruses to the Interferon-Induced Antiviral State and IFITMs. J. Virol. 2017, 91, e00246-17. [Google Scholar] [CrossRef]
- Boyd, D.F.; Sharma, A.; Humes, D.; Cheng-Mayer, C.; Overbaugh, J. Adapting SHIVs In Vivo Selects for Envelope-Mediated Interferon-alpha Resistance. PLoS Pathog. 2016, 12, e1005727. [Google Scholar] [CrossRef]
- Rihn, S.J.; Foster, T.L.; Busnadiego, I.; Aziz, M.A.; Hughes, J.; Neil, S.J.D.; Wilson, S.J. The Envelope Gene of Transmitted HIV-1 Resists a Late Interferon Gamma-Induced Block. J. Virol. 2017, 91, e02254-16. [Google Scholar] [CrossRef]
- Le Tortorec, A.; Neil, S.J.D. Antagonism to and Intracellular Sequestration of Human Tetherin by the Human Immunodeficiency Virus Type 2 Envelope Glycoprotein. J. Virol. 2009, 83, 11966–11978. [Google Scholar] [CrossRef]
- Lemaître, C.; Harper, F.; Pierron, G.; Heidmann, T.; Dewannieux, M.; Ross, S.R.; Palmer, S.G.; DeVito, I.; Jenkins, S.G.; Niewiesk, S.; et al. The HERV-K Human Endogenous Retrovirus Envelope Protein Antagonizes Tetherin Antiviral Activity. J. Virol. 2014, 88, 13626–13637. [Google Scholar] [CrossRef]
- Lopez, L.A.; Yang, S.J.; Hauser, H.; Exline, C.M.; Haworth, K.G.; Oldenburg, J.; Cannon, P.M. Ebola Virus Glycoprotein Counteracts BST-2/Tetherin Restriction in a Sequence-Independent Manner That Does Not Require Tetherin Surface Removal. J. Virol. 2010, 84, 7243–7255. [Google Scholar] [CrossRef]
- Van Damme, N.; Goff, D.; Katsura, C.; Jorgenson, R.L.; Mitchell, R.; Johnson, M.C.; Stephens, E.B.; Guatelli, J. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 2008, 3, 245–252. [Google Scholar] [CrossRef]
- Neil, S.J.D.; Zang, T.; Bieniasz, P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef]
- Zhang, F.; Wilson, S.J.; Langford, W.; Virgen, B.; Gregory, D.; Johnson, M.C.; Münch, J.; Kirchhoff, F.; Bieniasz, P.D.; Hatziioannou, T.; et al. Nef proteins from simian immunodeficiency viruses are tetherin antagonists. Cell Host Microbe 2009, 6, 54–67. [Google Scholar] [CrossRef]
- Gupta, R.K.; Mlcochova, P.; Pelchen-Matthews, A.; Petit, S.J.; Mattiuzzo, G.; Pillay, D.; Takeuchi, Y.; Marsh, M.; Towers, G.J.; Towers, G. Simian immunodeficiency virus envelope glycoprotein counteracts tetherin/BST-2/CD317 by intracellular sequestration. Proc. Natl. Acad. Sci. USA 2009, 106, 20889–20894. [Google Scholar] [CrossRef]
- Noble, B.; Abada, P.; Nunez-Iglesias, J.; Cannon, P.M. Recruitment of the Adaptor Protein 2 Complex by the Human Immunodeficiency Virus Type 2 Envelope Protein Is Necessary for High Levels of Virus Release. J. Virol. 2006, 80, 2924–2932. [Google Scholar] [CrossRef]
- Hauser, H.; A Lopez, L.; Yang, S.J.; E Oldenburg, J.; Exline, C.M.; Guatelli, J.C.; Cannon, P.M. HIV-1 Vpu and HIV-2 Env counteract BST-2/tetherin by sequestration in a perinuclear compartment. Retrovirology 2010, 7, 51. [Google Scholar] [CrossRef]
- Chen, C.-Y.; Shingai, M.; Welbourn, S.; Martin, M.A.; Borrego, P.; Taveira, N.; Strebel, K. Antagonism of BST-2/Tetherin Is a Conserved Function of the Env Glycoprotein of Primary HIV-2 Isolates. J. Virol. 2016, 90, 11062–11074. [Google Scholar] [CrossRef]
- González-Hernández, M.; Hoffmann, M.; Brinkmann, C.; Nehls, J.; Winkler, M.; Schindler, M.; Pöhlmann, S. A GXXXA Motif in the Transmembrane Domain of the Ebola Virus Glycoprotein Is Required for Tetherin Antagonism. J. Virol. 2018, 92, e00403-18. [Google Scholar] [CrossRef]
- Nishimura, Y.; Martin, M.A. Of Mice, Macaques, and Men: Broadly Neutralizing Antibody Immunotherapy for HIV-1. Cell Host Microbe 2017, 22, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.; Akkina, R. TRIM5alpharh expression restricts HIV-1 infection in lentiviral vector-transduced CD34+-cell-derived macrophages. Mol. Ther. 2005, 12, 687–696. [Google Scholar] [CrossRef]
- Chan, E.; Towers, G.J.; Qasim, W. Gene Therapy Strategies to Exploit TRIM Derived Restriction Factors against HIV-1. Viruses 2014, 6, 243–263. [Google Scholar] [CrossRef]


{kind=link}
| Restriction Factor | Impact on HIV-1 Env | Other Enveloped Viruses Affected | Virus Escape Mechanism | References |
|---|---|---|---|---|
| ErManI | Decrease Env expression via ERAD pathway; modulate glycosylation of HIV-1 Env | IAV | HIV Vpr increases Env expression | [24,26,27] |
| GBP5 | Impair cleavage of gp160; alter glycolysation of HIV-1 Env | MLV | Viral trade-off mechanism to increase Env expression by shutting down Vpu expression | [31,38] |
| 90K | Prevent gp160 processing; decrease mature gp120/gp41 in virions | EBOV | TBD * | [32,47] |
| IFITM2/3 | Deter viral entry into virus target cells; impair gp160 processing; promote gp120 shedding; decrease mature gp120/gp41 in virions; incorporate into virions and impair viral entry; | MLV, WNV, MPMV, EBOV, EBV, MeV, DENV | Overcome by HIV-1 Env | [33,61] |
| MARCH1/2/8 | Downregulate Env from the plasma membrane | HIV-2, SIV, MLV, VSV | TBD | [55,56,57] |
| SERINC5 | Impair virus infectivity; incorporate into virus particles; affect the conformation of the MPER region of Env | MLV, EIAV, EBOV | Downregulated by Nef from plasma membrane; countered by HIV-1 Env | [63,64,65,67] |
| 25-HC | Modify the secondary structure of the HIV-fusion peptide; prevents membrane fusion | VSV, ZIKV, EBOV, NiV, HCV, RVF | TBD | [83,84,85,91,92,93] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beitari, S.; Wang, Y.; Liu, S.-L.; Liang, C. HIV-1 Envelope Glycoprotein at the Interface of Host Restriction and Virus Evasion. Viruses 2019, 11, 311. https://doi.org/10.3390/v11040311
Beitari S, Wang Y, Liu S-L, Liang C. HIV-1 Envelope Glycoprotein at the Interface of Host Restriction and Virus Evasion. Viruses. 2019; 11(4):311. https://doi.org/10.3390/v11040311
Chicago/Turabian StyleBeitari, Saina, Yimeng Wang, Shan-Lu Liu, and Chen Liang. 2019. "HIV-1 Envelope Glycoprotein at the Interface of Host Restriction and Virus Evasion" Viruses 11, no. 4: 311. https://doi.org/10.3390/v11040311
APA StyleBeitari, S., Wang, Y., Liu, S.-L., & Liang, C. (2019). HIV-1 Envelope Glycoprotein at the Interface of Host Restriction and Virus Evasion. Viruses, 11(4), 311. https://doi.org/10.3390/v11040311