Arthritogenic Alphavirus-Induced Immunopathology and Targeting Host Inflammation as A Therapeutic Strategy for Alphaviral Disease


{kind=link}
Abstract
1. Introduction
2. Innate Immune Response to Acute Arthritogenic Alphavirus Infection
2.1. Induction of Interferon and Alphavirus Detection
2.2. Anti-Alphavirus Interferon Response Pathways
3. Proinflammatory Cellular Responses During Acute Alphavirus-Induced Arthritic Disease
3.1. Macrophage-Derived Immune Responses
3.2. T-Cell-Derived Immune Responses
4. Immune Responses During Chronic Alphavirus-Induced Arthritic Disease
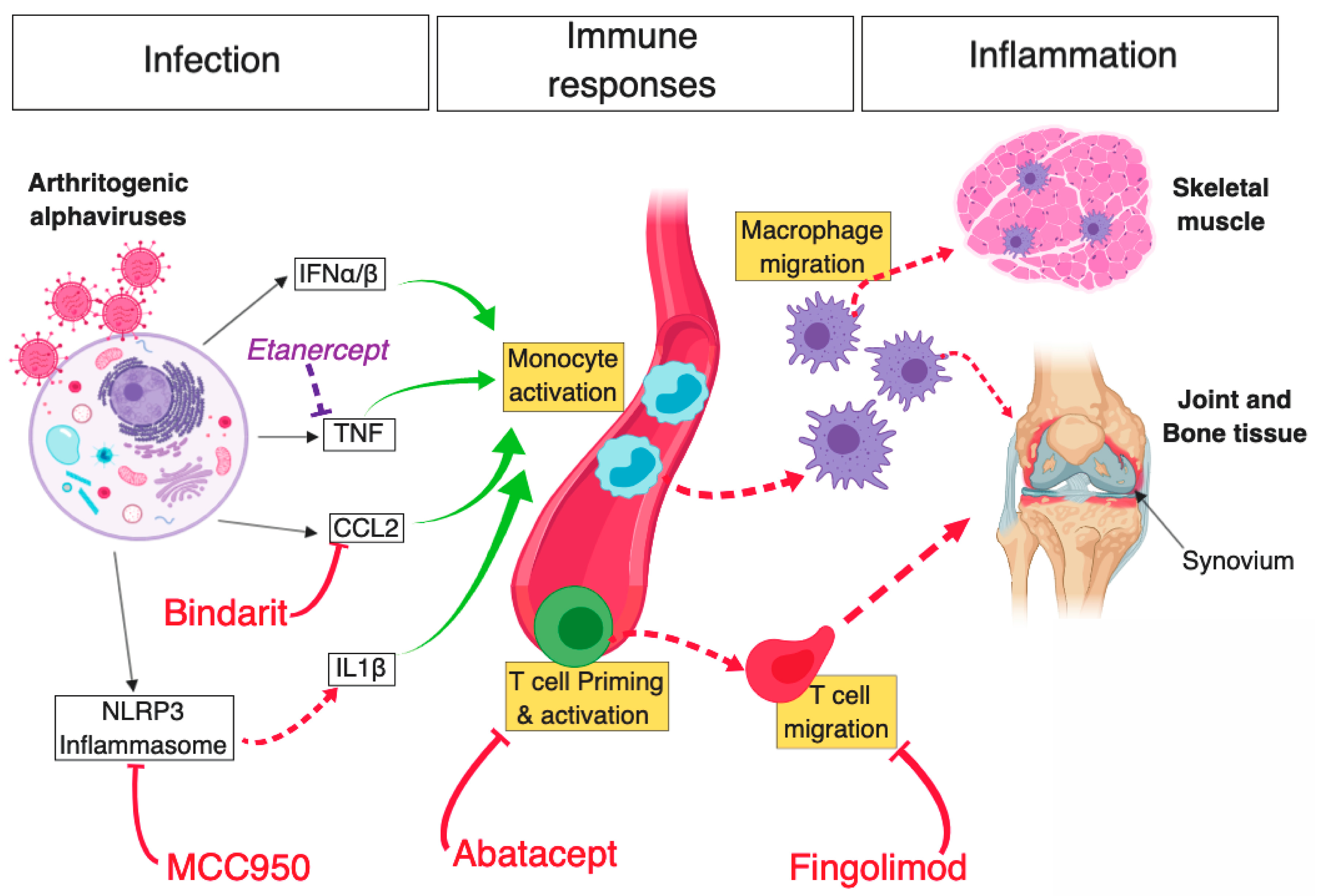
5. Treatment Strategies Targeting Alphavirus-Induced Inflammation
6. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Powers, A.M.; Brault, A.C.; Shirako, Y.; Strauss, E.G.; Kang, W.L.; Strauss, J.H.; Weaver, S.C. Evolutionary relationships and systematics of the alphaviruses. J. Virol. 2001, 75, 10118–10131. [Google Scholar] [CrossRef]
- Seyler, T.; Hutin, Y.; Ramanchandran, V.; Ramakrishnan, R.; Manickam, P.; Murhekar, M. Estimating the burden of disease and the economic cost attributable to chikungunya, Andhra Pradesh, India, 2005–2006. Trans. R. Soc. Trop. Med. Hyg. 2010, 104, 133–138. [Google Scholar] [CrossRef]
- Renault, P.; Solet, J.L.; Sissoko, D.; Balleydier, E.; Larrieu, S.; Filleul, L.; Lassalle, C.; Thiria, J.; Rachou, E.; de Valk, H.; et al. A major epidemic of chikungunya virus infection on Reunion Island, France, 2005–2006. Am. J. Trop. Med. Hyg. 2007, 77, 727–731. [Google Scholar] [CrossRef]
- Beesoon, S.; Funkhouser, E.; Kotea, N.; Spielman, A.; Robicht, R.M. Chikungunya fever, Mauritius, 2006. Emerg. Infect. Dis. 2008, 14, 337–338. [Google Scholar] [CrossRef]
- Van Bortel, W.; Dorleans, F.; Rosine, J.; Blateau, A.; Rousset, D.; Matheus, S.; Leparc-Goffart, I.; Flusin, O.; Prat, C.M.; Cesaire, R.; et al. Chikungunya outbreak in the Caribbean region, December 2013 to March 2014, and the significance for Europe. Eurosurveillance 2014, 19, 17–27. [Google Scholar] [CrossRef]
- Tsetsarkin, K.A.; Vanlandingham, D.L.; McGee, C.E.; Higgs, S. A single mutation in chikungunya virus affects vector specificity and epidemic potential. PLoS Pathog. 2007, 3, e201. [Google Scholar] [CrossRef] [PubMed]
- Randolph, S.E.; Rogers, D.J. The arrival, establishment and spread of exotic diseases: Patterns and predictions. Nat. Rev. Microbiol. 2010, 8, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Miner, J.J.; Yeang, H.X.A.; Fox, J.M.; Taffner, S.; Malkova, O.N.; Oh, S.T.; Kim, A.H.J.; Diamond, M.S.; Lenschow, D.J.; Yokoyama, W.M. Chikungunya Viral Arthritis in the United States A Mimic of Seronegative Rheumatoid Arthritis. Arthritis Rheumatol. 2015, 67, 1214–1220. [Google Scholar] [CrossRef]
- Blettery, M.; Brunier, L.; Mary, J.; Polomat, K.; Moinet, F.; Deligny, C.; Arfi, S.; Jean Baptiste, G.; De Bandt, M. Management of Chronic Post-Chikungunya Rheumatic Disease: The Martinican Experience. Arthritis Rheumatol. 2016, 68, 2817–2824. [Google Scholar] [CrossRef] [PubMed]
- Oehler, E.; Fournier, E.; Leparc-Goffart, I.; Larre, P.; Cubizolle, S.; Sookhareea, C.; Lastere, S.; Ghawche, F. Increase in cases of Guillain-Barre syndrome during a Chikungunya outbreak, French Polynesia, 2014 to 2015. Eurosurveillance 2015, 20, 13–14. [Google Scholar] [CrossRef]
- Cerny, T.; Schwarz, M.; Schwarz, U.; Lemant, J.; Gerardin, P.; Keller, E. The Range of Neurological Complications in Chikungunya Fever. Neurocrit. Care 2017, 27, 447–457. [Google Scholar] [CrossRef]
- Katze, M.G.; He, Y.P.; Gale, M. Viruses and interferon: A fight for supremacy. Nat. Rev. Immunol. 2002, 2, 675–687. [Google Scholar] [CrossRef]
- Sen, G.C. Viruses and interferons. Annu. Rev. Microbiol. 2001, 55, 255–281. [Google Scholar] [CrossRef]
- Wauquier, N.; Becquart, P.; Nkoghe, D.; Padilla, C.; Ndjoyi-Mbiguino, A.; Leroy, E.M. The Acute Phase of Chikungunya Virus Infection in Humans Is Associated with Strong Innate Immunity and T CD8 Cell Activation. J. Infect. Dis. 2011, 204, 115–123. [Google Scholar] [CrossRef]
- Ryman, K.D.; Klimstra, W.B. Host responses to alphavirus infection. Immunol. Rev. 2008, 225, 27–45. [Google Scholar] [CrossRef]
- Couderc, T.; Chretien, F.; Schilte, C.; Disson, O.; Brigitte, M.; Guivel-Benhassine, F.; Touret, Y.; Barau, G.; Cayet, N.; Schuffenecker, I.; et al. A mouse model for Chikungunya: Young age and inefficient type-I interferon signaling are risk factors for severe disease. PLoS Pathog. 2008, 4, e29. [Google Scholar] [CrossRef]
- Ryman, K.D.; Klimstra, W.B.; Nguyen, K.B.; Biron, C.A.; Johnston, R.E. Alpha/beta interferon protects adult mice from fatal Sindbis virus infection and is an important determinant of cell and tissue tropism. J. Virol. 2000, 74, 3366–3378. [Google Scholar] [CrossRef]
- Seymour, R.L.; Rossi, S.L.; Bergren, N.A.; Plante, K.S.; Weaver, S.C. The Role of Innate versus Adaptive Immune Responses in a Mouse Model of O’Nyong-Nyong Virus Infection. Am. J. Trop. Med. Hyg. 2013, 88, 1170–1179. [Google Scholar] [CrossRef]
- Werneke, S.W.; Schilte, C.; Rohatgi, A.; Monte, K.J.; Michault, A.; Arenzana-Seisdedos, F.; Vanlandingham, D.L.; Higgs, S.; Fontanet, A.; Albert, M.L.; et al. ISG15 is critical in the control of Chikungunya virus infection independent of UbE1L mediated conjugation. PLoS Pathog. 2011, 7, e1002322. [Google Scholar] [CrossRef]
- Ziegler, S.A.; Lu, L.; da Rosa, A.P.; Xiao, S.Y.; Tesh, R.B. An animal model for studying the pathogenesis of chikungunya virus infection. Am. J. Trop. Med. Hyg. 2008, 79, 133–139. [Google Scholar] [CrossRef]
- Weise, W.J.; Hermance, M.E.; Forrester, N.; Adams, A.P.; Langsjoen, R.; Gorchakov, R.; Wang, E.; Alcorn, M.D.; Tsetsarkin, K.; Weaver, S.C. A Novel Live-Attenuated Vaccine Candidate for Mayaro Fever. PLoS Negl. Trop. Dis. 2014, 8, e2969. [Google Scholar] [CrossRef]
- Schilte, C.; Couderc, T.; Chretien, F.; Sourisseau, M.; Gangneux, N.; Guivel-Benhassine, F.; Kraxner, A.; Tschopp, J.; Higgs, S.; Michault, A.; et al. Type I IFN controls chikungunya virus via its action on nonhematopoietic cells. J. Exp. Med. 2010, 207, 429–442. [Google Scholar] [CrossRef]
- Hirsch, R.L.; Griffin, D.E.; Winkelstein, J.A. The effect of complement depletion on the course of Sindbis virus infection in mice. J. Immunol. 1978, 121, 1276–1278. [Google Scholar]
- Morrison, T.E.; Fraser, R.J.; Smith, P.N.; Mahalingam, S.; Heise, M.T. Complement contributes to inflammatory tissue destruction in a mouse model of Ross River virus-induced disease. J. Virol. 2007, 81, 5132–5143. [Google Scholar] [CrossRef]
- Morrison, T.E.; Simmons, J.D.; Heise, M.T. Complement Receptor 3 Promotes Severe Ross River Virus-Induced Disease. J. Virol. 2008, 82, 11263–11272. [Google Scholar] [CrossRef]
- Gunn, B.M.; Jones, J.E.; Shabman, R.S.; Whitmore, A.C.; Sarkar, S.; Blevins, L.K.; Morrison, T.E.; Heise, M.T. Ross River virus envelope glycans contribute to disease through activation of the host complement system. Virology 2018, 515, 250–260. [Google Scholar] [CrossRef]
- Gunn, B.M.; Morrison, T.E.; Whitmore, A.C.; Blevins, L.K.; Hueston, L.; Fraser, R.J.; Herrero, L.J.; Ramirez, R.; Smith, P.N.; Mahalingam, S.; et al. Mannose Binding Lectin Is Required for Alphavirus-Induced Arthritis/Myositis. PLoS Pathog. 2012, 8. [Google Scholar] [CrossRef]
- Kozaki, T.; Komano, J.; Kanbayashi, D.; Takahama, M.; Misawa, T.; Satoh, T.; Takeuchi, O.; Kawai, T.; Shimizu, S.; Matsuura, Y.; et al. Mitochondrial damage elicits a TCDD-inducible poly(ADP-ribose) polymerase- mediated antiviral response. Proc. Natl. Acad. Sci. USA 2017, 114, 2681–2686. [Google Scholar] [CrossRef]
- Nair, S.; Poddar, S.; Shimak, R.M.; Diamond, M.S. Interferon regulatory factor-1 (IRF-1) protects against chikungunya virus induced immunopathology by restricting infection in muscle cells. J. Virol. 2017. [Google Scholar] [CrossRef]
- Ekchariyawat, P.; Hamel, R.; Bernard, E.; Wichit, S.; Surasombatpattana, P.; Talignani, L.; Thomas, F.; Choumet, V.; Yssel, H.; Despres, P.; et al. Inflammasome signaling pathways exert antiviral effect against Chikungunya virus in human dermal fibroblasts. Infect. Genet. Evol. 2015, 32, 401–408. [Google Scholar] [CrossRef]
- Chen, W.; Foo, S.S.; Zaid, A.; Teng, T.S.; Herrero, L.J.; Wolf, S.; Tharmarajah, K.; Vu, L.D.; van Vreden, C.; Taylor, A.; et al. Specific inhibition of NLRP3 in chikungunya disease reveals a role for inflammasomes in alphavirus-induced inflammation. Nat. Microbiol. 2017, 2, 1435–1445. [Google Scholar] [CrossRef]
- Her, Z.S.; Teng, T.S.; Tan, J.J.L.; Teo, T.H.; Kam, Y.W.; Lum, F.M.; Lee, W.W.L.; Gabriel, C.; Melchiotti, R.; Andiappan, A.K.; et al. Loss of TLR3 aggravates CHIKV replication and pathology due to an altered virus-specific neutralizing antibody response. EMBO Mol. Med. 2015, 7, 24–41. [Google Scholar] [CrossRef]
- Esen, N.; Blakely, P.K.; Rainey-Barger, E.K.; Irani, D.N. Complexity of the microglial activation pathways that drive innate host responses during lethal alphavirus encephalitis in mice. ASN Neuro 2012, 4, 207–221. [Google Scholar] [CrossRef]
- McKimmie, C.S.; Johnson, N.; Fooks, A.R.; Fazakerley, J.K. Viruses selectively upregulate Toll-like receptors in the central nervous system. Biochem. Biophys. Res. Commun. 2005, 336, 925–933. [Google Scholar] [CrossRef]
- Priya, R.; Patro, I.K.; Parida, M.M. TLR3 mediated innate immune response in mice brain following infection with Chikungunya virus. Virus Res. 2014, 189, 194–205. [Google Scholar] [CrossRef]
- McKimmie, C.S.; Fazakerley, J.K. In response to pathogens, glial cells dynamically and differentially regulate Toll-like receptor gene expression. J. Neuroimmunol. 2005, 169, 116–125. [Google Scholar] [CrossRef]
- Wollish, A.C.; Ferris, M.T.; Blevins, L.K.; Loo, Y.M.; Gale, M.; Heise, M.T. An attenuating mutation in a neurovirulent Sindbis virus strain interacts with the IPS-1 signaling pathway in vivo. Virology 2013, 435, 269–280. [Google Scholar] [CrossRef]
- Pryke, K.M.; Abraham, J.; Sali, T.M.; Gall, B.J.; Archer, I.; Liu, A.; Bambina, S.; Baird, J.; Gough, M.; Chakhtoura, M.; et al. A Novel Agonist of the TRIF Pathway Induces a Cellular State Refractory to Replication of Zika, Chikungunya, and Dengue Viruses. mBio 2017, 8. [Google Scholar] [CrossRef]
- Rudd, P.A.; Wilson, J.; Gardner, J.; Larcher, T.; Babarit, C.; Le, T.T.; Anraku, I.; Kumagai, Y.; Loo, Y.M.; Gale, M.; et al. Interferon Response Factors 3 and 7 Protect against Chikungunya Virus Hemorrhagic Fever and Shock. J. Virol. 2012, 86, 9888–9898. [Google Scholar] [CrossRef]
- Neighbours, L.M.; Long, K.; Whitmore, A.C.; Heise, M.T. Myd88-Dependent Toll-Like Receptor 7 Signaling Mediates Protection from Severe Ross River Virus-Induced Disease in Mice. J. Virol. 2012, 86, 10675–10685. [Google Scholar] [CrossRef]
- Akhrymuk, I.; Frolov, I.; Frolova, E.I. Both RIG-I and MDA5 detect alphavirus replication in concentration-dependent mode. Virology 2016, 487, 230–241. [Google Scholar] [CrossRef]
- Nikonov, A.; Molder, T.; Sikut, R.; Kiiver, K.; Mannik, A.; Toots, U.; Lulla, A.; Lulla, V.; Utt, A.; Merits, A.; et al. RIG-I and MDA-5 Detection of Viral RNA-dependent RNA Polymerase Activity Restricts Positive-Strand RNA Virus Replication. PLoS Pathog. 2013, 9. [Google Scholar] [CrossRef]
- David, R.Y.S.; Combredet, C.; Sismeiro, O.; Dillies, M.A.; Jagla, B.; Coppee, J.Y.; Mura, M.; Galla, M.G.; Despres, P.; Tangy, F.; et al. Comparative analysis of viral RNA signatures on different RIG-I-like receptors. eLife 2016, 5. [Google Scholar] [CrossRef]
- Olagnier, D.; Scholte, F.E.M.; Chiang, C.; Albulescu, I.C.; Nichols, C.; He, Z.; Lin, R.T.; Snijder, E.J.; van Hemert, M.J.; Hiscott, J. Inhibition of Dengue and Chikungunya Virus Infections by RIG-I-Mediated Type I Interferon-Independent Stimulation of the Innate Antiviral Response. J. Virol. 2014, 88, 4180–4194. [Google Scholar] [CrossRef]
- Liu, X.; Mutso, M.; Utt, A.; Lepland, A.; Herrero, L.J.; Taylor, A.; Bettadapura, J.; Rudd, P.A.; Merits, A.; Mahalingam, S. Decreased Virulence of Ross River Virus Harboring a Mutation in the First Cleavage Site of Nonstructural Polyprotein Is Caused by a Novel Mechanism Leading to Increased Production of Interferon-Inducing RNAs. mBio 2018, 9. [Google Scholar] [CrossRef]
- Sali, T.M.; Pryke, K.M.; Abraham, J.; Liu, A.; Archer, I.; Broeckel, R.; Staverosky, J.A.; Smith, J.L.; Al-Shammari, A.; Amsler, L.; et al. Characterization of a Novel Human-Specific STING Agonist that Elicits Antiviral Activity Against Emerging Alphaviruses. PLoS Pathog. 2015, 11. [Google Scholar] [CrossRef]
- White, L.K.; Sali, T.; Alvarado, D.; Gatti, E.; Pierre, P.; Streblow, D.; DeFilippis, V.R. Chikungunya Virus Induces IPS-1-Dependent Innate Immune Activation and Protein Kinase R-Independent Translational Shutoff. J. Virol. 2011, 85, 606–620. [Google Scholar] [CrossRef]
- Schultz, K.L.W.; Troisi, E.M.; Baxter, V.K.; Glowinski, R.; Griffin, D.E. Interferon regulatory factors 3 and 7 have distinct roles in the pathogenesis of alphavirus encephalomyelitis. J. Gen. Virol. 2019, 100, 46–62. [Google Scholar] [CrossRef]
- Lane, W.C.; Dunn, M.D.; Gardner, C.L.; Lam, L.K.M.; Watson, A.M.; Hartman, A.L.; Ryman, K.D.; Klimstra, W.B. The Efficacy of the Interferon Alpha/Beta Response versus Arboviruses Is Temperature Dependent. mBio 2018, 9. [Google Scholar] [CrossRef]
- Prow, N.A.; Tang, B.; Gardner, J.; Le, T.T.; Taylor, A.; Poo, Y.S.; Nakayama, E.; Hirata, T.D.C.; Nakaya, H.I.; Slonchak, A.; et al. Lower temperatures reduce type I interferon activity and promote alphaviral arthritis. PLoS Pathog. 2017, 13. [Google Scholar] [CrossRef]
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.; Rice, C.M. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 2011, 472, 481–485. [Google Scholar] [CrossRef]
- Schoggins, J.W.; MacDuff, D.A.; Imanaka, N.; Gainey, M.D.; Shrestha, B.; Eitson, J.L.; Mar, K.B.; Richardson, R.B.; Ratushny, A.V.; Litvak, V.; et al. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 2014, 505, 691–695. [Google Scholar] [CrossRef]
- Wilson, J.A.; Prow, N.A.; Schroder, W.A.; Ellis, J.J.; Cumming, H.E.; Gearing, L.J.; Poo, Y.S.; Taylor, A.; Hertzog, P.J.; Di Giallonardo, F.; et al. RNA-Seq analysis of chikungunya virus infection and identification of granzyme A as a major promoter of arthritic inflammation. PLoS Pathog. 2017, 13, e1006155. [Google Scholar] [CrossRef]
- Zhang, Y.G.; Burke, C.W.; Ryman, K.D.; Klimstra, W.B. Identification and characterization of interferon-induced proteins that inhibit alphavirus replication. J. Virol. 2007, 81, 11246–11255. [Google Scholar] [CrossRef]
- Ryman, K.D.; White, L.J.; Johnston, R.E.; Klimstra, W.B. Effects of PKR/RNase L-dependent and alternative antiviral pathways on alphavirus replication and pathogenesis. Viral Immunol. 2002, 15, 53–76. [Google Scholar] [CrossRef]
- Nair, S.R.; Abraham, R.; Sundaram, S.; Sreekumar, E. Interferon regulated gene (IRG) expression-signature in a mouse model of chikungunya virus neurovirulence. J. Neurovirol. 2017, 23, 886–902. [Google Scholar] [CrossRef]
- Brehin, A.C.; Casademont, I.; Frenkiel, M.P.; Julier, C.; Sakuntabhai, A.; Despres, P. The large form of human 2′,5′-Oligoadenylate Synthetase (OAS3) exerts antiviral effect against Chikungunya virus. Virology 2009, 384, 216–222. [Google Scholar] [CrossRef]
- Li, Y.Z.; Banerjee, S.; Wang, Y.Y.; Goldstein, S.A.; Dong, B.H.; Gaughan, C.; Silverman, R.H.; Weiss, S.R. Activation of RNase L is dependent on OAS3 expression during infection with diverse human viruses. Proc. Natl. Acad. Sci. USA 2016, 113, 2241–2246. [Google Scholar] [CrossRef]
- Ryman, K.D.; Meier, K.C.; Nangle, E.M.; Ragsdale, S.L.; Korneeva, N.L.; Rhoads, R.E.; MacDonald, M.R.; Klimstra, W.B. Sindbis virus translation is inhibited by a PKR/RNase L-independent effector induced by alpha/beta interferon priming of dendritic cells. J. Virol. 2005, 79, 1487–1499. [Google Scholar] [CrossRef]
- Weiss, C.M.; Trobaugh, D.W.; Sun, C.Q.; Lucas, T.M.; Diamond, M.S.; Ryman, K.D.; Klimstra, W.B. The Interferon-Induced Exonuclease ISG20 Exerts Antiviral Activity through Upregulation of Type I Interferon Response Proteins. mSphere 2018, 3. [Google Scholar] [CrossRef]
- Bick, M.J.; Carroll, J.W.N.; Gao, G.X.; Goff, S.P.; Rice, C.M.; MacDonald, M.R. Expression of the zinc-finger antiviral protein inhibits alphavirus replication. J. Virol. 2003, 77, 11555–11562. [Google Scholar] [CrossRef] [PubMed]
- Li, M.M.H.; Lau, Z.; Cheung, P.; Aguilar, E.G.; Schneider, W.M.; Bozzacco, L.; Molina, H.; Buehler, E.; Takaoka, A.; Rice, C.M.; et al. TRIM25 Enhances the Antiviral Action of Zinc-Finger Antiviral Protein (ZAP). PLoS Pathog. 2017, 13. [Google Scholar] [CrossRef]
- Karki, S.; Li, M.M.H.; Schoggins, J.W.; Tian, S.Y.; Rice, C.M.; MacDonald, M.R. Multiple Interferon Stimulated Genes Synergize with the Zinc Finger Antiviral Protein to Mediate Anti-Alphavirus Activity. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Tesfay, M.Z.; Yin, J.; Gardner, C.L.; Khoretonenko, M.V.; Korneeva, N.L.; Rhoads, R.E.; Ryman, K.D.; Klimstra, W.B. Alpha/beta interferon inhibits cap-dependent translation of viral but not cellular mRNA by a PKR-independent mechanism. J. Virol. 2008, 82, 2620–2630. [Google Scholar] [CrossRef] [PubMed]
- Atasheva, S.; Akhrymuk, M.; Frolova, E.I.; Frolov, I. New PARP Gene with an Anti-Alphavirus Function. J. Virol. 2012, 86, 8147–8160. [Google Scholar] [CrossRef] [PubMed]
- Lenschow, D.J.; Giannakopoulos, N.V.; Gunn, L.J.; Johnston, C.; O’Guin, A.K.; Schmidt, R.E.; Levine, B.; Virgin, H.W. Identification of interferon-stimulated gene 15 as an antiviral molecule during Sindbis virus infection in vivo. J. Virol. 2005, 79, 13974–13983. [Google Scholar] [CrossRef]
- Teng, T.S.; Foo, S.S.; Simamarta, D.; Lum, F.M.; Teo, T.H.; Lulla, A.; Yeo, N.K.W.; Koh, E.G.L.; Chow, A.; Leo, Y.S.; et al. Viperin restricts chikungunya virus replication and pathology. J. Clin. Investig. 2012, 122, 4447–4460. [Google Scholar] [CrossRef]
- Carissimo, G.; Teo, T.H.; Chan, Y.H.; Lee, C.Y.; Lee, B.; Torres-Ruesta, A.; Tan, J.J.; Chua, T.K.; Fong, S.W.; Lum, F.M.; et al. Viperin controls chikungunya virus-specific pathogenic T cell IFNgamma Th1 stimulation in mice. Life Sci Alliance 2019, 2. [Google Scholar] [CrossRef]
- Xu, D.K.; Holko, M.; Sadler, A.J.; Scott, B.; Higashiyama, S.; Berkofsky-Fessler, W.; McConnell, M.J.; Pandolfi, P.P.; Licht, J.D.; Williams, B.R.G. Promyelocytic Leukemia Zinc Finger Protein Regulates Interferon-Mediated Innate Immunity. Immunity 2009, 30, 802–816. [Google Scholar] [CrossRef] [PubMed]
- Lenschow, D.J.; Lai, C.; Frias-Staheli, N.; Giannakopoulos, N.V.; Lutz, A.; Wolff, T.; Osiak, A.; Levine, B.; Schmidt, R.E.; Garcia-Sastre, A.; et al. IFN-stimulated gene 15 functions as a critical antiviral molecule against influenza, herpes, and Sindbis viruses. Proc. Natl. Acad. Sci. USA 2007, 104, 1371–1376. [Google Scholar] [CrossRef]
- Poddar, S.; Hyde, J.L.; Gorman, M.J.; Farzan, M.; Diamond, M.S. The Interferon-Stimulated Gene IFITM3 Restricts Infection and Pathogenesis of Arthritogenic and Encephalitic Alphaviruses. J. Virol. 2016, 90, 8780–8794. [Google Scholar] [CrossRef]
- Ooi, Y.S.; Dube, M.; Kielian, M. BST2/Tetherin Inhibition of Alphavirus Exit. Viruses 2015, 7, 2147–2167. [Google Scholar] [CrossRef]
- Mahauad-Fernandez, W.D.; Jones, P.H.; Okeoma, C.M. Critical role for bone marrow stromal antigen 2 in acute Chikungunya virus infection. J. Gen. Virol. 2014, 95, 2450–2461. [Google Scholar] [CrossRef]
- Assunção-Miranda, I.; Cruz-Oliveira, C.; Poian, A.T. Molecular Mechanisms Involved in the Pathogenesis of Alphavirus-Induced Arthritis. Biomed Res. Int. 2013, 2013, 973516. [Google Scholar] [CrossRef]
- MacDonald, G.H.; Johnston, R.E. Role of Dendritic Cell Targeting in Venezuelan Equine Encephalitis Virus Pathogenesis. J. Virol. 2000, 74, 914–922. [Google Scholar] [CrossRef]
- Benaglio, F.; Vitolo, B.; Scarabelli, M.; Binda, E.; Bugatti, S.; Caporali, R.; Montecucco, C.; Manzo, A. The Draining Lymph Node in Rheumatoid Arthritis: Current Concepts and Research Perspectives. Biomed Res. Int. 2015, 2015, 420251. [Google Scholar] [CrossRef]
- Labadie, K.; Larcher, T.; Joubert, C.; Mannioui, A.; Delache, B.; Brochard, P.; Guigand, L.; Dubreil, L.; Lebon, P.; Verrier, B.; et al. Chikungunya disease in nonhuman primates involves long-term viral persistence in macrophages. J. Clin. Investig. 2010, 120, 894–906. [Google Scholar] [CrossRef]
- Her, Z.; Malleret, B.; Chan, M.; Ong, E.K.S.; Wong, S.-C.; Kwek, D.J.C.; Tolou, H.; Lin, R.T.P.; Tambyah, P.; Rénia, L.; et al. Active Infection of Human Blood Monocytes by Chikungunya Virus Triggers an Innate Immune Response. J. Immunol. 2010, 184, 5903–5913. [Google Scholar] [CrossRef]
- Hazelton, R.A.; Hughes, C.; Aaskov, J.G. The Inflammatory Response in the Synovium of a Patient with Ross River Arbovirus Infection. Aust. N. Z. J. Med. 1985, 15. [Google Scholar] [CrossRef]
- Poo, Y.; Nakaya, H.; Gardner, J.; Larcher, T.; Schroder, W.A.; Le, T.T.; Major, L.D.; Suhrbier, A. CCR2 Deficiency Promotes Exacerbated Chronic Erosive Neutrophil-Dominated Chikungunya Virus Arthritis. J. Virol. 2014, 88, 6862–6872. [Google Scholar] [CrossRef]
- Morrison, T.E.; Oko, L.; Montgomery, S.A.; Whitmore, A.C.; Lotstein, A.R.; Gunn, B.M.; Elmore, S.A.; Heise, M.T. A Mouse Model of Chikungunya Virus–Induced Musculoskeletal Inflammatory Disease Evidence of Arthritis, Tenosynovitis, Myositis, and Persistence. Am. J. Pathol. 2011, 178, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Morrison, T.E.; Whitmore, A.C.; Shabman, R.S.; Lidbury, B.A.; Mahalingam, S.; Heise, M.T. Characterization of Ross River Virus Tropism and Virus-Induced Inflammation in a Mouse Model of Viral Arthritis and Myositis. J. Virol. 2006, 80, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Harley, D.; Sleigh, A.; Ritchie, S. Ross River Virus Transmission, Infection, and Disease: A Cross-Disciplinary Review. Clin. Microbiol. Rev. 2001, 14, 909–932. [Google Scholar] [CrossRef] [PubMed]
- Mateo, L.; Linn, L.M.; McColl, S.R.; Cross, S.; Gardner, J.; Suhrbier, A. An Arthrogenic Alphavirus Induces Monocyte Chemoattractant Protein-1 and Interleukin-8. Intervirology 2000, 43, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Chow, A.; Her, Z.; Ong, E.K.S.; Chen, J.-M.; Dimatatac, F.; Kwek, D.J.C.; Barkham, T.; Yang, H.; Rénia, L.; Leo, Y.-S.; et al. Persistent Arthralgia Induced by Chikungunya Virus Infection is Associated with Interleukin-6 and Granulocyte Macrophage Colony-Stimulating Factor. J. Infect. Dis. 2011, 203, 149–157. [Google Scholar] [CrossRef]
- Kelvin, A.A.; Banner, D.; Silvi, G.; Moro, M.; Spataro, N.; Gaibani, P.; Cavrini, F.; Pierro, A.; Rossini, G.; Cameron, M.J.; et al. Inflammatory Cytokine Expression Is Associated with Chikungunya Virus Resolution and Symptom Severity. PLoS Negl. Trop. Dis. 2011, 5. [Google Scholar] [CrossRef] [PubMed]
- Chaaitanya, I.; Muruganandam, N.; Sundaram, S.G.; Kawalekar, O.; Sugunan, A.P.; Manimunda, S.P.; Ghosal, S.R.; Muthumani, K.; Vijayachari, P. Role of proinflammatory cytokines and chemokines in chronic arthropathy in CHIKV infection. Viral Immunol. 2011, 24, 265–271. [Google Scholar] [CrossRef]
- Teng, T.-S.; Kam, Y.-W.; Lee, B.; Hapuarachchi, H.; Wimal, A.; Ng, L.-C.; Ng, L.F.P. A Systematic Meta-analysis of Immune Signatures in Patients with Acute Chikungunya Virus Infection. J. Infect. Dis. 2015, 211, 1925–1935. [Google Scholar] [CrossRef]
- Tappe, D.; Pérez-Girón, J.; Gómez-Medina, S.; Günther, S.; Muñoz-Fontela, C.; Schmidt-Chanasit, J. Increased Proinflammatory Cytokine Levels in Prolonged Arthralgia in Ross River Virus Infection. Emerg. Infect. Dis. 2017, 23, 702–704. [Google Scholar] [CrossRef]
- Lidbury, B.A.; Rulli, N.E.; Suhrbier, A.; Smith, P.N.; McColl, S.R.; Cunningham, A.L.; Tarkowski, A.; van Rooijen, N.; Fraser, R.J.; Mahalingam, S. Macrophage-Derived Proinflammatory Factors Contribute to the Development of Arthritis and Myositis after Infection with an Arthrogenic Alphavirus. J. Infect. Dis. 2008, 197, 1585–1593. [Google Scholar] [CrossRef]
- Ng, L.F.P.; Chow, A.; Sun, Y.-J.; Kwek, D.J.C.; Lim, P.-L.; Dimatatac, F.; Ng, L.-C.; Ooi, E.-E.; Choo, K.-H.; Her, Z.; et al. IL-1β, IL-6, and RANTES as Biomarkers of Chikungunya Severity. PLoS ONE 2009, 4. [Google Scholar] [CrossRef]
- Tappe, D.; Pérez-Girón, J.; Just-Nübling, G.; Schuster, G.; Gómez-Medina, S.; Günther, S.; Muñoz-Fontela, C.; Schmidt-Chanasit, J. Sustained Elevated Cytokine Levels during Recovery Phase of Mayaro Virus Infection. Emerg. Infect. Dis. 2016, 22, 750–752. [Google Scholar] [CrossRef]
- Thanapati, S.; Das, R.; Tripathy, A.S. Phenotypic and functional analyses of NK and NKT-like populations during the early stages of chikungunya infection. Front. Microbiol. 2015, 6, 895. [Google Scholar] [CrossRef]
- Chang, A.Y.; Tritsch, S.; Reid, S.; Martins, K.; Encinales, L.; Pacheco, N.; Amdur, R.L.; Porras-Ramirez, A.; Rico-Mendoza, A.; Li, G.; et al. The Cytokine Profile in Acute Chikungunya Infection is Predictive of Chronic Arthritis 20 Months Post Infection. Diseases 2018, 6, 95. [Google Scholar] [CrossRef]
- Rulli, N.E.; Guglielmotti, A.; Mangano, G.; Rolph, M.S.; Apicella, C.; Zaid, A.; Suhrbier, A.; Mahalingam, S. Amelioration of alphavirus-induced arthritis and myositis in a mouse model by treatment with bindarit, an inhibitor of monocyte chemotactic proteins. Arthritis Rheum. 2009, 60, 2513–2523. [Google Scholar] [CrossRef]
- Rulli, N.E.; Rolph, M.S.; Srikiatkhachorn, A.; Anantapreecha, S.; Guglielmotti, A.; Mahalingam, S. Protection from Arthritis and Myositis in a Mouse Model of Acute Chikungunya Virus Disease by Bindarit, an Inhibitor of Monocyte Chemotactic Protein-1 Synthesis. J. Infect. Dis. 2011, 204, 1026–1030. [Google Scholar] [CrossRef]
- Fox, J.M.; Roy, V.; Gunn, B.M.; Huang, L.; Edeling, M.A.; Mack, M.; Fremont, D.H.; Doranz, B.J.; Johnson, S.; Alter, G.; et al. Optimal therapeutic activity of monoclonal antibodies against chikungunya virus requires Fc-FcgammaR interaction on monocytes. Sci. Immunol. 2019, 4. [Google Scholar] [CrossRef]
- Linn, M.L.; Mateo, L.; Gardner, J.; Suhrbier, A. Alphavirus-specific cytotoxic T lymphocytes recognize a cross-reactive epitope from the capsid protein and can eliminate virus from persistently infected macrophages. J. Virol. 1998, 72, 5146–5153. [Google Scholar]
- Haist, K.C.; Burrack, K.S.; Davenport, B.J.; Morrison, T.E. Inflammatory monocytes mediate control of acute alphavirus infection in mice. PLoS Pathog. 2017, 13. [Google Scholar] [CrossRef]
- Burrack, K.A.S.; Hawman, D.W.; Jupille, H.J.; Oko, L.; Minor, M.; Shives, K.D.; Gunn, B.M.; Long, K.M.; Morrison, T.E. Attenuating Mutations in nsP1 Reveal Tissue-Specific Mechanisms for Control of Ross River Virus Infection. J. Virol. 2014, 88, 3719–3732. [Google Scholar] [CrossRef]
- Gardner, J.; Anraku, I.; Le, T.T.; Larcher, T.; Major, L.; Roques, P.; Schroder, W.A.; Higgs, S.; Suhrbier, A. Chikungunya Virus Arthritis in Adult Wild-Type Mice. J. Virol. 2010, 84, 8021–8032. [Google Scholar] [CrossRef]
- Teo, T.-H.; Lum, F.-M.; Claser, C.; Lulla, V.; Lulla, A.; Merits, A.; Rénia, L.; Ng, L.F.P. A Pathogenic Role for CD4+ T Cells during Chikungunya Virus Infection in Mice. J. Immunol. 2013, 190, 259–269. [Google Scholar] [CrossRef]
- Burrack, K.S.; Montgomery, S.A.; Homann, D.; Morrison, T.E. CD8+ T cells control Ross River virus infection in musculoskeletal tissues of infected mice. J. Immunol. 2014, 194, 678–689. [Google Scholar] [CrossRef]
- Hoarau, J.-J.; Bandjee, M.-C.; Trotot, P.; Das, T.; Li-Pat-Yuen, G.; Dassa, B.; Denizot, M.; Guichard, E.; Ribera, A.; Henni, T.; et al. Persistent Chronic Inflammation and Infection by Chikungunya Arthritogenic Alphavirus in Spite of a Robust Host Immune Response. J. Immunol. 2010, 184, 5914–5927. [Google Scholar] [CrossRef]
- De Dias, C.; Gois, B.; Lima, V.; Guerra-Gomes, I.; Araújo, J.; de Gomes, J.; Araújo, D.; Medeiros, I.; de Azevedo, F.; Veras, R.; et al. Human CD8 T-cell activation in acute and chronic chikungunya infection. Immunology 2018, 155, 499–504. [Google Scholar] [CrossRef]
- Soden, M.; Vasudevan, H.; Roberts, B. Detection of viral ribonucleic acid and histologic analysis of inflamed synovium in Ross River virus infection. Arthritis Rheum. 2000, 43, 365–369. [Google Scholar] [CrossRef]
- Alves, E.; da, F. Characterization of the immune response following in vitro mayaro and chikungunya viruses (Alphavirus, Togaviridae) infection of mononuclear cells. Virus Res. 2018, 256. [Google Scholar] [CrossRef]
- Lee, W.W.L.; Teo, T.-H.; Her, Z.; Lum, F.-M.; Kam, Y.-W.; Haase, D.; Rénia, L.; Rötzschke, O.; Ng, L.F.P. Expanding Regulatory T Cells Alleviates Chikungunya Virus-Induced Pathology in Mice. J. Virol. 2015, 89, 7893–7904. [Google Scholar] [CrossRef]
- Hawman, D.W.; Fox, J.M.; Ashbrook, A.W.; May, N.A.; Schroeder, K.M.S.; Torres, R.M.; Crowe, J.E.; Dermody, T.S.; Diamond, M.S.; Morrison, T.E. Pathogenic Chikungunya Virus Evades B Cell Responses to Establish Persistence. Cell Rep. 2016, 16, 1326–1338. [Google Scholar] [CrossRef]
- Palmqvist, P.; Persson, E.; Conaway, H.H.; Lerner, U.H. IL-6, Leukemia Inhibitory Factor, and Oncostatin M Stimulate Bone Resorption and Regulate the Expression of Receptor Activator of NF-κB Ligand, Osteoprotegerin, and Receptor Activator of NF-κB in Mouse Calvariae. J. Immunol. 2002, 169, 3353–3362. [Google Scholar] [CrossRef]
- Liu, X.-H.; Kirschenbaum, A.; Yao, S.; Levine, A.C. Cross-talk between the interleukin-6 and prostaglandin E2 signaling systems results in enhancement of osteoclastogenesis through effects on the osteoprotegerin/receptor activator of nuclear factor-κB (RANK) ligand/RANK system. Endocrinology 2004, 146, 1991–1998. [Google Scholar] [CrossRef]
- Takahashi, N.; Maeda, K.; Ishihara, A.; Uehara, S.; Kobayashi, Y. Regulatory mechanism of osteoclastogenesis by RANKL and Wnt signals. Front. Biosci. 2011, 16, 21–30. [Google Scholar] [CrossRef]
- Chen, W.; Foo, S.-S.; Rulli, N.E.; Taylor, A.; Sheng, K.-C.; Herrero, L.J.; Herring, B.L.; Lidbury, B.A.; Li, R.W.; Walsh, N.C.; et al. Arthritogenic alphaviral infection perturbs osteoblast function and triggers pathologic bone loss. Proc. Natl. Acad. Sci. USA 2014, 111, 6040–6045. [Google Scholar] [CrossRef]
- Suhrbier, A.; Linn, M. Clinical and pathologic aspects of arthritis due to Ross River virus and other alphaviruses. Curr. Opin. Rheumatol. 2004. [Google Scholar] [CrossRef]
- Stocks, N.; Selden, S.; Cameron, S. Ross River virus infection. Diagnosis and treatment by general practitioners in South Australia. Aust. Fam. Physician 1997, 26, 710–717. [Google Scholar]
- Bancos, S.; Bernard, M.P.; Topham, D.J.; Phipps, R.P. Ibuprofen and other widely used non-steroidal anti-inflammatory drugs inhibit antibody production in human cells. Cell. Immunol. 2009, 258, 18–28. [Google Scholar] [CrossRef]
- Chen, W.; Foo, S.-S.; Taylor, A.; Lulla, A.; Merits, A.; Hueston, L.; Forwood, M.R.; Walsh, N.C.; Sims, N.A.; Herrero, L.J.; et al. Bindarit, an Inhibitor of Monocyte Chemotactic Protein Synthesis, Protects against Bone Loss Induced by Chikungunya Virus Infection. J. Virol. 2015, 89, 581–593. [Google Scholar] [CrossRef]
- Parsons, L.C.; Mulholland, G.S. Successful Therapy of Interstitial Cystitis with Pentosanpolysulfate. J. Urol. 1987, 138, 513–516. [Google Scholar] [CrossRef]
- Kumagai, K.; Shirabe, S.; Miyata, N.; Murata, M.; Yamauchi, A.; Kataoka, Y.; Niwa, M. Sodium pentosan polysulfate resulted in cartilage improvement in knee osteoarthritis—An open clinical trial. BMC Clin. Pharmacol. 2010, 10, 7. [Google Scholar] [CrossRef]
- Herrero, L.J.; Foo, S.-S.; Sheng, K.-C.; Chen, W.; Forwood, M.R.; Bucala, R.; Mahalingam, S. Pentosan Polysulfate: A Novel Glycosaminoglycan-Like Molecule for Effective Treatment of Alphavirus-Induced Cartilage Destruction and Inflammatory Disease. J. Virol. 2015, 89, 8063–8076. [Google Scholar] [CrossRef]
- Wiens, A.; Venson, R.; Correr, C.J.; Otuki, M.; Pontarolo, R. Meta-analysis of the Efficacy and Safety of Adalimumab, Etanercept, and Infliximab for the Treatment of Rheumatoid Arthritis. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2010, 30, 339–353. [Google Scholar] [CrossRef]
- Zaid, A.; Rulli, N.E.; Rolph, M.S.; Suhrbier, A.; Mahalingam, S. Disease exacerbation by etanercept in a mouse model of alphaviral arthritis and myositis. Arthritis Rheum. 2011, 63, 488–491. [Google Scholar] [CrossRef]
- Taylor, A.; Sheng, K.C.; Herrero, L.J.; Chen, W.; Rulli, N.E.; Mahalingam, S. Methotrexate treatment causes early onset of disease in a mouse model of Ross River virus-induced inflammatory disease through increased monocyte production. PLoS ONE 2013, 8, e71146. [Google Scholar] [CrossRef]
- Ganu, M.A.; Ganu, A.S. Post-chikungunya chronic arthritis—our experience with DMARDs over two year follow up. J. Assoc. Physicians India 2011, 59, 83–86. [Google Scholar]
- Teo, T.-H.; Chan, Y.-H.; Lee, W.W.L.; Lum, F.-M.; Amrun, S.; Her, Z.; Rajarethinam, R.; Merits, A.; Rötzschke, O.; Rénia, L.; et al. Fingolimod treatment abrogates chikungunya virus–induced arthralgia. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Miner, J.J.; Cook, L.E.; Hong, J.P.; Smith, A.M.; Richner, J.M.; Shimak, R.M.; Young, A.R.; Monte, K.; Poddar, S.; Crowe, J.E.; et al. Therapy with CTLA4-Ig and an antiviral monoclonal antibody controls chikungunya virus arthritis. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Cardona-Ospina, J.A.; Villamil-Gomez, W.E.; Jimenez-Canizales, C.E.; Castaneda-Hernandez, D.M.; Rodriguez-Morales, A.J. Estimating the burden of disease and the economic cost attributable to chikungunya, Colombia, 2014. Trans. R. Soc. Trop. Med. Hyg. 2015, 109, 793–802. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mostafavi, H.; Abeyratne, E.; Zaid, A.; Taylor, A. Arthritogenic Alphavirus-Induced Immunopathology and Targeting Host Inflammation as A Therapeutic Strategy for Alphaviral Disease. Viruses 2019, 11, 290. https://doi.org/10.3390/v11030290
Mostafavi H, Abeyratne E, Zaid A, Taylor A. Arthritogenic Alphavirus-Induced Immunopathology and Targeting Host Inflammation as A Therapeutic Strategy for Alphaviral Disease. Viruses. 2019; 11(3):290. https://doi.org/10.3390/v11030290
Chicago/Turabian StyleMostafavi, Helen, Eranga Abeyratne, Ali Zaid, and Adam Taylor. 2019. "Arthritogenic Alphavirus-Induced Immunopathology and Targeting Host Inflammation as A Therapeutic Strategy for Alphaviral Disease" Viruses 11, no. 3: 290. https://doi.org/10.3390/v11030290
APA StyleMostafavi, H., Abeyratne, E., Zaid, A., & Taylor, A. (2019). Arthritogenic Alphavirus-Induced Immunopathology and Targeting Host Inflammation as A Therapeutic Strategy for Alphaviral Disease. Viruses, 11(3), 290. https://doi.org/10.3390/v11030290