Localization of Frog Virus 3 Conserved Viral Proteins 88R, 91R, and 94L

Abstract
1. Introduction
2. Materials and Methods
2.1. Isolation of FV3 DNA
2.2. Cloning FV3 88R, 91R, and 94L into pGEM-T Easy Vector
2.3. Cloning of FV3 88R, 91R, and 94L into a Eukaryotic Expression Vector
2.4. Transfection of Eukaryotic Expression Vector into Eukaryotic Cells
2.5. Indirect Immunofluorescence
3. Results
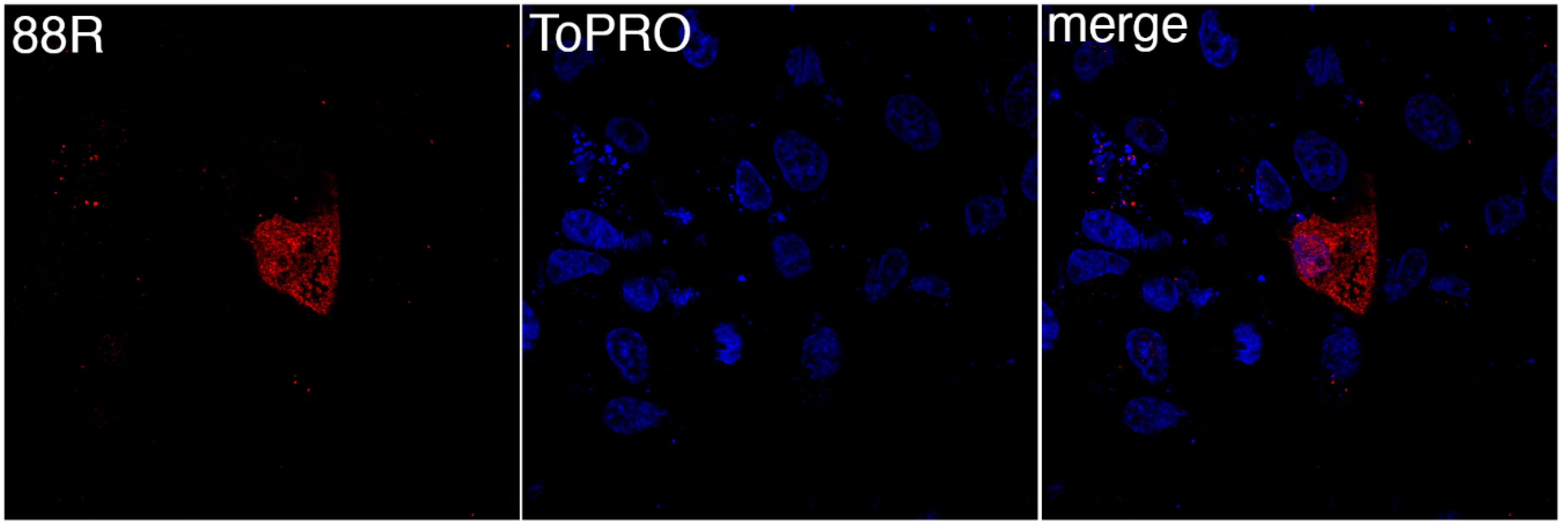
3.1. FV3 ORF 88R Localize to the Cytoplasm of Transfected Cells
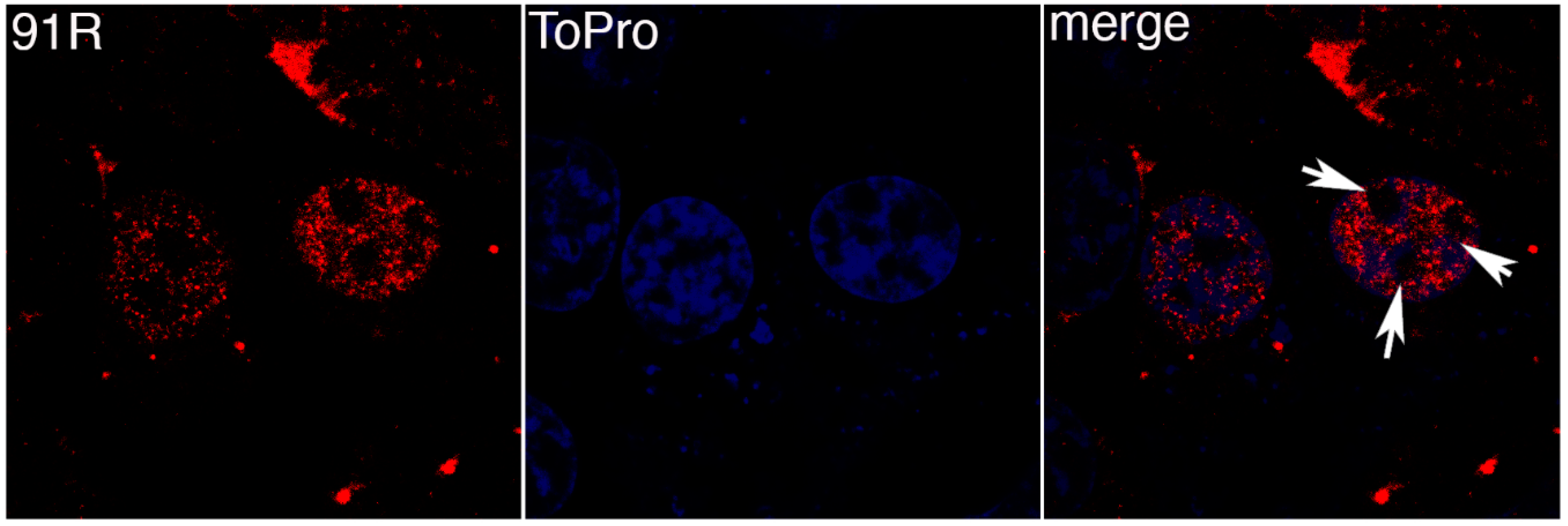
3.2. FV3 ORF 91R Localizes to the Nucleus of Transfected Cells
3.3. FV3 ORF 94L Localizes to the ER in Transfected Cells
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Goorha, R. Frog virus 3 DNA replication occurs in two stages. J. Virol. 1982, 43, 519–528. [Google Scholar] [PubMed]
- He, J.G.; Deng, M.; Weng, S.P.; Li, Z.; Zhou, S.Y.; Long, Q.X.; Wang, X.Z.; Chan, S.M. Complete genome analysis of the mandarin fish infectious spleen and kidney necrosis iridovirus. Virology 2001, 291, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Goorha, R.; Murti, K.G. The genome of frog virus 3, an animal DNA virus, is circularly permuted and terminally redundant. Proc. Natl. Acad. Sci. USA 1982, 79, 248–252. [Google Scholar] [CrossRef] [PubMed]
- Jakob, N.J.; Muller, K.; Bahr, U.; Darai, G. Analysis of the first complete DNA sequence of an invertebrate iridovirus: Coding strategy of the genome of chilo iridescent virus. Virology 2001, 286, 182–196. [Google Scholar] [CrossRef] [PubMed]
- Williams, T. The iridoviruses. Adv. Virus Res. 1996, 46, 345–412. [Google Scholar] [PubMed]
- Chinchar, V.G.; Waltzek, T.B.; Subramaniam, K. Ranaviruses and other members of the family iridoviridae: Their place in the virosphere. Virology 2017, 511, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Chinchar, V.G.; Hick, P.; Ince, I.A.; Jancovich, J.K.; Marschang, R.; Qin, Q.; Subramaniam, K.; Waltzek, T.B.; Whittington, R.; Williams, T.; et al. Ictv virus taxonomy profile: Iridoviridae. J. Gen. Virol. 2017, 98, 890–891. [Google Scholar] [CrossRef]
- Chinchar, V.G.; Yu, K.H.; Jancovich, J.K. The molecular biology of frog virus 3 and other iridoviruses infecting cold-blooded vertebrates. Viruses 2011, 3, 1959–1985. [Google Scholar] [CrossRef]
- Eaton, H.E.; Metcalf, J.; Penny, E.; Tcherepanov, V.; Upton, C.; Brunetti, C.R. Comparative genomic analysis of the family iridoviridae: Re-annotating and defining the core set of iridovirus genes. Virol. J. 2007, 4, 11. [Google Scholar] [CrossRef]
- Delhon, G.; Tulman, E.R.; Afonso, C.L.; Lu, Z.; Becnel, J.J.; Moser, B.A.; Kutish, G.F.; Rock, D.L. Genome of invertebrate iridescent virus type 3 (mosquito iridescent virus). J. Virol. 2006, 80, 8439–8449. [Google Scholar] [CrossRef]
- Gubser, C.; Hue, S.; Kellam, P.; Smith, G.L. Poxvirus genomes: A phylogenetic analysis. J. Gen. Virol. 2004, 85, 105–117. [Google Scholar] [CrossRef]
- Iyer, L.M.; Aravind, L.; Koonin, E.V. Common origin of four diverse families of large eukaryotic DNA viruses. J. Virol. 2001, 75, 11720–11734. [Google Scholar] [CrossRef]
- McGeoch, D.J. Molecular evolution of the gamma-herpesvirinae. Philos. Trans. R. Soc. Lond B Biol. Sci. 2001, 356, 421–435. [Google Scholar] [CrossRef]
- Song, W.J.; Qin, Q.W.; Qiu, J.; Huang, C.H.; Wang, F.; Hew, C.L. Functional genomics analysis of singapore grouper iridovirus: Complete sequence determination and proteomic analysis. J. Virol. 2004, 78, 12576–12590. [Google Scholar] [CrossRef]
- Tan, W.G.; Barkman, T.J.; Gregory Chinchar, V.; Essani, K. Comparative genomic analyses of frog virus 3, type species of the genus ranavirus (family iridoviridae). Virology 2004, 323, 70–84. [Google Scholar] [CrossRef]
- Zhang, Q.; Gui, J.F. Virus genomes and virus-host interactions in aquaculture animals. Sci. China Life Sci. 2015, 58, 156–169. [Google Scholar] [CrossRef]
- Long, F.W.; Liang, S.Y.; Liu, Z.J.; Chen, Y.; Tu, Z.D.; Shi, Y.J.; Bao, J.; Gong, J. Augmentor of liver regeneration ameliorates renal tubular epithelial cell injury after rat liver transplantation. Transplant Proc. 2008, 40, 2696–2699. [Google Scholar] [CrossRef]
- Su, H.P.; Lin, D.Y.; Garboczi, D.N. The structure of g4, the poxvirus disulfide oxidoreductase essential for virus maturation and infectivity. J. Virol. 2006, 80, 7706–7713. [Google Scholar] [CrossRef]
- Senkevich, T.G.; White, C.L.; Weisberg, A.; Granek, J.A.; Wolffe, E.J.; Koonin, E.V.; Moss, B. Expression of the vaccinia virus a2.5l redox protein is required for virion morphogenesis. Virology 2002, 300, 296–303. [Google Scholar] [CrossRef]
- Senkevich, T.G.; White, C.L.; Koonin, E.V.; Moss, B. A viral member of the erv1/alr protein family participates in a cytoplasmic pathway of disulfide bond formation. Proc. Natl. Acad. Sci. USA 2000, 97, 12068–12073. [Google Scholar] [CrossRef]
- Lotti, L.V.; Mottola, G.; Torrisi, M.R.; Bonatti, S. A different intracellular distribution of a single reporter protein is determined at steady state by kkxx or kdel retrieval signals. J. Biol. Chem. 1999, 274, 10413–10420. [Google Scholar] [CrossRef]
- Akagi, S.; Yamamoto, A.; Yoshimori, T.; Masaki, R.; Ogawa, R.; Tashiro, Y. Distribution of protein disulfide isomerase in rat hepatocytes. J. Histochem. Cytochem. 1988, 36, 1533–1542. [Google Scholar] [CrossRef]
- Senkevich, T.G.; Koonin, E.V.; Bugert, J.J.; Darai, G.; Moss, B. The genome of molluscum contagiosum virus: Analysis and comparison with other poxviruses. Virology 1997, 233, 19–42. [Google Scholar] [CrossRef]
- White, C.L.; Senkevich, T.G.; Moss, B. Vaccinia virus g4l glutaredoxin is an essential intermediate of a cytoplasmic disulfide bond pathway required for virion assembly. J. Virol. 2002, 76, 467–472. [Google Scholar] [CrossRef]
- Lewis, T.; Zsak, L.; Burrage, T.G.; Lu, Z.; Kutish, G.F.; Neilan, J.G.; Rock, D.L. An african swine fever virus erv1-alr homologue, 9gl, affects virion maturation and viral growth in macrophages and viral virulence in swine. J. Virol. 2000, 74, 1275–1285. [Google Scholar] [CrossRef]
- Majji, S.; Thodima, V.; Sample, R.; Whitley, D.; Deng, Y.; Mao, J.; Chinchar, V.G. Transcriptome analysis of frog virus 3, the type species of the genus ranavirus, family iridoviridae. Virology 2009, 391, 293–303. [Google Scholar] [CrossRef]
- Husain, M.; Weisberg, A.S.; Moss, B. Existence of an operative pathway from the endoplasmic reticulum to the immature poxvirus membrane. Proc. Natl. Acad. Sci. USA 2006, 103, 19506–19511. [Google Scholar] [CrossRef]
- Netherton, C.; Rouiller, I.; Wileman, T. The subcellular distribution of multigene family 110 proteins of african swine fever virus is determined by differences in c-terminal kdel endoplasmic reticulum retention motifs. J. Virol. 2004, 78, 3710–3721. [Google Scholar] [CrossRef]
- Tolonen, N.; Doglio, L.; Schleich, S.; Krijnse Locker, J. Vaccinia virus DNA replication occurs in endoplasmic reticulum-enclosed cytoplasmic mini-nuclei. Mol. Biol. Cell 2001, 12, 2031–2046. [Google Scholar] [CrossRef]
- Ring, B.A.; Ferreira Lacerda, A.; Drummond, D.J.; Wangen, C.; Eaton, H.E.; Brunetti, C.R. Frog virus 3 open reading frame 97r localizes to the endoplasmic reticulum and induces nuclear invaginations. J. Virol. 2013, 87, 9199–9207. [Google Scholar] [CrossRef]
- Eaton, H.E.; Ferreira Lacerda, A.; Desrochers, G.; Metcalf, J.; Angers, A.; Brunetti, C.R. Cellular litaf interacts with frog virus 3 75l protein and alters its subcellular localization. J. Virol. 2013, 87, 716–723. [Google Scholar] [CrossRef]
- Eaton, H.E.; Metcalf, J.; Brunetti, C.R. Expression of frog virus 3 genes is impaired in mammalian cell lines. Virol. J. 2008, 5, 83. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Name | Sequence |
|---|---|
| 88R-F | 5′-AAGCTTAAAATGCACGGTTGCAATTG-3′ |
| 88R-R | 5′-CTCGAGGTTAAAAGTGCTCGTATTTG-3′ |
| 91R-F | 5′-AAGCTTAACATGGCAAACTTTGTGAC-3′ |
| 91R-R | 5′-CTCGAGGGCTCTGACCACAAACAG-3′ |
| 94L-F | 5′-AAGCTTGCAATGGATCCAGAAGGAATG-3′ |
| 94L-R | 5′-CTCGAGCAGCACCTTTCTCAGGTAC-3′ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Penny, E.; Brunetti, C.R. Localization of Frog Virus 3 Conserved Viral Proteins 88R, 91R, and 94L. Viruses 2019, 11, 276. https://doi.org/10.3390/v11030276
Penny E, Brunetti CR. Localization of Frog Virus 3 Conserved Viral Proteins 88R, 91R, and 94L. Viruses. 2019; 11(3):276. https://doi.org/10.3390/v11030276
Chicago/Turabian StylePenny, Emily, and Craig R. Brunetti. 2019. "Localization of Frog Virus 3 Conserved Viral Proteins 88R, 91R, and 94L" Viruses 11, no. 3: 276. https://doi.org/10.3390/v11030276
APA StylePenny, E., & Brunetti, C. R. (2019). Localization of Frog Virus 3 Conserved Viral Proteins 88R, 91R, and 94L. Viruses, 11(3), 276. https://doi.org/10.3390/v11030276