Characterization of the Filovirus-Resistant Cell Line SH-SY5Y Reveals Redundant Role of Cell Surface Entry Factors

 , ,
, ,  , ,
, ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Material and Methods
2.1. Cell Lines
2.2. DNA Plasmid Constructs
2.3. Ebolavirus
2.4. Reagents and Antibodies
2.5. Pseudovirion Production and Transduction
2.6. Authentic Filovirus Infection
2.7. rVSVΔG-EBOVGP Infection
2.8. Flow Cytometry
2.9. Expression of EBOV GP1-Fc Fusion Protein
2.10. Western Blot
2.11. RNA Extraction and Microarray Analysis
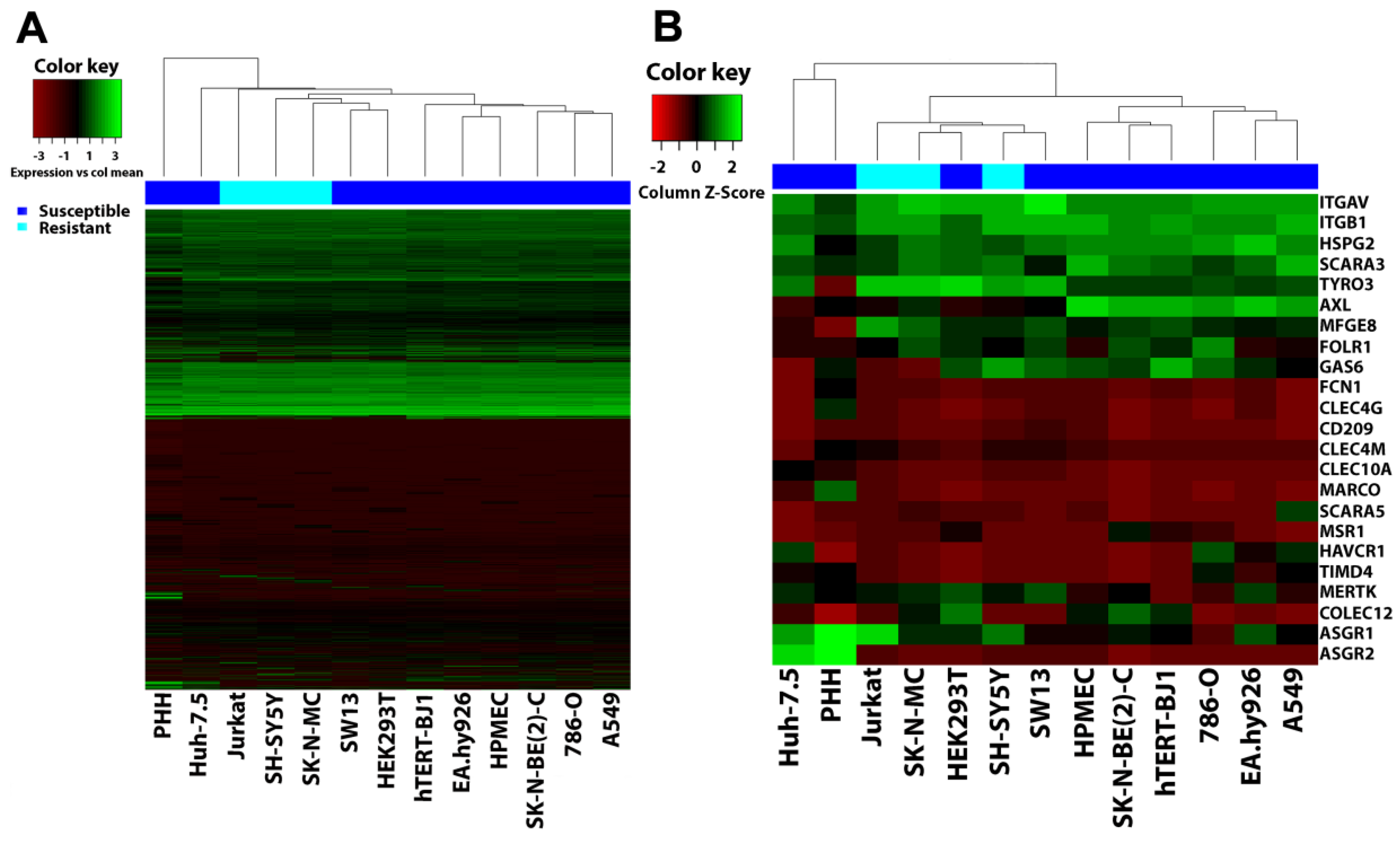
2.12. Hierarchical Clustering Analysis (HCA)
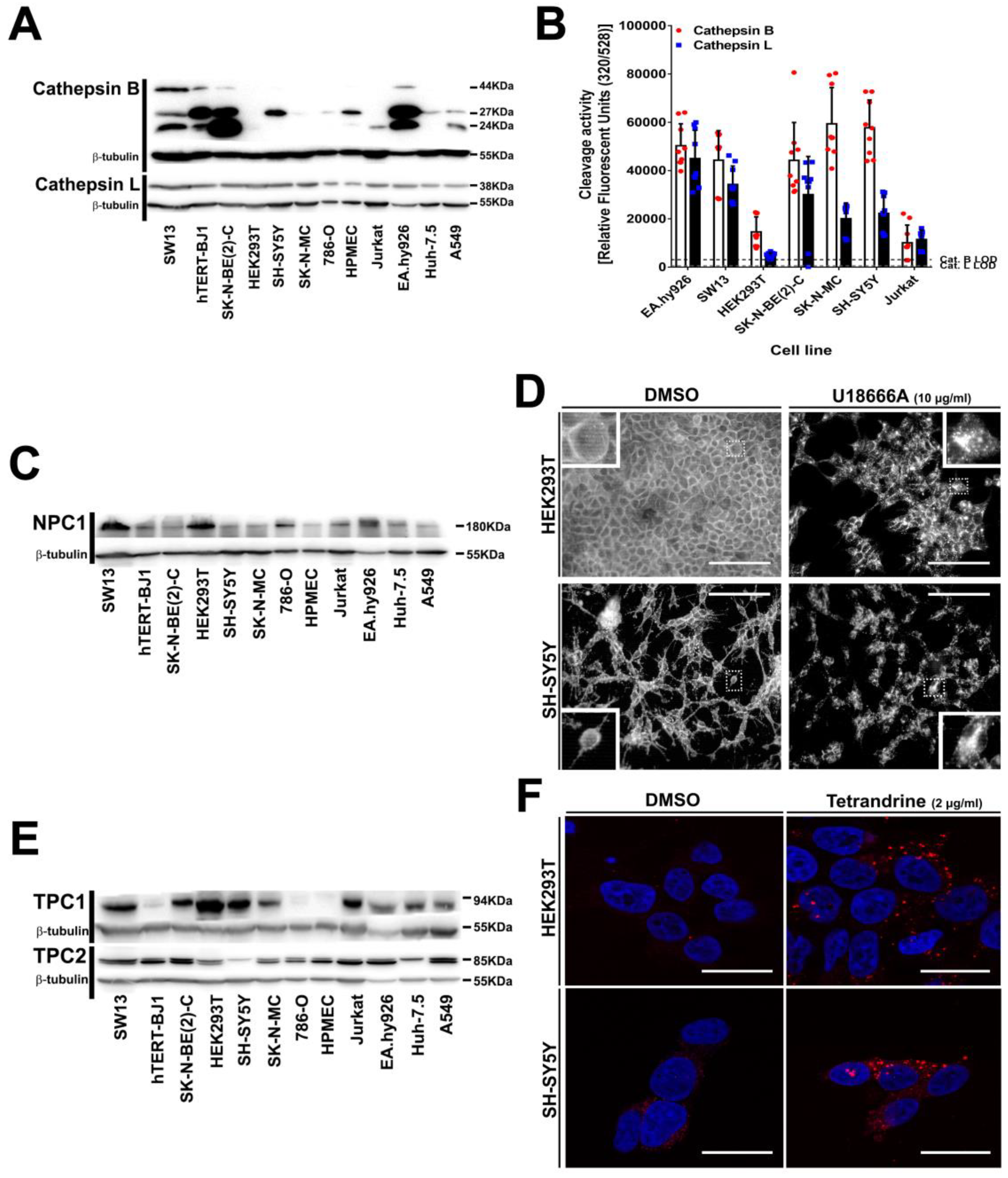
2.13. Cathepsin B and L Activity Assay
2.14. Cholesterol and EGF Endosomal Accumulation Assays
2.15. Light Microscopy
2.16. Polyethylene Glycol (PEG) Mediated Cell–Cell Fusion
2.17. Statistical Analysis
3. Results
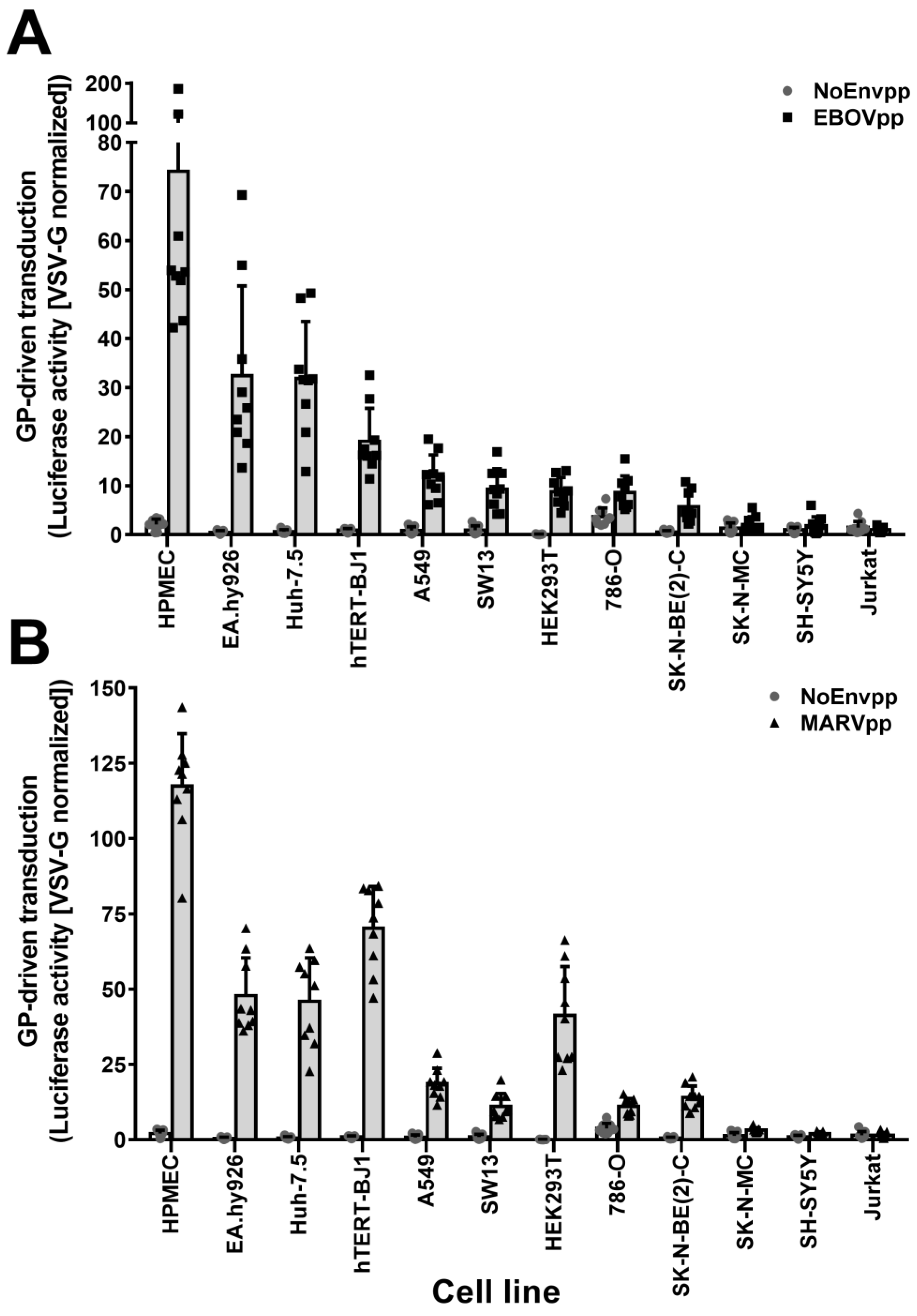
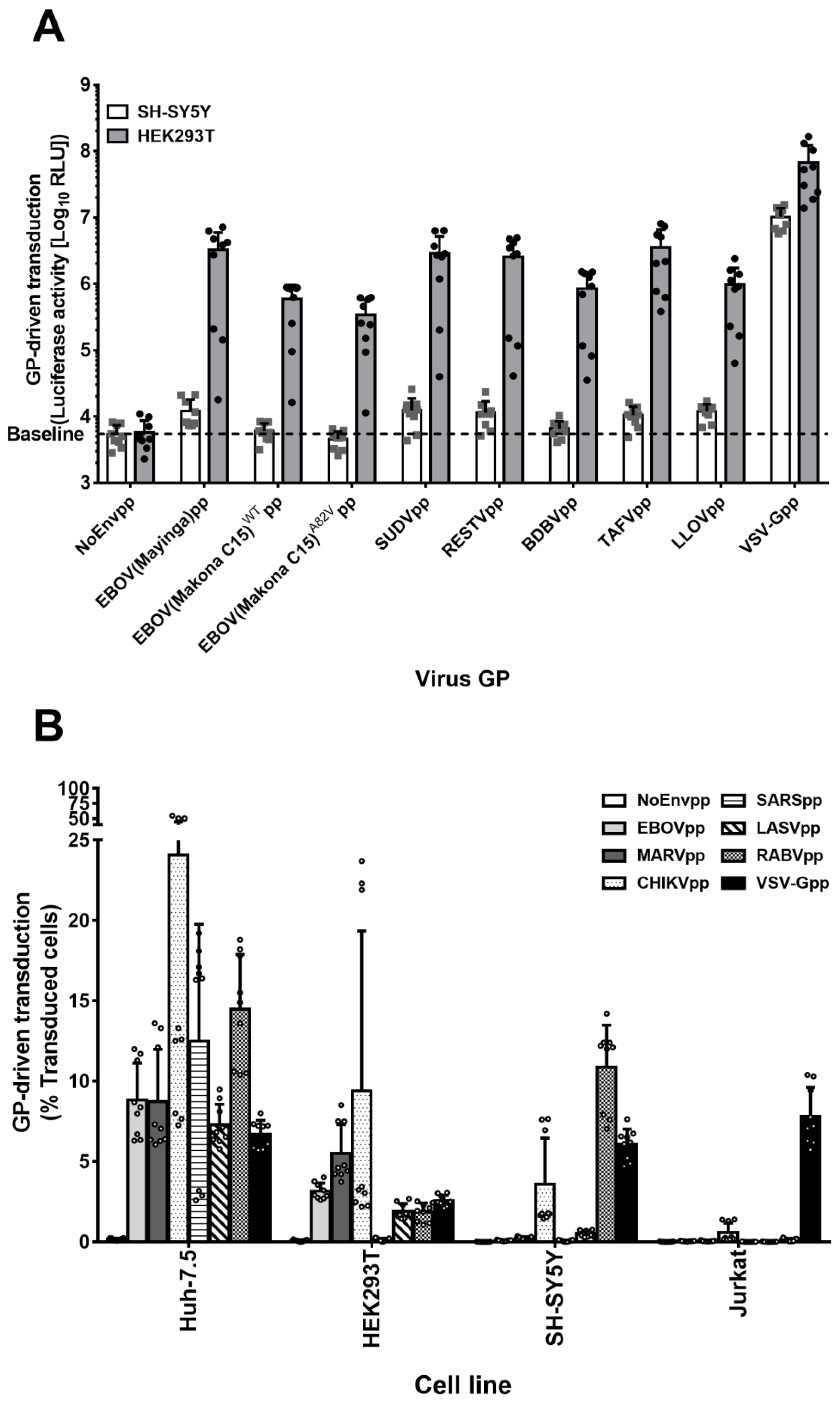
3.1. SH-SY5Y and SK-N-MC Cells Are Resistant to Filovirus GP-Driven Lentiviral Transduction
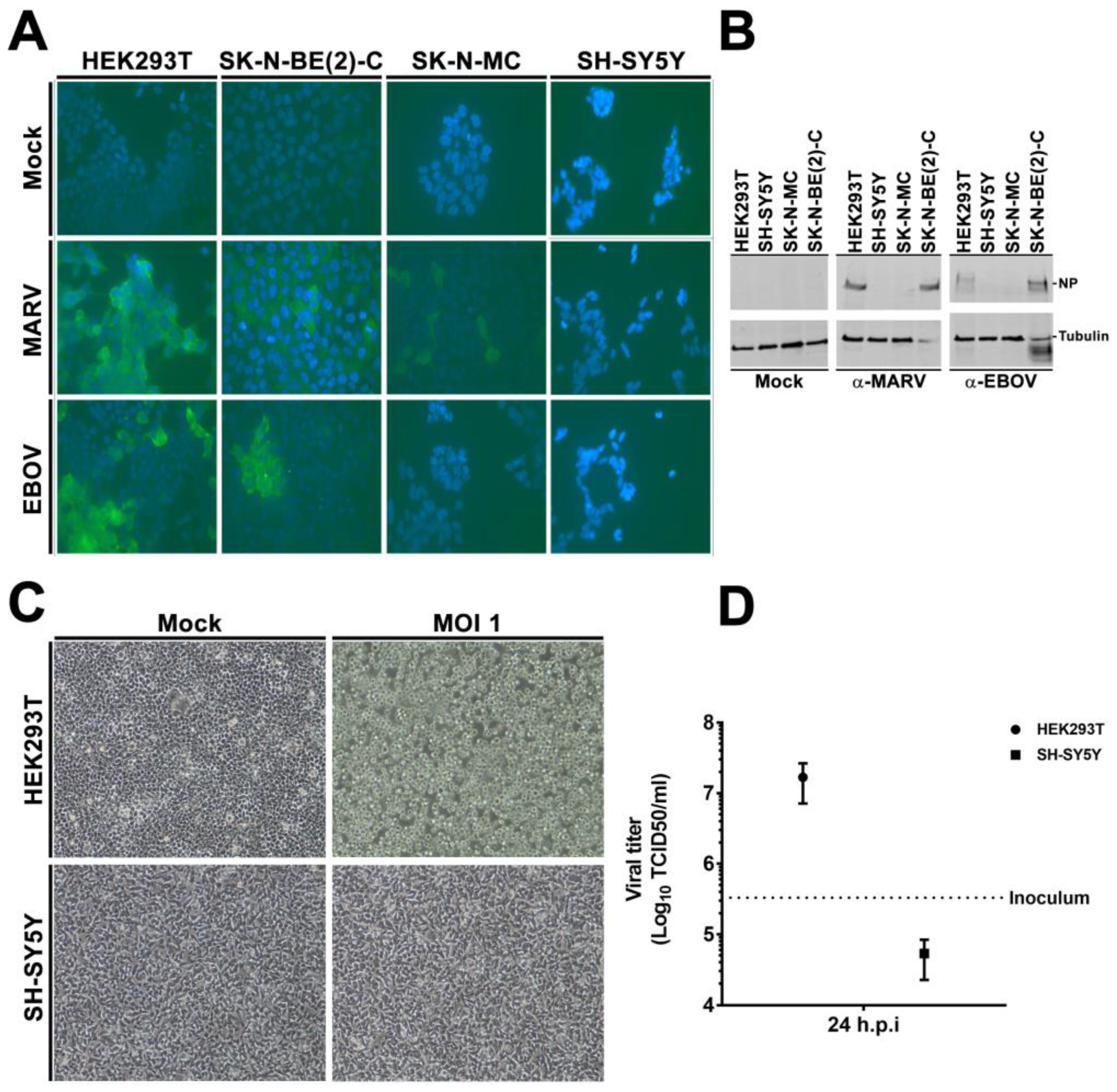
3.2. rVSVΔG-EBOV-GP and Authentic Filovirus Cannot Infect SH-SY5Y Cells
3.3. SH-SY5Y Cells Can Be Transduced by Many Viral Envelope Proteins but Not by GP of Filoviruses
3.4. SH-SY5Y Cells Do Not Express a Dominant Restriction Factor
3.5. Intracellular Filovirus Entry Factors are Expressed and Functional in SH-SY5Y Cells
3.6. Filovirus Resistance Is Neither Explained by SH-SY5Y Cells’ Transcriptome nor by the Expression Profile of Filovirus Attachment Factors
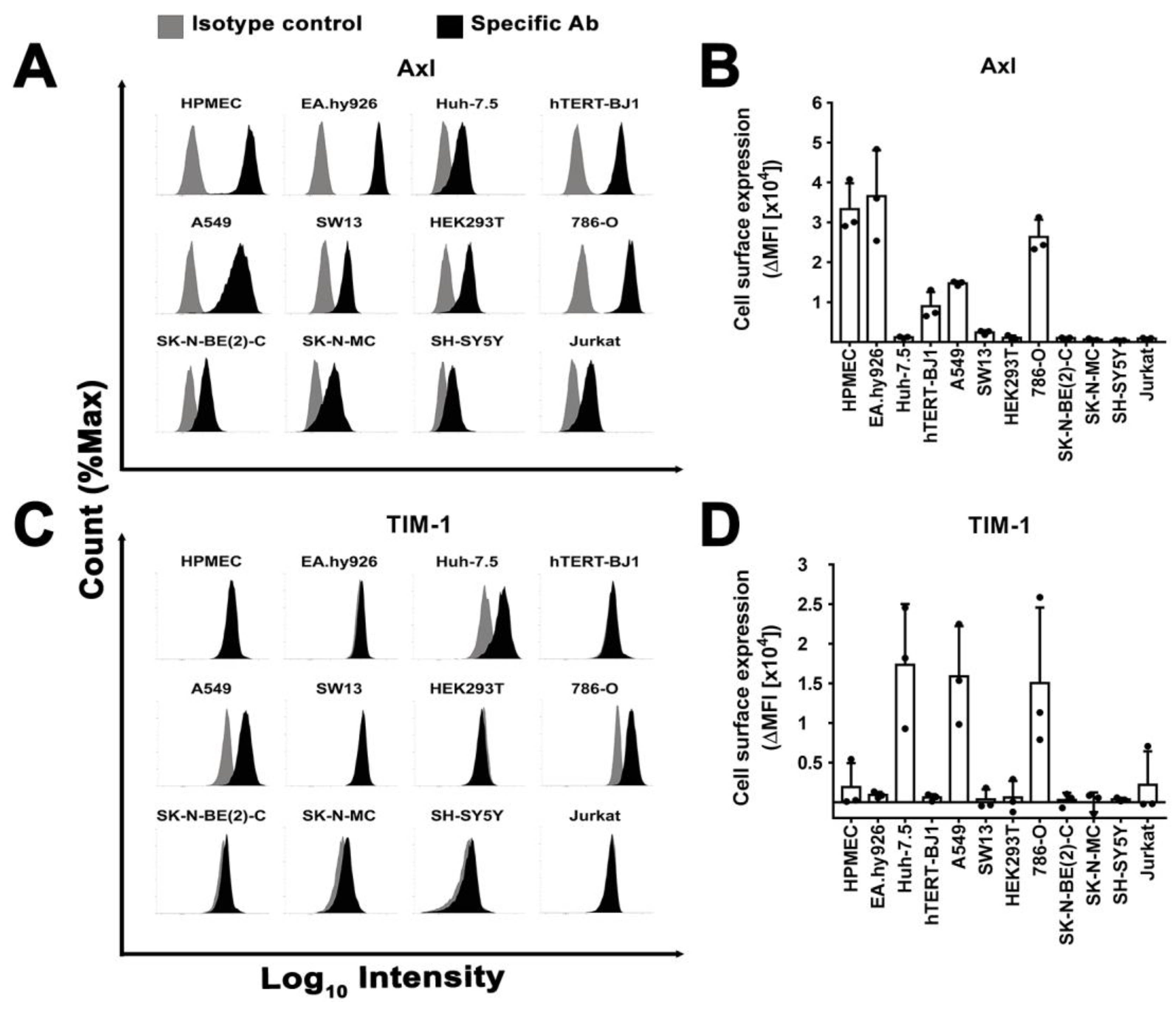
3.7. Surface Expression of Plasma Membrane Filovirus Entry Factors Does Not Correlate with Susceptibility
3.8. Attachment Limits GP-Driven Entry into SH-SY5Y Cells
3.9. Diverse Cell Surface Factors Can Overcome the Block to Filovirus GP-Driven Cell Entry in SH-SY5Y Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kuhn, J.H.; Becker, S.; Ebihara, H.; Geisbert, T.W.; Johnson, K.M.; Kawaoka, Y.; Lipkin, W.I.; Negredo, A.I.; Netesov, S.V.; Nichol, S.T.; et al. Proposal for a revised taxonomy of the family Filoviridae: Classification, names of taxa and viruses, and virus abbreviations. Arch. Virol. 2010, 155, 2083–2103. [Google Scholar] [CrossRef]
- Siegert, R.; Shu, H.L.; Slenczka, H.L.; Peters, D.; Muller, G. The aetiology of an unknown human infection transmitted by monkeys (preliminary communication). Ger. Med. Mon. 1968, 13, 1–2. [Google Scholar] [PubMed]
- Bowen, E.T.W.; Lloyd, G.; Harris, W.J.; Platt, G.S.; Baskerville, A.; Vella, E.E. Viral haemorraghic fever in southern Sudan and northern Zaire. Preliminary Studies on the Aetiological Agent. Lancet 1977, 309, 571–573. [Google Scholar] [CrossRef]
- CDC. Outbreaks Chronology: Ebola Virus Disease|Ebola Hemorrhagic Fever|CDC. 2018. Available online: https://www.cdc.gov/vhf/ebola/history/chronology.html.Cdc-Ebola (accessed on 13 August 2018).
- CDC. Outbreaks Chronology: Marburg Hemorrhagic Fever. 2018. Available online: https://www.cdc.gov/vhf/marburg/outbreaks/chronology.html (accessed on 13 August 2018).
- Warfield, K.L.; Bradfute, S.B.; Wells, J.; Lofts, L.; Cooper, M.T.; Alves, D.A.; Reed, D.K.; VanTongeren, S.A.; Mech, C.A.; Bavari, S. Development and characterization of a mouse model for Marburg hemorrhagic fever. J. Virol. 2009, 83, 6404–6415. [Google Scholar] [CrossRef] [PubMed]
- Volchkov, V.E.; Chepurnov, A.A.; Volchkova, V.A.; Ternovoj, V.A.; Klenk, H.D. Molecular characterization of guinea pig-adapted variants of Ebola virus. Virology 2000, 277, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Towner, J.S.; Amman, B.R.; Sealy, T.K.; Reeder Carroll, S.A.; Comer, J.A.; Kemp, A.; Swanepoel, R.; Paddock, C.D.; Balinandi, S.; Khristova, M.L.; et al. Isolation of genetically diverse Marburg viruses from Egyptian fruit bats. PLoS Pathog. 2009, 5, e1000536. [Google Scholar] [CrossRef] [PubMed]
- Allela, L.; Bourry, O.; Pouillot, R.; Délicat, A.; Yaba, P.; Kumulungui, B.; Rouquet, P.; Gonzalez, J.P.; Leroy, E.M. Ebola virus antibody prevalence in dogs and human risk. Emerg. Infect. Dis. 2005, 11, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Leroy, E.M.; Rouquet, P.; Formenty, P.; Souquière, S.; Kilbourne, A.; Froment, J.-M.; Bermejo, M.; Smit, S.; Karesh, W.; Swanepoel, R.; et al. Multiple Ebola virus transmission events and rapid decline of central African wildlife. Science 2004, 303, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Barrette, R.W.; Metwally, S.A.; Rowland, J.M.; Xu, L.; Zaki, S.R.; Nichol, S.T.; Rollin, P.E.; Towner, J.S.; Shieh, W.-J.; Batten, B.; et al. Discovery of Swine as a Host for the Reston ebolavirus. Source Sci. New Ser. 2009, 325, 204–206. [Google Scholar] [CrossRef]
- Cross, R.W.; Mire, C.E.; Borisevich, V.; Geisbert, J.B.; Fenton, K.A.; Geisbert, T.W. The Domestic Ferret (Mustela putorius furo) as a Lethal Infection Model for 3 Species of Ebolavirus. J. Infect. Dis. 2016, 214, 565–569. [Google Scholar] [CrossRef]
- Yang, Z.Y.; Delgado, R.; Xu, L.; Todd, R.F.; Nabel, E.G.; Sanchez, A.; Nabel, G.J. Distinct cellular interactions of secreted and transmembrane Ebola virus glycoproteins. Science 1998, 279, 1034–1037. [Google Scholar] [CrossRef] [PubMed]
- Geisbert, T.W.; Hensley, L.E.; Gibb, T.R.; Steele, K.E.; Jaax, N.K.; Jahrling, P.B. Apoptosis Induced In Vitro and In Vivo During Infection by Ebola and Marburg Viruses. Lab. Investig. 2000, 80, 171–186. [Google Scholar] [CrossRef]
- Ryabchikova, E.I.; Kolesnikova, L.V.; Luchko, S.V. An analysis of features of pathogenesis in two animal models of Ebola virus infection. J. Infect. Dis. 1999, 179, S199–S202. [Google Scholar] [CrossRef]
- Wool-Lewis, R.J.; Bates, P. Characterization of Ebola virus entry by using pseudotyped viruses: Identification of receptor-deficient cell lines. J. Virol. 1998, 72, 3155–3160. [Google Scholar]
- Chan, S.Y.; Speck, R.F.; Ma, M.C.; Goldsmith, M.A. Distinct Mechanisms of Entry by Envelope Glycoproteins of Marburg and Ebola (Zaire) Viruses. J. Virol. 2000, 74, 4933–4937. [Google Scholar] [CrossRef]
- Dube, D.; Schornberg, K.L.; Stantchev, T.S.; Bonaparte, M.I.; Delos, S.E.; Bouton, A.H.; Broder, C.C.; White, J.M. Cell adhesion promotes Ebola virus envelope glycoprotein-mediated binding and infection. J. Virol. 2008, 82, 7238–7242. [Google Scholar] [CrossRef]
- Dube, D.; Schornberg, K.L.; Shoemaker, C.J.; Delos, S.E.; Stantchev, T.S.; Clouse, K.A.; Broder, C.C.; White, J.M. Cell adhesion-dependent membrane trafficking of a binding partner for the ebolavirus glycoprotein is a determinant of viral entry. Proc. Natl. Acad. Sci. USA 2010, 107, 16637–16642. [Google Scholar] [CrossRef]
- Feldmann, H.; Geisbert, T.W. Ebola haemorrhagic fever. Lancet 2011, 377, 849–862. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Hensley, L.E. Ebola virus: New insights into disease aetiopathology and possible therapeutic interventions. Expert Rev. Mol. Med. 2004, 6, 1–24. [Google Scholar] [CrossRef]
- Takada, A.; Robison, C.; Goto, H.; Sanchez, A.; Murti, K.G.; Whitt, M.A.; Kawaoka, Y. A system for functional analysis of Ebola virus glycoprotein. Proc. Natl. Acad. Sci. USA 1997, 94, 14764–14769. [Google Scholar] [CrossRef]
- Jemielity, S.; Wang, J.J.; Chan, Y.K.; Ahmed, A.A.; Li, W.; Monahan, S.; Bu, X.; Farzan, M.; Freeman, G.J.; Umetsu, D.T.; et al. TIM-family Proteins Promote Infection of Multiple Enveloped Viruses through Virion-associated Phosphatidylserine. PLoS Pathog. 2013, 9, e1003232. [Google Scholar] [CrossRef]
- Gramberg, T.; Hofmann, H.; Möller, P.; Lalor, P.F.; Marzi, A.; Geier, M.; Krumbiegel, M.; Winkler, T.; Kirchhoff, F.; Adams, D.H.; et al. LSECtin interacts with filovirus glycoproteins and the spike protein of SARS coronavirus. Virology 2005, 340, 224–236. [Google Scholar] [CrossRef]
- Takada, A.; Fujioka, K.; Tsuiji, M.; Morikawa, A.; Higashi, N.; Ebihara, H.; Kobasa, D.; Feldmann, H.; Irimura, T.; Kawaoka, Y. Human Macrophage C-Type Lectin Specific for Galactose and N-Acetylgalactosamine Promotes Filovirus Entry. J. Virol. 2004, 78, 2943–2947. [Google Scholar] [CrossRef]
- Simmons, G.; Reeves, J.D.; Grogan, C.C.; Vandenberghe, L.H.; Baribaud, F.; Whitbeck, J.C.; Burke, E.; Buchmeier, M.J.; Soilleux, E.J.; Riley, J.L.; et al. DC-SIGN and DC-SIGNR bind ebola glycoproteins and enhance infection of macrophages and endothelial cells. Virology 2003, 305, 115–123. [Google Scholar] [CrossRef]
- Alvarez, C.P.; Lasala, F.; Carrillo, J.; Muñiz, O.; Corbí, A.L.; Delgado, R. C-type lectins DC-SIGN and L-SIGN mediate cellular entry by Ebola virus in cis and in trans. J. Virol. 2002, 76, 6841–6844. [Google Scholar] [CrossRef]
- Becker, S.; Spiess, M.; Klenk, H.D. The asialoglycoprotein receptor is a potential liver-specific receptor for Marburg virus. J. Gen. Virol. 1995, 76, 393–399. [Google Scholar] [CrossRef]
- Rhein, B.A.; Brouillette, R.B.; Schaack, G.A.; Chiorini, J.A.; Maury, W. Characterization of Human and Murine T-Cell Immunoglobulin Mucin Domain 4 (TIM-4) IgV Domain Residues Critical for Ebola Virus Entry. J. Virol. 2016, 90, 6097–6111. [Google Scholar] [CrossRef]
- Kondratowicz, A.S.; Lennemann, N.J.; Sinn, P.L.; Davey, R.A.; Hunt, C.L.; Moller-Tank, S.; Meyerholz, D.K.; Rennert, P.; Mullins, R.F.; Brindley, M.; et al. T-cell immunoglobulin and mucin domain 1 (TIM-1) is a receptor for Zaire Ebolavirus and Lake Victoria Marburgvirus. Proc. Natl. Acad. Sci. USA 2011, 108, 8426–8431. [Google Scholar] [CrossRef]
- Shimojima, M.; Takada, A.; Ebihara, H.; Neumann, G.; Fujioka, K.; Irimura, T.; Jones, S.; Feldmann, H.; Kawaoka, Y. Tyro3 family-mediated cell entry of Ebola and Marburg viruses. J. Virol. 2006, 80, 10109–10116. [Google Scholar] [CrossRef]
- Takada, A.; Watanabe, S.; Ito, H.; Okazaki, K.; Kida, H.; Kawaoka, Y. Downregulation of beta1 integrins by Ebola virus glycoprotein: Implication for virus entry. Virology 2000, 278, 20–26. [Google Scholar] [CrossRef]
- Schornberg, K.L.; Shoemaker, C.J.; Dube, D.; Abshire, M.Y.; Delos, S.E.; Bouton, A.H.; White, J.M. Alpha5beta1-integrin controls ebolavirus entry by regulating endosomal cathepsins. Proc. Natl. Acad. Sci. USA 2009, 106, 8003–8008. [Google Scholar] [CrossRef]
- Matsuno, K.; Nakayama, E.; Noyori, O.; Marzi, A.; Ebihara, H.; Irimura, T.; Feldmann, H.; Takada, A. C-type lectins do not act as functional receptors for filovirus entry into cells. Biochem. Biophys. Res. Commun. 2010, 403, 144–148. [Google Scholar] [CrossRef]
- Simmons, G.; Rennekamp, A.J.; Chai, N.; Vandenberghe, L.H.; Riley, J.L.; Bates, P. Folate Receptor Alpha and Caveolae Are Not Required for Ebola Virus Glycoprotein-Mediated Viral Infection. J. Virol. 2003, 77, 13433–13438. [Google Scholar] [CrossRef]
- Ichimura, T.; Asseldonk, E.J.P.V.; Humphreys, B.D.; Gunaratnam, L.; Duffield, J.S.; Bonventre, J.V. Kidney injury molecule-1 is a phosphatidylserine receptor that confers a phagocytic phenotype on epithelial cells. J. Clin. Investig. 2008, 118, 1657–1668. [Google Scholar] [CrossRef]
- Hunt, C.L.; Kolokoltsov, A.A.; Davey, R.A.; Maury, W. The Tyro3 receptor kinase Axl enhances macropinocytosis of Zaire ebolavirus. J. Virol. 2011, 85, 334–347. [Google Scholar] [CrossRef]
- Chandran, K.; Sullivan, N.J.; Felbor, U.; Whelan, S.P.; Cunningham, J.M. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science 2005, 308, 1643–1645. [Google Scholar] [CrossRef]
- Sakurai, Y.; Kolokoltsov, A.A.; Chen, C.C.; Tidwell, M.W.; Bauta, W.E.; Klugbauer, N.; Grimm, C.; Wahl-Schott, C.; Biel, M.; Davey, R.A. Two-pore channels control Ebola virus host cell entry and are drug targets for disease treatment. Science 2015, 347, 995–998. [Google Scholar] [CrossRef]
- Côté, M.; Misasi, J.; Ren, T.; Bruchez, A.; Lee, K.; Filone, C.M.; Hensley, L.; Li, Q.; Ory, D.; Chandran, K.; et al. Small molecule inhibitors reveal Niemann-Pick C1 is essential for Ebola virus infection. Nature 2011, 477, 344–348. [Google Scholar] [CrossRef]
- Carette, J.E.; Raaben, M.; Wong, A.C.; Herbert, A.S.; Obernosterer, G.; Mulherkar, N.; Kuehne, A.I.; Kranzusch, P.J.; Griffin, A.M.; Ruthel, G.; et al. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature 2011, 477, 340–343. [Google Scholar] [CrossRef]
- Wang, H.; Shi, Y.; Song, J.; Qi, J.; Lu, G.; Yan, J.; Gao, G.F. Ebola Viral Glycoprotein Bound to Its Endosomal Receptor Niemann-Pick C1. Cell 2016, 164, 258–268. [Google Scholar] [CrossRef]
- Evans, M.J.; von Hahn, T.; Tscherne, D.M.; Syder, A.J.; Panis, M.; Wölk, B.; Hatziioannou, T.; McKeating, J.A.; Bieniasz, P.D.; Rice, C.M. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 2007, 446, 801–805. [Google Scholar] [CrossRef]
- Pham, H.M.; Argañaraz, E.R.; Groschel, B.; Trono, D.; Lama, J. Lentiviral Vectors Interfering with Virus-Induced CD4 Down-Modulation Potently Block Human Immunodeficiency Virus Type 1 Replication in Primary Lymphocytes. J. Virol. 2004, 78, 13072–13081. [Google Scholar] [CrossRef]
- Simmons, G.; Bertram, S.; Glowacka, I.; Steffen, I.; Chaipan, C.; Agudelo, J.; Lu, K.; Rennekamp, A.J.; Hofmann, H.; Bates, P.; et al. Different host cell proteases activate the SARS-coronavirus spike-protein for cell–cell and virus–cell fusion. Virology 2011, 413, 265–274. [Google Scholar] [CrossRef]
- Birkmann, A.; Mahr, K.; Ensser, A.; Yag, S.; Titgemeyer, F.; Fleckenstein, B.; Neipel, F. Cell Surface Heparan Sulfate Is a Receptor for Human Herpesvirus 8 and Interacts with Envelope Glycoprotein K8.1. J. Virol. 2001, 75, 11583–11593. [Google Scholar] [CrossRef]
- Pohlmann, S.; Baribaud, F.; Lee, B.; Leslie, G.J.; Sanchez, M.D.; Hiebenthal-Millow, K.; Munch, J.; Kirchhoff, F.; Doms, R.W. DC-SIGN Interactions with Human Immunodeficiency Virus Type 1 and 2 and Simian Immunodeficiency Virus. J. Virol. 2001, 75, 4664–4672. [Google Scholar] [CrossRef]
- Ciesek, S.; Westhaus, S.; Wicht, M.; Wappler, I.; Henschen, S.; Sarrazin, C.; Hamdi, N.; Abdelaziz, A.I.; Strassburg, C.P.; Wedemeyer, H.; et al. Impact of intra- and interspecies variation of occludin on its function as coreceptor for authentic hepatitis C virus particles. J. Virol. 2011, 85, 7613–7621. [Google Scholar] [CrossRef]
- Dolnik, O.; Kolesnikova, L.; Welsch, S.; Strecker, T.; Schudt, G.; Becker, S. Interaction with Tsg101 Is Necessary for the Efficient Transport and Release of Nucleocapsids in Marburg Virus-Infected Cells. PLoS Pathog. 2014, 10, e1004463. [Google Scholar] [CrossRef]
- Kuhn, J.H.; Radoshitzky, S.R.; Guth, A.C.; Warfield, K.L.; Li, W.; Vincent, M.J.; Towner, J.S.; Nichol, S.T.; Bavari, S.; Choe, H.; et al. Conserved receptor-binding domains of Lake Victoria marburgvirus and Zaire ebolavirus bind a common receptor. J. Biol. Chem. 2006, 281, 15951–15958. [Google Scholar] [CrossRef]
- Warnes, G.R.; Bolker, B.; Bonebakker, L.; Gentleman, R.; Liaw, W.H.A.; Lumley, T.; Maechler, M.; Magnusson, A.; Moeller, S.; Schwartz, M.; et al. Gplots: Various R Programming Tools for Plotting Data. R Package version 2170. 2015. Available online: https://rdrr.io/cran/gplots/ (accessed on 15 August 2015).
- Döhner, K.; Wolfstein, A.; Prank, U.; Echeverri, C.; Dujardin, D.; Vallee, R.; Sodeik, B. Function of dynein and dynactin in herpes simplex virus capsid transport. Mol. Biol. Cell 2002, 13, 2795–2809. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Frentzen, A.; Hüging, K.; Bitzegeio, J.; Friesland, M.; Haid, S.; Gentzsch, J.; Hoffmann, M.; Lindemann, D.; Zimmer, G.; Zielecki, F.; et al. Completion of hepatitis C virus replication cycle in heterokaryons excludes dominant restrictions in human non-liver and mouse liver cell lines. PLoS Pathog. 2011, 7, e1002029. [Google Scholar] [CrossRef]
- Encinas, M.; Iglesias, M.; Liu, Y.; Wang, H.; Muhaisen, A.; Ceña, V.; Gallego, C.; Comella, J.X. Sequential treatment of SH-SY5Y cells with retinoic acid and brain-derived neurotrophic factor gives rise to fully differentiated, neurotrophic factor-dependent, human neuron-like cells. J. Neurochem. 2000, 75, 991–1003. [Google Scholar] [CrossRef]
- Biedler, J.L.; Helson, L.; Spengler, B.A. Morphology and Growth, Tumorigenicity, and Cytogenetics of Human Neuroblastoma Cells in Continuous Culture. Cancer Res. 1973, 33, 2643–2652. [Google Scholar]
- Yang, Z.Y.; Duckers, H.J.; Sullivan, N.J.; Sanchez, A.; Nabel, E.G.; Nabel, G.J. Identification of the Ebola virus glycoprotein as the main viral determinant of vascular cell cytotoxicity and injury. Nat. Med. 2000, 6, 886–889. [Google Scholar] [CrossRef]
- Negredo, A.; Palacios, G.; Vázquez-Morón, S.; González, F.; Dopazo, H.; Molero, F.; Juste, J.; Quetglas, J.; Savji, N.; de la Cruz Martínez, M.; et al. Discovery of an ebolavirus-like filovirus in europe. PLoS Pathog. 2011, 7, e1002304. [Google Scholar] [CrossRef]
- Diehl, W.E.; Lin, A.E.; Grubaugh, N.D.; Carvalho, L.M.; Kim, K.; Kyawe, P.P.; McCauley, S.M.; Donnard, E.; Kucukural, A.; McDonel, P.; et al. Ebola Virus Glycoprotein with Increased Infectivity Dominated the 2013–2016 Epidemic. Cell 2016, 167, 1088–1098.e6. [Google Scholar] [CrossRef]
- Urbanowicz, R.A.; McClure, C.P.; Sakuntabhai, A.; Sall, A.A.; Kobinger, G.; Müller, M.A.; Holmes, E.C.; Rey, F.A.; Simon-Loriere, E.; Ball, J.K. Human Adaptation of Ebola Virus during the West African Outbreak. Cell 2016, 167, 1079–1087. [Google Scholar] [CrossRef]
- Malim, M.H.; Bieniasz, P.D. HIV restriction factors and mechanisms of evasion. Cold Spring Harb. Perspect. Med. 2012, 2, a006940. [Google Scholar] [CrossRef]
- Kaletsky, R.L.; Francica, J.R.; Agrawal-Gamse, C.; Bates, P. Tetherin-mediated restriction of filovirus budding is antagonized by the Ebola glycoprotein. Proc. Natl. Acad. Sci. USA 2009, 106, 2886–2891. [Google Scholar] [CrossRef]
- Gossen, M.; Bujard, H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl. Acad. Sci. USA 2006, 89, 5547–5551. [Google Scholar] [CrossRef]
- Shaw, E. Peptidyl diazomethanes as inhibitors of cysteine and serine proteinases. Methods Enzymol. 1994, 244, 649–656. [Google Scholar] [PubMed]
- Lu, F.; Liang, Q.; Abi-Mosleh, L.; Das, A.; de Brabander, J.K.; Goldstein, J.L.; Brown, M.S. Identification of NPC1 as the target of U18666A, an inhibitor of lysosomal cholesterol export and Ebola infection. eLife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Wojtanik, K.M.; Liscum, L. The transport of low density lipoprotein-derived cholesterol to the plasma membrane is defective in NPC1 cells. J. Biol. Chem. 2003, 278, 14850–14856. [Google Scholar] [CrossRef] [PubMed]
- Liscum, L.; Faust, J.R. The intracellular transport of low density lipoprotein-derived cholesterol is inhibited in Chinese hamster ovary cells cultured with 3-beta-[2-(diethylamino)ethoxy]androst-5-en-17-one. J. Biol. Chem. 1989, 264, 11796–11806. [Google Scholar] [PubMed]
- Dahlmann, F.; Biedenkopf, N.; Babler, A.; Jahnen-Dechent, W.; Karsten, C.B.; Gnirß, K.; Schneider, H.; Wrensch, F.; O’Callaghan, C.A.; Bertram, S.; et al. Analysis of Ebola Virus Entry Into Macrophages. J. Infect. Dis. 2015, 212, S247–S257. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Zagórska, A.; Lew, E.D.; Shrestha, B.; Rothlin, C.V.; Naughton, J.; Diamond, M.S.; Lemke, G.; Young, J.A.T. Enveloped Viruses Disable Innate Immune Responses in Dendritic Cells by Direct Activation of TAM Receptors. Cell Host Microbe 2013, 14, 136–147. [Google Scholar] [CrossRef]
- Ross, R.A.; Spengler, B.A.; Biedler, J.L. Coordinate Morphological and Biochemical Interconversion of Human Neuroblastoma Cells. J. Natl. Cancer Inst. 1983, 71, 741–747. [Google Scholar]
- Christensen, J.; Steain, M.; Slobedman, B.; Abendroth, A. Differentiated neuroblastoma cells provide a highly efficient model for studies of productive varicella-zoster virus infection of neuronal cells. J. Virol. 2011, 85, 8436–8442. [Google Scholar] [CrossRef]
- La Monica, N.; Racaniello, V.R. Differences in replication of attenuated and neurovirulent polioviruses in human neuroblastoma cell line SH-SY5Y. J. Virol. 1989, 63, 2357–2360. [Google Scholar]
- Castellanos, J.E.; Neissa, J.I.; Camacho, S.J. Dengue virus induces apoptosis in SH-SY5Y human neuroblastoma cells. Biomedica 2016, 36, 156–158. [Google Scholar] [CrossRef]
- Dhanwani, R.; Khan, M.; Bhaskar, A.S.B.; Singh, R.; Patro, I.K.; Rao, P.V.L.; Parida, M.M. Characterization of Chikungunya virus infection in human neuroblastoma SH-SY5Y cells: Role of apoptosis in neuronal cell death. Virus Res. 2012, 163, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Cambi, A.; Koopman, M.; Figdor, C.G. How C-type lectins detect pathogens. Cell Microbiol. 2005, 7, 481–488. [Google Scholar] [CrossRef]
- Kuroda, M.; Fujikura, D.; Nanbo, A.; Marzi, A.; Noyori, O.; Kajihara, M.; Maruyama, J.; Matsuno, K.; Miyamoto, H.; Yoshida, R.; et al. Interaction between TIM-1 and NPC1 Is Important for Cellular Entry of Ebola Virus. J. Virol. 2015, 89, 6481–6493. [Google Scholar] [CrossRef] [PubMed]
- Brindley, M.A.; Hunt, C.L.; Kondratowicz, A.S.; Bowman, J.; Sinn, P.L.; McCray, P.B.; Quinn, K.; Weller, M.L.; Chiorini, J.A.; Maury, W. Tyrosine kinase receptor Axl enhances entry of Zaire ebolavirus without direct interactions with the viral glycoprotein. Virology 2011, 415, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Younan, P.; Iampietro, M.; Nishida, A.; Ramanathan, P.; Santos, R.I.; Dutta, M.; Lubaki, N.M.; Koup, R.A.; Katze, M.G.; Bukreyev, A. Ebola virus binding to Tim-1 on T lymphocytes induces a cytokine storm. MBio 2017, 8, e00845-17. [Google Scholar] [CrossRef] [PubMed]
- Volchkov, V.E.; Becker, S.; Volchkova, V.A.; Ternovoj, V.A.; Kotov, A.N.; Netesov, S.V.; Klenk, H.D. GP mRNA of Ebola virus is edited by the Ebola virus polymerase and by T7 and vaccinia virus polymerases. Virology 1995, 214, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Marzi, A.; Gramberg, T.; Simmons, G.; Möller, P.; Rennekamp, A.J.; Krumbiegel, M.; Geier, M.; Eisemann, J.; Turza, N.; Saunier, B.; et al. DC-SIGN and DC-SIGNR Interact with the Glycoprotein of Marburg Virus and the S Protein, of Severe Acute Respiratory Syndrome Coronavirus. J. Virol. 2004, 78, 12090–12095. [Google Scholar] [CrossRef]
- Morizono, K.; Chen, I.S. Role of phosphatidylserine receptors in enveloped virus infection. J. Virol. 2014, 88, 4275–4290. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zapatero-Belinchón, F.J.; Dietzel, E.; Dolnik, O.; Döhner, K.; Costa, R.; Hertel, B.; Veselkova, B.; Kirui, J.; Klintworth, A.; Manns, M.P.; et al. Characterization of the Filovirus-Resistant Cell Line SH-SY5Y Reveals Redundant Role of Cell Surface Entry Factors. Viruses 2019, 11, 275. https://doi.org/10.3390/v11030275
Zapatero-Belinchón FJ, Dietzel E, Dolnik O, Döhner K, Costa R, Hertel B, Veselkova B, Kirui J, Klintworth A, Manns MP, et al. Characterization of the Filovirus-Resistant Cell Line SH-SY5Y Reveals Redundant Role of Cell Surface Entry Factors. Viruses. 2019; 11(3):275. https://doi.org/10.3390/v11030275
Chicago/Turabian StyleZapatero-Belinchón, Francisco J., Erik Dietzel, Olga Dolnik, Katinka Döhner, Rui Costa, Barbara Hertel, Barbora Veselkova, Jared Kirui, Anneke Klintworth, Michael P. Manns, and et al. 2019. "Characterization of the Filovirus-Resistant Cell Line SH-SY5Y Reveals Redundant Role of Cell Surface Entry Factors" Viruses 11, no. 3: 275. https://doi.org/10.3390/v11030275
APA StyleZapatero-Belinchón, F. J., Dietzel, E., Dolnik, O., Döhner, K., Costa, R., Hertel, B., Veselkova, B., Kirui, J., Klintworth, A., Manns, M. P., Pöhlmann, S., Pietschmann, T., Krey, T., Ciesek, S., Gerold, G., Sodeik, B., Becker, S., & von Hahn, T. (2019). Characterization of the Filovirus-Resistant Cell Line SH-SY5Y Reveals Redundant Role of Cell Surface Entry Factors. Viruses, 11(3), 275. https://doi.org/10.3390/v11030275