Gram-Positive Bacteria-Like DNA Binding Machineries Involved in Replication Initiation and Termination Mechanisms of Mimivirus


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
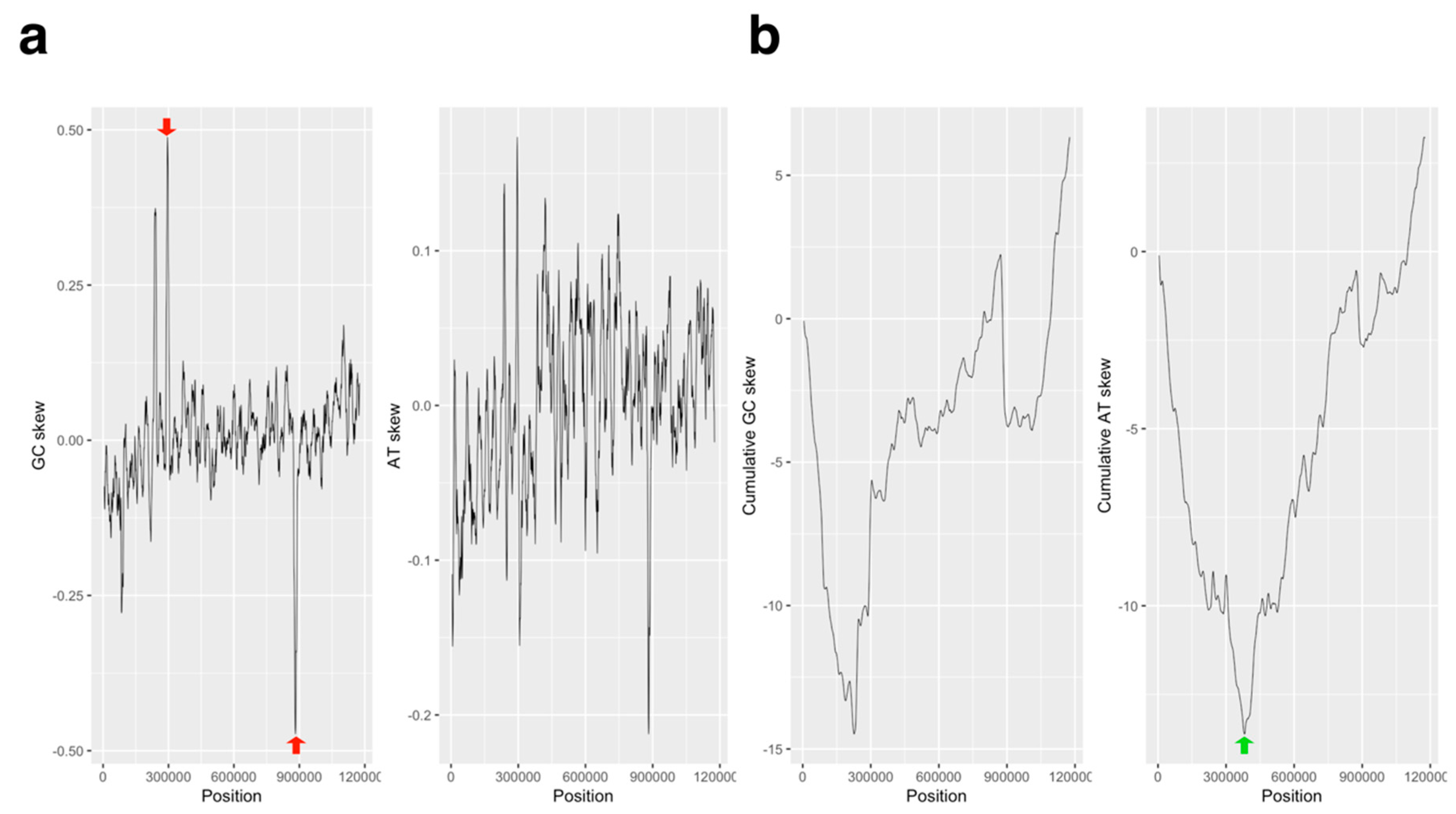
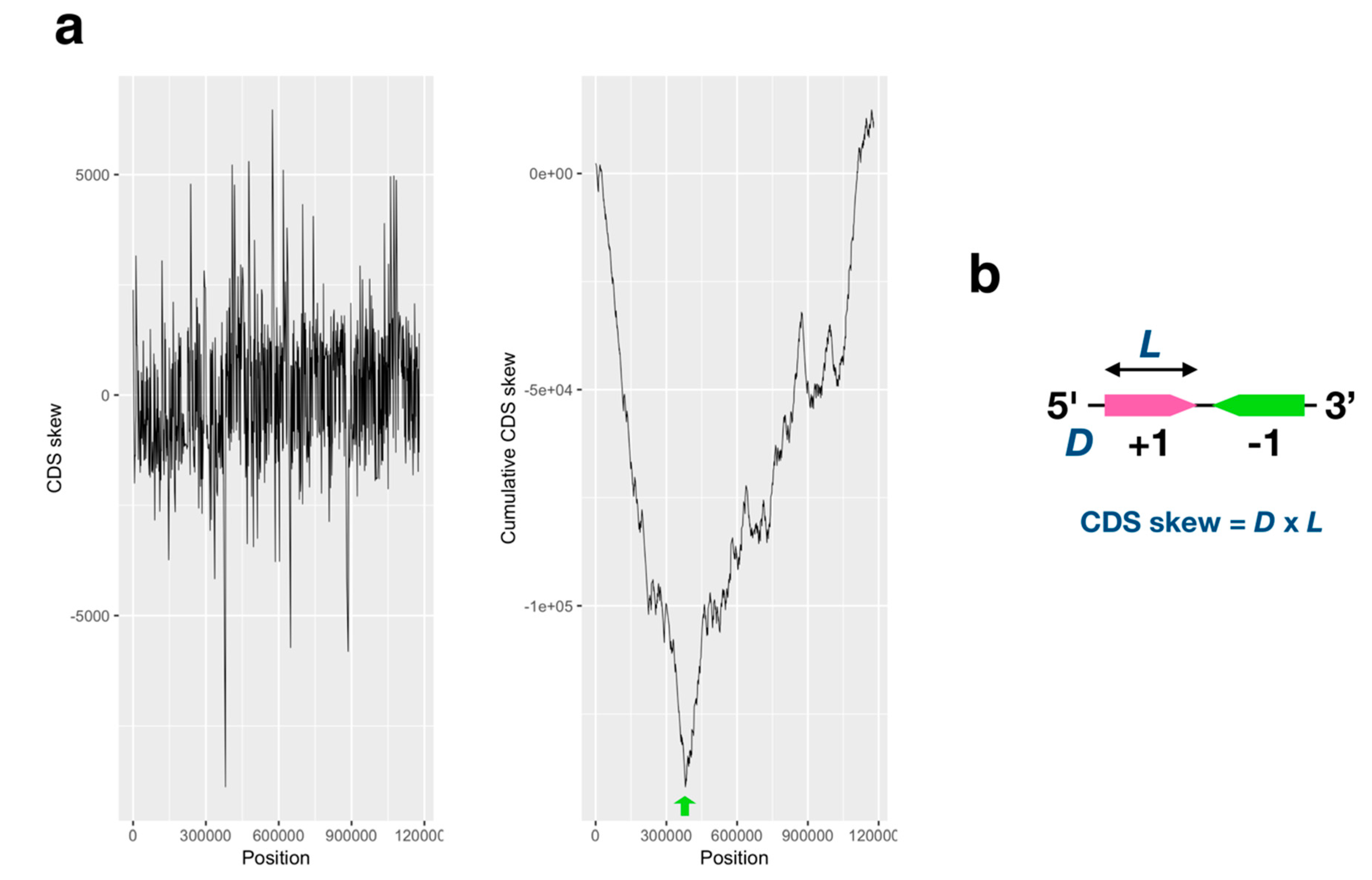
2.1. GC/AT, Coding Sequence (CDS) Skew Analysis
2.2. Correlation Analysis of the CDS Length of Left/Right Side from the Estimated Ori Region
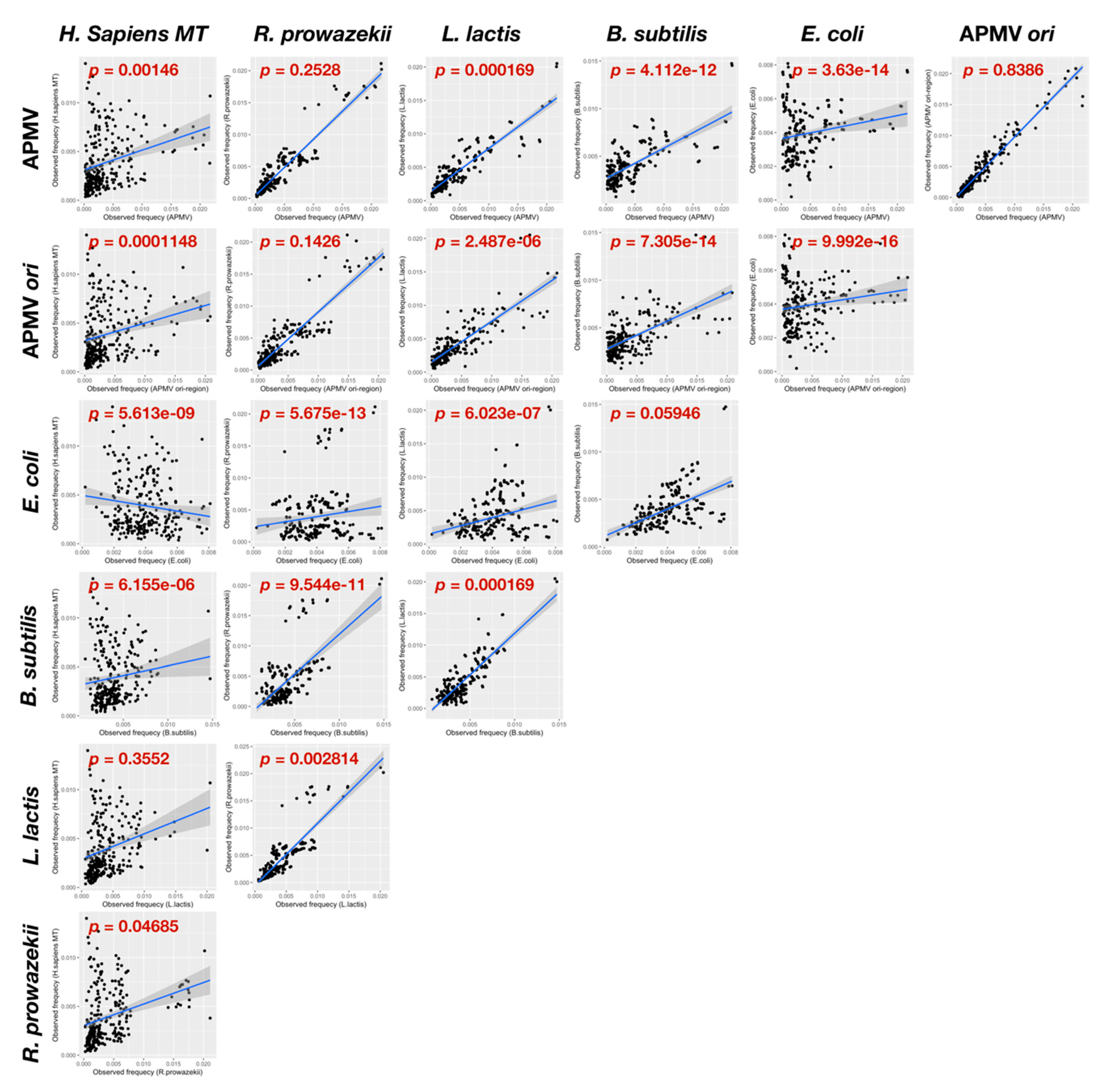
2.3. Paralogous Gene Localization Analysis
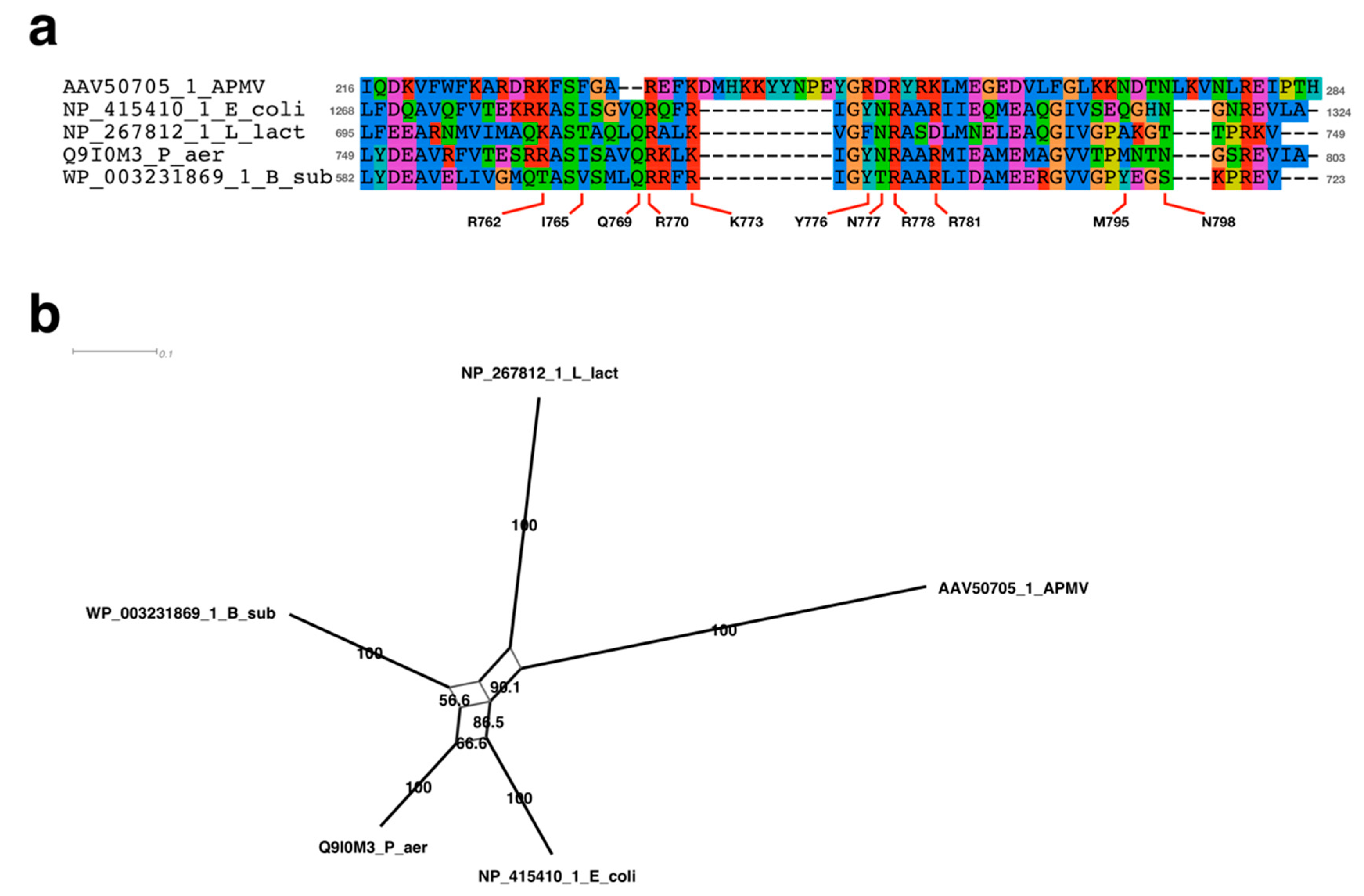
2.4. Sequence Alignment Analysis Neighbor-Net Network Analysis
2.5. KOPS Distribution in the Genome
2.6. Quartet Sequence Composition Analysis
2.7. Homology Modeling Analysis
3. Results
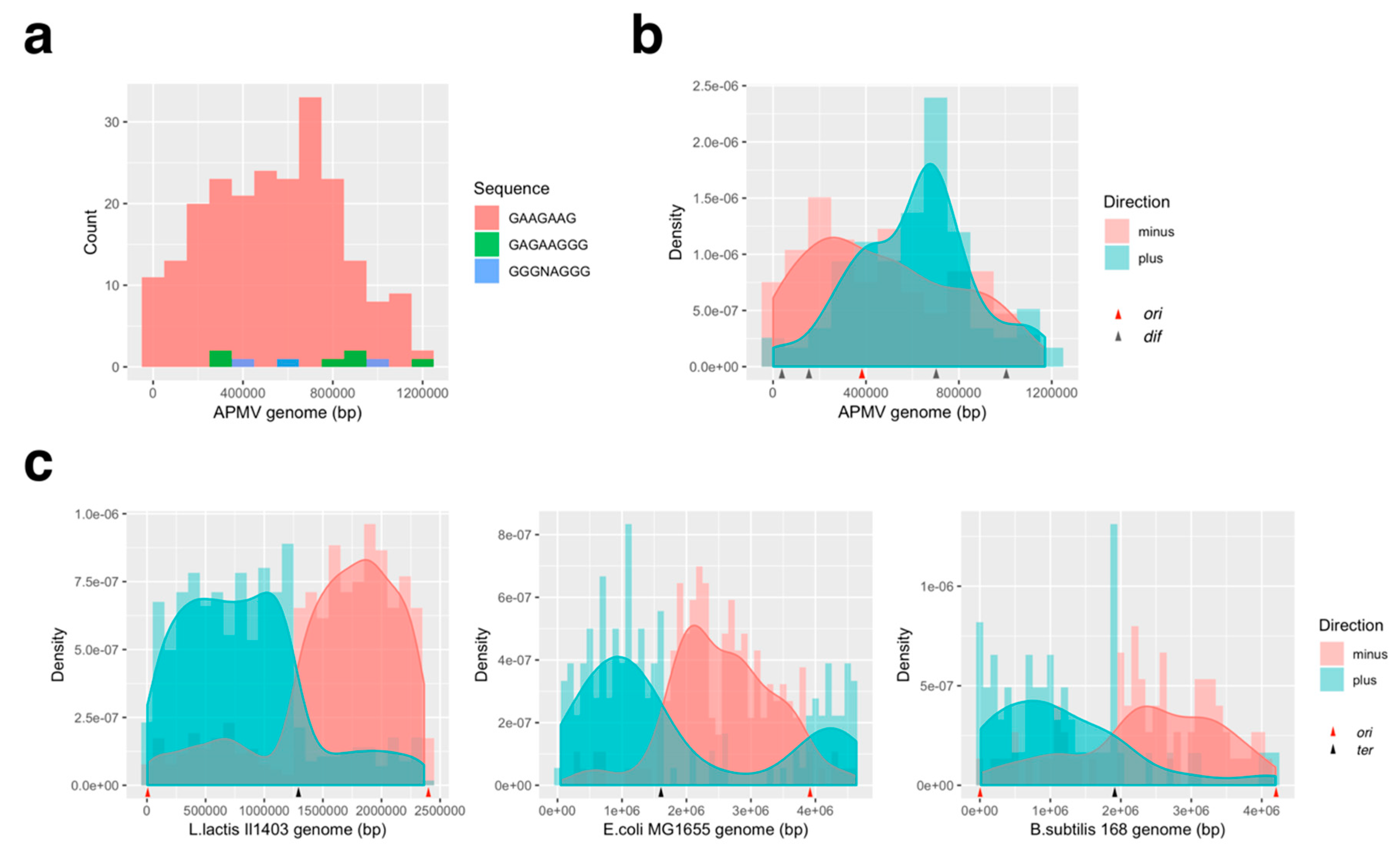
3.1. GC/AT Skew and CDS Skew Analyses
3.2. Initiation of DNA Replication
3.2.1. Sequence Analysis of UvrD/Rep-Like Helicase
3.2.2. Signal Sequence of the Initiation of DNA Replication
3.3. Termination of DNA Replication and Chromosome Segregation
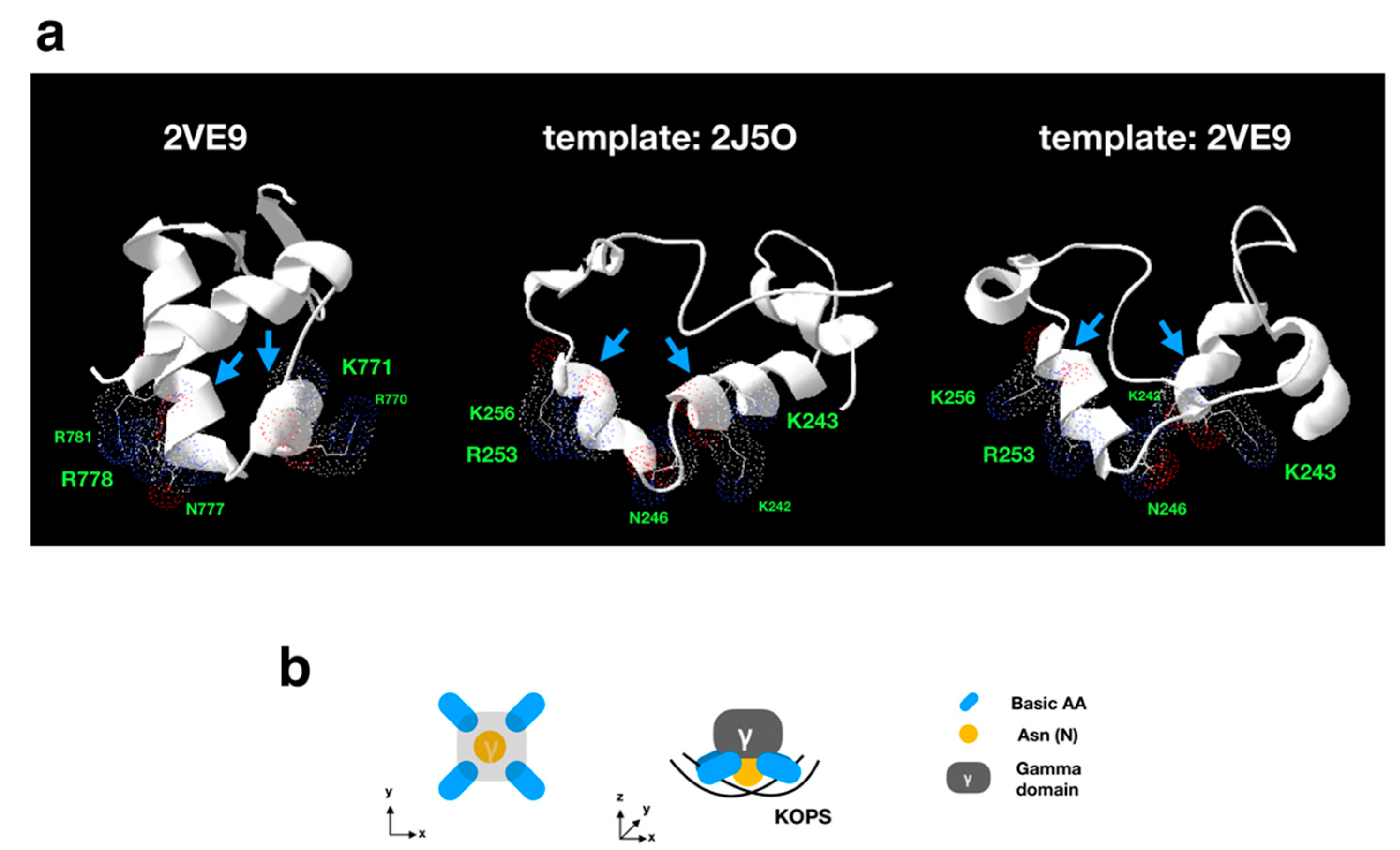
3.3.1. Sequence Analysis of FtsK-Like Protein in APMV
3.3.2. Distribution Pattern of KOPS on APMV Genome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- La Scola, B.; Audic, S.; Robert, C.; Jungang, L.; de Lamballerie, X.; Drancourt, M.; Birtles, R.; Claverie, J.M.; Raoult, D. A giant virus in amoebae. Science 2003, 299, 2033. [Google Scholar] [CrossRef] [PubMed]
- Raoult, D.; Audic, S.; Robert, C.; Abergel, C.; Renesto, P.; Ogata, H.; La Scola, B.; Suzan, M.; Claverie, J.M. The 1.2-Megabase Genome Sequence of Mimivirus. Science 2004, 306, 1344–1350. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Claverie, J.-M.; Ogata, H. Mimivirus reveals Mre11/Rad50 fusion proteins with a sporadic distribution in eukaryotes, bacteria, viruses and plasmids. Virol. J. 2011, 8, 427. [Google Scholar] [CrossRef] [PubMed]
- Kreuzer, K.N. Recombination-dependent DNA replication in phage T4. Trends Biochem. Sci. 2000, 25, 165–173. [Google Scholar] [CrossRef]
- Chelikani VRanjan, T.; Zade, A.; Shukla, A.; Kondabagil, K. Genome segregation and packaging machinery in Acanthamoeba polyphaga mimivirus is reminiscent of bacterial apparatus. J. Virol. 2014, 88, 6069–6075. [Google Scholar] [CrossRef] [PubMed]
- Nolivos, S.; Touzain, F.; Pages, C.; Coddeville, M.; Rousseau, P.; El Karoui, M.; Le Bourgeois, P.; Cornet, F. Co-evolution of segregation guide DNA motifs and the FtsK translocase in bacteria: Identification of the atypical Lactococcus lactis KOPS motif. Nucleic Acids Res. 2012, 40, 5535–5545. [Google Scholar] [CrossRef]
- Ptacin, J.L.; Nollmann, M.; Becker, E.C.; Cozzarelli, N.R.; Pogliano, K.; Bustamante, C. Sequence-directed DNA export guides chromosome translocation during sporulation in Bacillus subtilis. Nat. Struct. Mol. Biol. 2008, 15, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Bigot, S.; Saleh, O.A.; Lesterlin, C.; Pages, C.; El Karoui, M.; Dennis, C.; Grigoriev, M.; Allemand, J.F.; Barre, F.X.; Cornet, F. KOPS: DNA motifs that control E. coli chromosome segregation by orienting the FtsK translocase. EMBO J. 2005, 24, 3770–3780. [Google Scholar] [CrossRef]
- Kaguni, J.M. Replication initiation at the Escherichia coli chromosomal origin. Curr. Opin. Chem. Biol. 2011, 15, 606–613. [Google Scholar] [CrossRef]
- Lohman, T.M. Escherichia coli DNA helicases: Mechanisms of DNA unwinding. Mol. Microbiol. 1992, 6, 5–14. [Google Scholar] [CrossRef]
- Lane, H.E.; Denhardt, D.T. The rep mutation. IV. Slower movement of replication forks in Escherichia coli rep strains. J. Mol. Biol. 1975, 97, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Chédin, F.; Noirot, P.; Biaudet, V.; Ehrlich, S.D. A five-nucleotide sequence protects DNA from exonucleolytic degradation by AddAB, the RecBCD analogue of Bacillus subtilis. Mol. Microbiol. 1998, 29, 1369–1377. [Google Scholar] [CrossRef] [PubMed]
- Velankar, S.S.; Soultanas, P.; Dillingham, M.S.; Subramanya, H.S.; Wigley, D.B. Crystal structures of complexes of PcrA DNA helicase with a DNA substrate indicate an inchworm mechanism. Cell 1999, 97, 75–84. [Google Scholar] [CrossRef]
- Petit, M.A.; Dervyn, E.; Rose, M.; Entian, K.D.; McGovern, S.; Ehrlich, S.D.; Bruand, C. PcrA is an essential DNA helicase of Bacillus subtilis fulfilling functions both in repair and rolling-circle replication. Mol. Microbiol. 1998, 29, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, T.; Fujimura, Y. Recombinational rescue of the stalled DNA replication fork: A model based on analysis of an Escherichia coli strain with a chromosome region difficult to replicate. J. Bacteriol. 1995, 177, 783–791. [Google Scholar] [CrossRef]
- Amundsen, S.K.; Sharp, J.W.; Smith, G.R. RecBCD Enzyme “Chi Recognition” Mutants Recognize Chi Recombination Hotspots in the Right DNA Context. Genetics 2016, 204, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Biswas, I.; Maguin, E.; Ehrlich, S.D.; Gruss, A. A 7-base-pair sequence protects DNA from exonucleolytic degradation in Lactococcus lactis. Proc. Natl. Acad. Sci. USA 1995, 92, 2244–2248. [Google Scholar] [CrossRef]
- Frank, A.C.; Lobry, J.R. Asymmetric substitution patterns: A review of possible underlying mutational or selective mechanisms. Gene 1999, 238, 65–77. [Google Scholar] [CrossRef]
- Arakawa, K.; Tomita, M. The GC skew index: A measure of genomic compositional asymmetry and the degree of replicational selection. Evol. Bioinform. 2007, 3, 159–168. [Google Scholar] [CrossRef]
- Freeman, J.M. Patterns of Genome Organization in Bacteria. Science 1998, 279, 1827. [Google Scholar] [CrossRef]
- Grigoriev, A. Analyzing genomes with cumulative skew diagrams. Nucleic Acids Res. 1998, 26, 2286–2290. [Google Scholar] [CrossRef] [PubMed]
- Lobry, J.R.; Sueoka, N. Asymmetric directional mutation pressures in bacteria. Genome Biol. 2002, 3, RESEARCH0058.1. [Google Scholar] [CrossRef]
- Lobry, J.R.; Louarn, J.-M. Polarisation of prokaryotic chromosomes. Curr. Opin. Microbiol. 2003, 6, 101–108. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Whelan, S.; Irisarri, I.; Burki, F. PREQUAL: Detecting non-homologous characters in sets of unaligned homologous sequences. Bioinformatics 2018, 34, 3929–3930. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Nakhleh, L. Evolutionary Phylogenetic Networks: Models and Issues. In Problem Solving Handbook in Computational Biology and Bioinformatics; Springer: Boston, MA, USA, 2010; pp. 125–158. [Google Scholar] [CrossRef]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef]
- Arakawa, K.; Tamaki, S.; Kono, N.; Kido, N.; Ikegami, K.; Ogawa, R.; Tomita, M. Genome Projector: zoomable genome map with multiple views. BMC Bioinform. 2009, 10, 31. [Google Scholar] [CrossRef]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef]
- Löwe, J.; Ellonen, A.; Allen, M.D.; Atkinson, C.; Sherratt, D.J.; Grainge, I. Molecular mechanism of sequence-directed DNA loading and translocation by FtsK. Mol. Cell 2008, 31, 498–509. [Google Scholar] [CrossRef] [PubMed]
- Sivanathan, V.; Allen, M.D.; de Bekker, C.; Baker, R.; Arciszewska, L.K.; Freund, S.M.; Bycroft, M.; Löwe, J.; Sherratt, D.J. The FtsK gamma domain directs oriented DNA translocation by interacting with KOPS. Nat. Struct. Mol. Biol. 2006, 13, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Arai, N.; Arai, K.; Kornberg, A. Complexes of Rep protein with ATP and DNA as a basis for helicase action. J. Biol. Chem. 1981, 256, 5287–5293. [Google Scholar]
- Kooistra, J.; Haijema, B.J.; Hesseling-Meinders, A.; Venema, G. A conserved helicase motif of the AddA subunit of the Bacillus subtilis ATP-dependent nuclease (AddAB) is essential for DNA repair and recombination. Mol. Microbiol. 1997, 23, 137–149. [Google Scholar] [CrossRef]
- Krajewski, W.W.; Fu, X.; Wilkinson, M.; Cronin, N.B.; Dillingham, M.S.; Wigley, D.B. Structural basis for translocation by AddAB helicase-nuclease and its arrest at χ sites. Nature 2014, 508, 416–419. [Google Scholar] [CrossRef] [PubMed]
- Hixson, J.E.; Wong, T.W.; Clayton, D.A. Both the conserved stem-loop and divergent 5′-flanking sequences are required for initiation at the human mitochondrial origin of light-strand DNA replication. J. Biol. Chem. 1986, 261, 2384–2390. [Google Scholar]
- Andersson, S.G.; Zomorodipour, A.; Andersson, J.O.; Sicheritz-Pontén, T.; Alsmark, U.C.; Podowski, R.M.; Näslund, A.K.; Eriksson, A.S.; Winkler, H.H.; Kurland, C.G. The genome sequence of Rickettsia prowazekii and the origin of mitochondria. Nature 1998, 396, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Seligmann, H. Giant viruses as protein-coated amoeban mitochondria? Virus Res. 2018, 253, 77–86. [Google Scholar] [CrossRef]
- Massey, T.H.; Mercogliano, C.P.; Yates, J.; Sherratt, D.J.; Löwe, J. Double-stranded DNA translocation: Structure and mechanism of hexameric FtsK. Mol. Cell 2006, 23, 457–469. [Google Scholar] [CrossRef]
- Paul, S.; Million-Weaver, S.; Chattopadhyay, S.; Sokurenko, E.; Merrikh, H. Accelerated gene evolution through replication-transcription conflicts. Nature 2013, 495, 512–515. [Google Scholar] [CrossRef] [PubMed]
- Arslan, D.; Legendre, M.; Seltzer, V.; Abergel, C.; Claverie, J.-M. Distant Mimivirus relative with a larger genome highlights the fundamental features of Megaviridae. Proc. Natl. Acad. Sci. USA 2011, 108, 17486–17491. [Google Scholar] [CrossRef]
- Han, M.; Yagura, M.; Itoh, T. Specific interaction between the initiator protein (Rep) and origin of plasmid ColE2-P9. J. Bacteriol. 2007, 189, 1061–1071. [Google Scholar] [CrossRef] [PubMed]
- Seligmann, H. Giant viruses: Spore-like missing links between Rickettsia and mitochondria? Ann. N. Y. Acad. Sci. 2019. [Google Scholar] [CrossRef]










© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akashi, M.; Takemura, M. Gram-Positive Bacteria-Like DNA Binding Machineries Involved in Replication Initiation and Termination Mechanisms of Mimivirus. Viruses 2019, 11, 267. https://doi.org/10.3390/v11030267
Akashi M, Takemura M. Gram-Positive Bacteria-Like DNA Binding Machineries Involved in Replication Initiation and Termination Mechanisms of Mimivirus. Viruses. 2019; 11(3):267. https://doi.org/10.3390/v11030267
Chicago/Turabian StyleAkashi, Motohiro, and Masaharu Takemura. 2019. "Gram-Positive Bacteria-Like DNA Binding Machineries Involved in Replication Initiation and Termination Mechanisms of Mimivirus" Viruses 11, no. 3: 267. https://doi.org/10.3390/v11030267
APA StyleAkashi, M., & Takemura, M. (2019). Gram-Positive Bacteria-Like DNA Binding Machineries Involved in Replication Initiation and Termination Mechanisms of Mimivirus. Viruses, 11(3), 267. https://doi.org/10.3390/v11030267