Genetic Susceptibility to Human Norovirus Infection: An Update


Abstract
1. Introduction
2. Norovirus—A Family with Many Members
3. A Brief History of Host Genetics and Norovirus Susceptibility
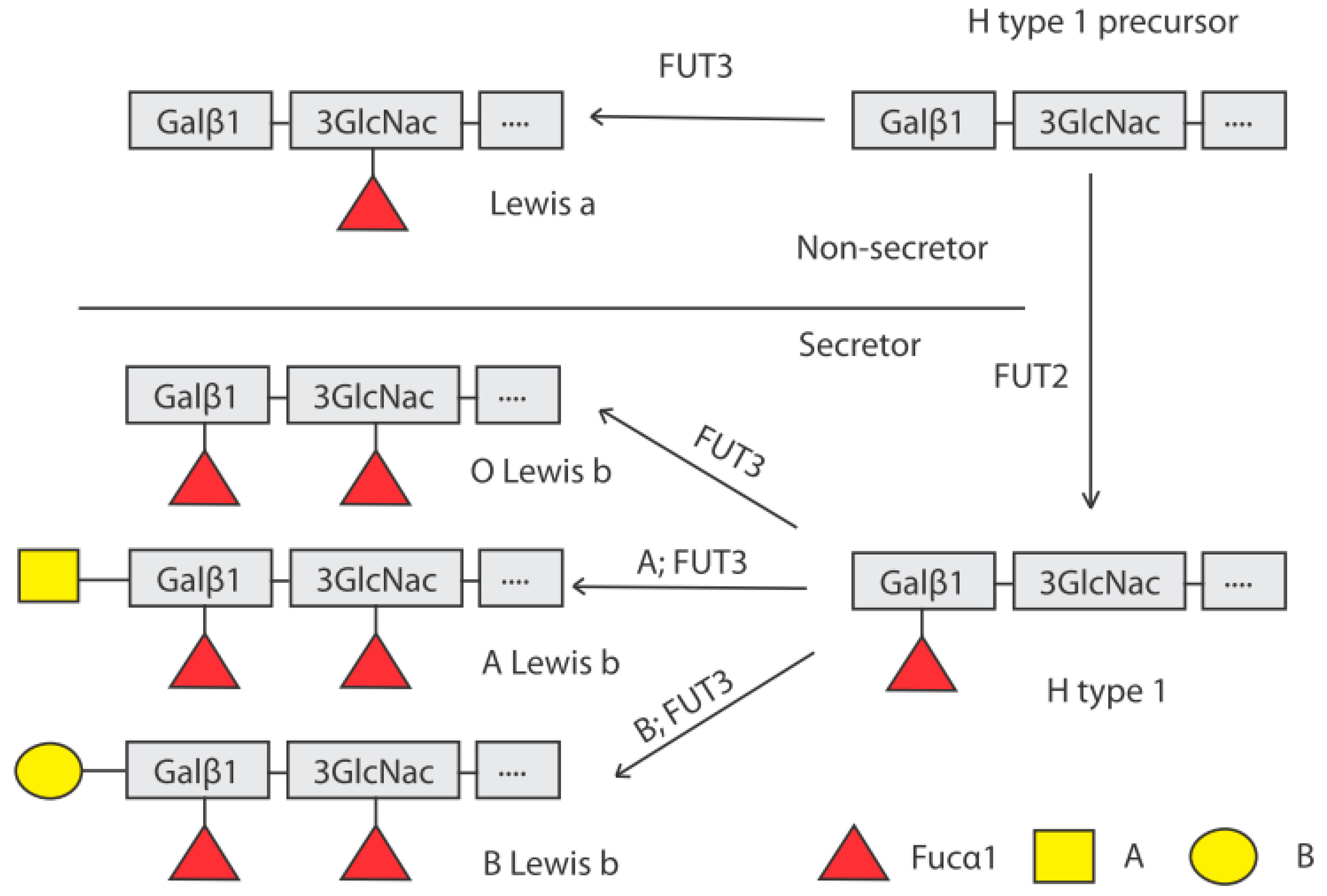
4. Genetic Control of HBGA Synthesis
5. Norovirus Challenge Studies
6. Norovirus Outbreak Studies
7. Norovirus Observational Studies
8. Population Genetics, Infection Rates and Epidemiology
9. Norovirus GII.4 Infection in Non-Secretors
10. Genetic Susceptibility and Asymptomatic Norovirus Infection
11. The Lewis Antigens as Mediators of Susceptibility
12. Norovirus GII.4 Evolution and Receptor Switching
13. Genetics vs. Immunity—Prolonged Norovirus Infection in Immunocompromised Hosts
14. Human Intestinal Enteroids: A Novel Model to Study Genetic Susceptibility to Norovirus
15. Impact of Secretor Status on Norovirus Vaccine Design
16. Relationships between Secretor Status, Microbiota and Susceptibility to Norovirus Infection
17. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ahmed, S.M.; Hall, A.J.; Robinson, A.E.; Verhoef, L.; Premkumar, P.; Parashar, U.D.; Koopmans, M.; Lopman, B.A. Global prevalence of norovirus in cases of gastroenteritis: A systematic review and meta-analysis. Lancet Infect. Dis. 2014, 14, 725–730. [Google Scholar] [CrossRef]
- Patel, M.M.; Widdowson, M.A.; Glass, R.I.; Akazawa, K.; Vinje, J.; Parashar, U.D. Systematic literature review of role of noroviruses in sporadic gastroenteritis. Emerg. Infect. Dis. 2008, 14, 1224–1231. [Google Scholar] [CrossRef] [PubMed]
- Becker-Dreps, S.; Bucardo, F.; Vilchez, S.; Zambrana, L.E.; Liu, L.; Weber, D.J.; Pena, R.; Barclay, L.; Vinje, J.; Hudgens, M.G.; et al. Etiology of childhood diarrhea after rotavirus vaccine introduction: A prospective, population-based study in Nicaragua. Pediatr. Infect. Dis. J. 2014, 33, 1156–1163. [Google Scholar] [CrossRef] [PubMed]
- Bucardo, F.; Reyes, Y.; Svensson, L.; Nordgren, J. Predominance of norovirus and sapovirus in Nicaragua after implementation of universal rotavirus vaccination. PLoS ONE 2014, 9, e98201. [Google Scholar] [CrossRef] [PubMed]
- Hemming, M.; Rasanen, S.; Huhti, L.; Paloniemi, M.; Salminen, M.; Vesikari, T. Major reduction of rotavirus, but not norovirus, gastroenteritis in children seen in hospital after the introduction of RotaTeq vaccine into the National Immunization Programme in Finland. Eur. J. Pediatr. 2013, 172, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Payne, D.C.; Vinje, J.; Szilagyi, P.G.; Edwards, K.M.; Staat, M.A.; Weinberg, G.A.; Hall, C.B.; Chappell, J.; Bernstein, D.I.; Curns, A.T.; et al. Norovirus and medically attended gastroenteritis in U.S. children. N. Engl. J. Med. 2013, 368, 1121–1130. [Google Scholar] [CrossRef] [PubMed]
- Teunis, P.F.; Moe, C.L.; Liu, P.; Miller, S.E.; Lindesmith, L.; Baric, R.S.; Le Pendu, J.; Calderon, R.L. Norwalk virus: How infectious is it? J. Med. Virol. 2008, 80, 1468–1476. [Google Scholar] [CrossRef] [PubMed]
- Mead, P.S.; Slutsker, L.; Dietz, V.; McCaig, L.F.; Bresee, J.S.; Shapiro, C.; Griffin, P.M.; Tauxe, R.V. Food-related illness and death in the United States. Emerg. Infect. Dis. 1999, 5, 607–625. [Google Scholar] [CrossRef] [PubMed]
- Ramani, S.; Atmar, R.L.; Estes, M.K. Epidemiology of human noroviruses and updates on vaccine development. Curr. Opin. Gastroenterol. 2014, 30, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Atmar, R.L.; Ramani, S.; Estes, M.K. Human noroviruses: Recent advances in a 50-year history. Curr. Opin. Infect. Dis. 2018, 31, 422–432. [Google Scholar] [CrossRef] [PubMed]
- de Rougemont, A.; Ruvoen-Clouet, N.; Simon, B.; Estienney, M.; Elie-Caille, C.; Aho, S.; Pothier, P.; Le Pendu, J.; Boireau, W.; Belliot, G. Qualitative and quantitative analysis of the binding of GII.4 norovirus variants onto human blood group antigens. J. Virol. 2011, 85, 4057–4070. [Google Scholar] [CrossRef] [PubMed]
- Nordgren, J.; Kindberg, E.; Lindgren, P.E.; Matussek, A.; Svensson, L. Norovirus gastroenteritis outbreak with a secretor-independent susceptibility pattern, Sweden. Emerg. Infect. Dis. 2010, 16, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Prasad, B.V.; Hardy, M.E.; Dokland, T.; Bella, J.; Rossmann, M.G.; Estes, M.K. X-ray crystallographic structure of the Norwalk virus capsid. Science 1999, 286, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.; Hedlund, K.O.; Thorhagen, M.; Larson, G.; Johansen, K.; Ekspong, A.; Svensson, L. Evolution of human calicivirus RNA in vivo: Accumulation of mutations in the protruding P2 domain of the capsid leads to structural changes and possibly a new phenotype. J. Virol. 2003, 77, 13117–13124. [Google Scholar] [CrossRef] [PubMed]
- Chakravarty, S.; Hutson, A.M.; Estes, M.K.; Prasad, B.V. Evolutionary trace residues in noroviruses: Importance in receptor binding, antigenicity, virion assembly, and strain diversity. J. Virol. 2005, 79, 554–568. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Xia, M.; Cao, S.; Huang, P.; Farkas, T.; Meller, J.; Hegde, R.S.; Li, X.; Rao, Z.; Jiang, X. Elucidation of strain-specific interaction of a GII-4 norovirus with HBGA receptors by site-directed mutagenesis study. Virology 2008, 379, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Huang, P.; Meller, J.; Zhong, W.; Farkas, T.; Jiang, X. Mutations within the P2 domain of norovirus capsid affect binding to human histo-blood group antigens: Evidence for a binding pocket. J. Virol. 2003, 77, 12562–12571. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.P.; Ando, T.; Fankhauser, R.L.; Beard, R.S.; Glass, R.I.; Monroe, S.S. Norovirus classification and proposed strain nomenclature. Virology 2006, 346, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Kroneman, A.; Vega, E.; Vennema, H.; Vinje, J.; White, P.A.; Hansman, G.; Green, K.; Martella, V.; Katayama, K.; Koopmans, M. Proposal for a unified norovirus nomenclature and genotyping. Arch. Virol. 2013, 158, 2059–2068. [Google Scholar] [CrossRef] [PubMed]
- Vinje, J. Advances in laboratory methods for detection and typing of norovirus. J. Clin. Microbiol. 2015, 53, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Chhabra, P.; Aswath, K.; Collins, N.; Ahmed, T.; Olortegui, M.P.; Kosek, M.; Cebelinski, E.; Cooper, P.J.; Bucardo, F.; Lopez, M.R.; et al. Near-Complete Genome Sequences of Several New Norovirus Genogroup II Genotypes. Genome Announc. 2018, 6, e00007-18. [Google Scholar] [CrossRef] [PubMed]
- Lopman, B.; Vennema, H.; Kohli, E.; Pothier, P.; Sanchez, A.; Negredo, A.; Buesa, J.; Schreier, E.; Reacher, M.; Brown, D.; et al. Increase in viral gastroenteritis outbreaks in Europe and epidemic spread of new norovirus variant. Lancet 2004, 363, 682–688. [Google Scholar] [CrossRef]
- Bull, R.A.; Tu, E.T.; McIver, C.J.; Rawlinson, W.D.; White, P.A. Emergence of a new norovirus genotype II.4 variant associated with global outbreaks of gastroenteritis. J. Clin. Microbiol. 2006, 44, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Lindesmith, L.C.; Donaldson, E.F.; Lobue, A.D.; Cannon, J.L.; Zheng, D.P.; Vinje, J.; Baric, R.S. Mechanisms of GII.4 norovirus persistence in human populations. PLoS Med. 2008, 5, e31. [Google Scholar] [CrossRef] [PubMed]
- Parrino, T.A.; Schreiber, D.S.; Trier, J.S.; Kapikian, A.Z.; Blacklow, N.R. Clinical immunity in acute gastroenteritis caused by Norwalk agent. N. Engl. J. Med. 1977, 297, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Simmons, K.; Gambhir, M.; Leon, J.; Lopman, B. Duration of immunity to norovirus gastroenteritis. Emerg. Infect. Dis. 2013, 19, 1260–1267. [Google Scholar] [CrossRef] [PubMed]
- Koopman, J.S.; Eckert, E.A.; Greenberg, H.B.; Strohm, B.C.; Isaacson, R.E.; Monto, A.S. Norwalk virus enteric illness acquired by swimming exposure. Am. J. Epidemiol. 1982, 115, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Lindesmith, L.; Moe, C.; Marionneau, S.; Ruvoen, N.; Jiang, X.; Lindblad, L.; Stewart, P.; LePendu, J.; Baric, R. Human susceptibility and resistance to Norwalk virus infection. Nat. Med. 2003, 9, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Marionneau, S.; Ruvoen, N.; Le Moullac-Vaidye, B.; Clement, M.; Cailleau-Thomas, A.; Ruiz-Palacois, G.; Huang, P.; Jiang, X.; Le Pendu, J. Norwalk virus binds to histo-blood group antigens present on gastroduodenal epithelial cells of secretor individuals. Gastroenterology 2002, 122, 1967–1977. [Google Scholar] [CrossRef] [PubMed]
- Kindberg, E.; Akerlind, B.; Johnsen, C.; Knudsen, J.D.; Heltberg, O.; Larson, G.; Bottiger, B.; Svensson, L. Host genetic resistance to symptomatic norovirus (GGII.4) infections in Denmark. J. Clin. Microbiol. 2007, 45, 2720–2722. [Google Scholar] [CrossRef] [PubMed]
- Kindberg, E.; Svensson, L. Genetic basis of host resistance to norovirus infection. Future Virol. 2009, 4, 369–382. [Google Scholar] [CrossRef]
- Pang, H.; Fujitani, N.; Soejima, M.; Koda, Y.; Islam, M.N.; Islam, A.K.; Kimura, H. Two distinct Alu-mediated deletions of the human ABO-secretor (FUT2) locus in Samoan and Bangladeshi populations. Hum. Mutat. 2000, 16, 274. [Google Scholar] [CrossRef]
- Frenck, R.; Bernstein, D.I.; Xia, M.; Huang, P.; Zhong, W.; Parker, S.; Dickey, M.; McNeal, M.; Jiang, X. Predicting susceptibility to norovirus GII.4 by use of a challenge model involving humans. J. Infect. Dis. 2012, 206, 1386–1393. [Google Scholar] [CrossRef] [PubMed]
- Hutson, A.M.; Airaud, F.; LePendu, J.; Estes, M.K.; Atmar, R.L. Norwalk virus infection associates with secretor status genotyped from sera. J. Med. Virol. 2005, 77, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Hutson, A.M.; Atmar, R.L.; Graham, D.Y.; Estes, M.K. Norwalk virus infection and disease is associated with ABO histo-blood group type. J. Infect. Dis. 2002, 185, 1335–1337. [Google Scholar] [CrossRef] [PubMed]
- Lindesmith, L.; Moe, C.; Lependu, J.; Frelinger, J.A.; Treanor, J.; Baric, R.S. Cellular and humoral immunity following Snow Mountain virus challenge. J. Virol. 2005, 79, 2900–2909. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.Y.; Jiang, X.; Tanaka, T.; Opekun, A.R.; Madore, H.P.; Estes, M.K. Norwalk virus infection of volunteers: New insights based on improved assays. J. Infect. Dis. 1994, 170, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Farkas, T.; Zhong, W.; Tan, M.; Thornton, S.; Morrow, A.L.; Jiang, X. Norovirus and histo-blood group antigens: Demonstration of a wide spectrum of strain specificities and classification of two major binding groups among multiple binding patterns. J. Virol. 2005, 79, 6714–6722. [Google Scholar] [CrossRef] [PubMed]
- Rockx, B.H.; Vennema, H.; Hoebe, C.J.; Duizer, E.; Koopmans, M.P. Association of histo-blood group antigens and susceptibility to norovirus infections. J. Infect. Dis. 2005, 191, 749–754. [Google Scholar] [CrossRef] [PubMed]
- Currier, R.L.; Payne, D.C.; Staat, M.A.; Selvarangan, R.; Shirley, S.H.; Halasa, N.; Boom, J.A.; Englund, J.A.; Szilagyi, P.G.; Harrison, C.J.; et al. Innate susceptibility to norovirus infections influenced by FUT2 genotype in a United States pediatric population. Clin. Infect. Dis. 2015, 60, 1631–1638. [Google Scholar] [CrossRef] [PubMed]
- Lopman, B.A.; Trivedi, T.; Vicuna, Y.; Costantini, V.; Collins, N.; Gregoricus, N.; Parashar, U.; Sandoval, C.; Broncano, N.; Vaca, M.; et al. Norovirus Infection and Disease in an Ecuadorian Birth Cohort: Association of Certain Norovirus Genotypes With Host FUT2 Secretor Status. J. Infect. Dis. 2014, 211, 1813–1821. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Jin, M.; Xie, H.; Duan, Z.; Jiang, X.; Fang, Z. Outbreak studies of a GII-3 and a GII-4 norovirus revealed an association between HBGA phenotypes and viral infection. J. Med. Virol. 2008, 80, 1296–1301. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; He, Y.; Li, H.; Huang, P.; Zhong, W.; Yang, H.; Zhang, H.; Tan, M.; Duan, Z.J. Two gastroenteritis outbreaks caused by GII Noroviruses: Host susceptibility and HBGA phenotypes. PLoS ONE 2013, 8, e58605. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Wang, X.; Lee, J.C.; Teunis, P.; Hu, S.; Paradise, H.T.; Moe, C. Genetic susceptibility to norovirus GII.3 and GII.4 infections in Chinese pediatric diarrheal disease. Pediatr. Infect. Dis. J. 2014, 33, e305–e309. [Google Scholar] [CrossRef] [PubMed]
- Ayouni, S.; Estienney, M.; Sdiri-Loulizi, K.; Ambert-Balay, K.; de Rougemont, A.; Aho, S.; Hammami, S.; Aouni, M.; Guediche, M.N.; Pothier, P.; et al. Relationship between GII.3 norovirus infections and blood group antigens in young children in Tunisia. Clin. Microbiol. Infect. 2015, 21, 874.e1–874.e8. [Google Scholar] [CrossRef] [PubMed]
- Van Trang, N.; Vu, H.T.; Le, N.T.; Huang, P.; Jiang, X.; Anh, D.D. Association between norovirus and rotavirus infection and histo-blood group antigen types in Vietnamese children. J. Clin. Microbiol. 2014, 52, 1366–1374. [Google Scholar] [CrossRef] [PubMed]
- Nordgren, J.; Nitiema, L.W.; Ouermi, D.; Simpore, J.; Svensson, L. Host genetic factors affect susceptibility to norovirus infections in Burkina Faso. PLoS ONE 2013, 8, e69557. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.F.; Huang, Q.; Long, Y.; Jiang, X.; Zhang, T.; Tan, M.; Zhang, Q.L.; Huang, Z.Y.; Li, Y.H.; Ding, Y.Q.; et al. An outbreak caused by GII.17 norovirus with a wide spectrum of HBGA-associated susceptibility. Sci. Rep. 2015, 5, 17687. [Google Scholar] [CrossRef] [PubMed]
- Thorven, M.; Grahn, A.; Hedlund, K.O.; Johansson, H.; Wahlfrid, C.; Larson, G.; Svensson, L. A homozygous nonsense mutation (428G→A) in the human secretor (FUT2) gene provides resistance to symptomatic norovirus (GGII) infections. J. Virol. 2005, 79, 15351–15355. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, B.; Kindberg, E.; Buesa, J.; Rydell, G.E.; Lidon, M.F.; Montava, R.; Abu Mallouh, R.; Grahn, A.; Rodriguez-Diaz, J.; Bellido, J.; et al. The G428A nonsense mutation in FUT2 provides strong but not absolute protection against symptomatic GII.4 Norovirus infection. PLoS ONE 2009, 4, e5593. [Google Scholar] [CrossRef] [PubMed]
- Costantini, V.P.; Cooper, E.M.; Hardaker, H.L.; Lee, L.E.; Bierhoff, M.; Biggs, C.; Cieslak, P.R.; Hall, A.J.; Vinje, J. Epidemiologic, Virologic, and Host Genetic Factors of Norovirus Outbreaks in Long-term Care Facilities. Clin. Infect. Dis. 2016, 62, 1–10. [Google Scholar] [CrossRef] [PubMed]
- de Graaf, M.; van Beek, J.; Vennema, H.; Podkolzin, A.T.; Hewitt, J.; Bucardo, F.; Templeton, K.; Mans, J.; Nordgren, J.; Reuter, G.; et al. Emergence of a novel GII.17 norovirus—End of the GII.4 era? Euro Surveill. 2015, 20, 21178. [Google Scholar] [CrossRef] [PubMed]
- Bucardo, F.; Kindberg, E.; Paniagua, M.; Grahn, A.; Larson, G.; Vildevall, M.; Svensson, L. Genetic susceptibility to symptomatic norovirus infection in Nicaragua. J. Med. Virol. 2009, 81, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Halperin, T.; Vennema, H.; Koopmans, M.; Kahila Bar-Gal, G.; Kayouf, R.; Sela, T.; Ambar, R.; Klement, E. No association between histo-blood group antigens and susceptibility to clinical infections with genogroup II norovirus. J. Infect. Dis. 2008, 197, 63–65. [Google Scholar] [CrossRef] [PubMed]
- Prystajecky, N.; Brinkman, F.S.; Auk, B.; Isaac-Renton, J.L.; Tang, P. Personalized genetic testing and norovirus susceptibility. Can. J. Infect. Dis. Med. Microbiol. 2014, 25, 222–224. [Google Scholar] [CrossRef] [PubMed]
- Le Pendu, J.; Ruvoen-Clouet, N.; Kindberg, E.; Svensson, L. Mendelian resistance to human norovirus infections. Semin. Immunol. 2006, 18, 375–386. [Google Scholar] [CrossRef] [PubMed]
- King, J.R.; Varade, J.; Hammarstrom, L. Fucosyltransferase Gene Polymorphisms and Lewisb-Negative Status Are Frequent in Swedish Newborns, With Implications for Infectious Disease Susceptibility and Personalized Medicine. J. Pediatr. Infect. Dis. Soc. 2018. [Google Scholar] [CrossRef] [PubMed]
- Nordgren, J.; Sharma, S.; Kambhampati, A.; Lopman, B.; Svensson, L. Innate Resistance and Susceptibility to Norovirus Infection. PLoS Pathog. 2016, 12, e1005385. [Google Scholar] [CrossRef] [PubMed]
- Bucardo, F.; Nordgren, J.; Carlsson, B.; Paniagua, M.; Lindgren, P.E.; Espinoza, F.; Svensson, L. Pediatric norovirus diarrhea in Nicaragua. J. Clin. Microbiol. 2008, 46, 2573–2580. [Google Scholar] [CrossRef] [PubMed]
- Bucardo, F.; Reyes, Y.; Becker-Dreps, S.; Bowman, N.; Gruber, J.F.; Vinje, J.; Espinoza, F.; Paniagua, M.; Balmaseda, A.; Svensson, L.; et al. Pediatric norovirus GII.4 infections in Nicaragua, 1999–2015. Infect. Genet. Evol. 2017, 55, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Huhti, L.; Szakal, E.D.; Puustinen, L.; Salminen, M.; Huhtala, H.; Valve, O.; Blazevic, V.; Vesikari, T. Norovirus GII-4 causes a more severe gastroenteritis than other noroviruses in young children. J. Infect. Dis. 2011, 203, 1442–1444. [Google Scholar] [CrossRef] [PubMed]
- Monedero, V.; Buesa, J.; Rodriguez-Diaz, J. The Interactions between Host Glycobiology, Bacterial Microbiota, and Viruses in the Gut. Viruses 2018, 10, 96. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.G.; Yang, T.Y.; Liu, T.C.; Lin, T.P.; Hu, C.J.; Kao, M.C.; Wang, N.M.; Tsai, F.J.; Peng, C.T.; Tsai, C.H. Molecular analysis of secretor type alpha(1,2)-fucosyltransferase gene mutations in the Chinese and Thai populations. Transfusion 1999, 39, 1013–1017. [Google Scholar] [CrossRef] [PubMed]
- Ruvoen-Clouet, N.; Magalhaes, A.; Marcos-Silva, L.; Breiman, A.; Figueiredo, C.; David, L.; Le Pendu, J. Increase in genogroup II.4 norovirus host spectrum by CagA-positive Helicobacter pylori infection. J. Infect. Dis. 2014, 210, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Miura, T.; Sano, D.; Suenaga, A.; Yoshimura, T.; Fuzawa, M.; Nakagomi, T.; Nakagomi, O.; Okabe, S. Histo-blood group antigen-like substances of human enteric bacteria as specific adsorbents for human noroviruses. J. Virol. 2013, 87, 9441–9451. [Google Scholar] [CrossRef] [PubMed]
- Bucardo, F.; Nordgren, J.; Carlsson, B.; Kindberg, E.; Paniagua, M.; Mollby, R.; Svensson, L. Asymptomatic norovirus infections in Nicaraguan children and its association with viral properties and histo-blood group antigens. Pediatr. Infect. Dis. J. 2010, 29, 934–939. [Google Scholar] [CrossRef] [PubMed]
- Phillips, G.; Tam, C.C.; Rodrigues, L.C.; Lopman, B. Prevalence and characteristics of asymptomatic norovirus infection in the community in England. Epidemiol. Infect. 2010, 138, 1454–1458. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, K.; Oka, T.; Takeda, N.; Hansman, G.S. Norovirus infections in symptomatic and asymptomatic food handlers in Japan. J. Clin. Microbiol. 2007, 45, 3996–4005. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Farkas, T.; Marionneau, S.; Zhong, W.; Ruvoen-Clouet, N.; Morrow, A.L.; Altaye, M.; Pickering, L.K.; Newburg, D.S.; LePendu, J.; et al. Noroviruses bind to human ABO, Lewis, and secretor histo-blood group antigens: Identification of 4 distinct strain-specific patterns. J. Infect. Dis. 2003, 188, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Larsson, M.M.; Rydell, G.E.; Grahn, A.; Rodriguez-Diaz, J.; Akerlind, B.; Hutson, A.M.; Estes, M.K.; Larson, G.; Svensson, L. Antibody prevalence and titer to norovirus (genogroup II) correlate with secretor (FUT2) but not with ABO phenotype or Lewis (FUT3) genotype. J. Infect. Dis. 2006, 194, 1422–1427. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Kumagai, A.; Ito, H.; Furukawa, S.; Someya, Y.; Takeda, N.; Ishii, K.; Wakita, T.; Narimatsu, H.; Shirato, H. Structural basis for the recognition of Lewis antigens by genogroup I norovirus. J. Virol. 2012, 86, 11138–11150. [Google Scholar] [CrossRef] [PubMed]
- Nasir, W.; Frank, M.; Koppisetty, C.A.; Larson, G.; Nyholm, P.G. Lewis histo-blood group alpha1,3/alpha1,4 fucose residues may both mediate binding to GII.4 noroviruses. Glycobiology 2012, 22, 1163–1172. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Xia, M.; Tan, M.; Huang, P.; Zhong, W.; Pang, X.L.; Lee, B.E.; Meller, J.; Wang, T.; Jiang, X. Genetic and phenotypic characterization of GII-4 noroviruses that circulated during 1987 to 2008. J. Virol. 2010, 84, 9595–9607. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Carlsson, B.; Czako, R.; Vene, S.; Haglund, M.; Ludvigsson, J.; Larson, G.; Hammarstrom, L.; Sosnovtsev, S.V.; Atmar, R.L.; et al. Human Sera Collected between 1979 and 2010 Possess Blocking-Antibody Titers to Pandemic GII.4 Noroviruses Isolated over Three Decades. J. Virol. 2017, 91, e00567-17. [Google Scholar] [CrossRef] [PubMed]
- Bull, R.A.; White, P.A. Mechanisms of GII.4 norovirus evolution. Trends Microbiol. 2011, 19, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Klement, E.; Halperin, T. Reply to Chan et al., Tan and Jiang, and St. Clair and Patel. J. Infect. Dis. 2008, 198, 942–943. [Google Scholar] [CrossRef]
- Gustavsson, L.; Norden, R.; Westin, J.; Lindh, M.; Andersson, L.M. Slow Clearance of Norovirus following Infection with Emerging Variants of Genotype GII.4 Strains. J. Clin. Microbiol. 2017, 55, 1533–1539. [Google Scholar] [CrossRef] [PubMed]
- Woodward, J.; Gkrania-Klotsas, E.; Kumararatne, D. Chronic norovirus infection and common variable immunodeficiency. Clin. Exp. Immunol. 2017, 188, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Echenique, I.A.; Stosor, V.; Gallon, L.; Kaufman, D.; Qi, C.; Zembower, T.R. Prolonged norovirus infection after pancreas transplantation: A case report and review of chronic norovirus. Transpl. Infect. Dis. 2016, 18, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Steyer, A.; Konte, T.; Sagadin, M.; Kolenc, M.; Skoberne, A.; Germ, J.; Dovc-Drnovsek, T.; Arnol, M.; Poljsak-Prijatelj, M. Intrahost Norovirus Evolution in Chronic Infection Over 5 Years of Shedding in a Kidney Transplant Recipient. Front. Microbiol. 2018, 9, 371. [Google Scholar] [CrossRef] [PubMed]
- van Beek, J.; van der Eijk, A.A.; Fraaij, P.L.; Caliskan, K.; Cransberg, K.; Dalinghaus, M.; Hoek, R.A.; Metselaar, H.J.; Roodnat, J.; Vennema, H.; et al. Chronic norovirus infection among solid organ recipients in a tertiary care hospital, The Netherlands, 2006–2014. Clin. Microbiol. Infect. 2017, 23, 265.e9–265.e13. [Google Scholar] [CrossRef] [PubMed]
- Lemes, L.G.; Correa, T.S.; Fiaccadori, F.S.; Cardoso, D.; Arantes Ade, M.; Souza, K.M.; Souza, M. Prospective study on Norovirus infection among allogeneic stem cell transplant recipients: Prolonged viral excretion and viral RNA in the blood. J. Clin. Virol. 2014, 61, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.; Vergoulidou, M.; Schreier, E.; Loddenkemper, C.; Reinwald, M.; Schmidt-Hieber, M.; Flegel, W.A.; Thiel, E.; Schneider, T. Norovirus gastroenteritis causes severe and lethal complications after chemotherapy and hematopoietic stem cell transplantation. Blood 2011, 117, 5850–5856. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, B.; Lindberg, A.M.; Rodriguez-Diaz, J.; Hedlund, K.O.; Persson, B.; Svensson, L. Quasispecies dynamics and molecular evolution of human norovirus capsid P region during chronic infection. J. Gen. Virol. 2009, 90, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Pietsch, C.; Ennuschat, N.; Hartel, S.; Liebert, U.G. Within-host evolution of virus variants during chronic infection with novel GII.P26-GII.26 norovirus. J. Clin. Virol. 2018, 108, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Doerflinger, S.Y.; Weichert, S.; Koromyslova, A.; Chan, M.; Schwerk, C.; Adam, R.; Jennewein, S.; Hansman, G.S.; Schroten, H. Human Norovirus Evolution in a Chronically Infected Host. mSphere 2017, 2, e00352-16. [Google Scholar] [CrossRef] [PubMed]
- Debbink, K.; Lindesmith, L.C.; Ferris, M.T.; Swanstrom, J.; Beltramello, M.; Corti, D.; Lanzavecchia, A.; Baric, R.S. Within-host evolution results in antigenically distinct GII.4 noroviruses. J. Virol. 2014, 88, 7244–7255. [Google Scholar] [CrossRef] [PubMed]
- Ettayebi, K.; Crawford, S.E.; Murakami, K.; Broughman, J.R.; Karandikar, U.; Tenge, V.R.; Neill, F.H.; Blutt, S.E.; Zeng, X.L.; Qu, L.; et al. Replication of human noroviruses in stem cell-derived human enteroids. Science 2016, 353, 1387–1393. [Google Scholar] [CrossRef] [PubMed]
- Costantini, V.; Morantz, E.K.; Browne, H.; Ettayebi, K.; Zeng, X.L.; Atmar, R.L.; Estes, M.K.; Vinje, J. Human Norovirus Replication in Human Intestinal Enteroids as Model to Evaluate Virus Inactivation. Emerg. Infect. Dis. 2018, 24, 1453–1464. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Hisaie, K.; Kurokawa, S.; Suzuki, A.; Sakon, N.; Uchida, Y.; Yuki, Y.; Kiyono, H. Human norovirus propagation in human induced pluripotent stem cell-derived intestinal epithelial cells. Cell. Mol. Gastroenterol. Hepatol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Treanor, J.J.; Atmar, R.L.; Frey, S.E.; Gormley, R.; Chen, W.H.; Ferreira, J.; Goodwin, R.; Borkowski, A.; Clemens, R.; Mendelman, P.M. A novel intramuscular bivalent norovirus virus-like particle vaccine candidate–reactogenicity, safety, and immunogenicity in a phase 1 trial in healthy adults. J. Infect. Dis. 2014, 210, 1763–1771. [Google Scholar] [CrossRef] [PubMed]
- Armah, G.E.; Cortese, M.M.; Dennis, F.E.; Yu, Y.; Morrow, A.L.; McNeal, M.M.; Lewis, K.D.C.; Awuni, D.A.; Armachie, J.; Parashar, U.D. Rotavirus Vaccine Take in Infants Is Associated With Secretor Status. J. Infect. Dis. 2018, 219, 746–749. [Google Scholar] [CrossRef] [PubMed]
- Bucardo, F.; Nordgren, J.; Reyes, Y.; Gonzalez, F.; Sharma, S.; Svensson, L. The Lewis A phenotype is a restriction factor for Rotateq and Rotarix vaccine-take in Nicaraguan children. Sci. Rep. 2018, 8, 1502. [Google Scholar] [CrossRef] [PubMed]
- Bry, L.; Falk, P.G.; Midtvedt, T.; Gordon, J.I. A model of host-microbial interactions in an open mammalian ecosystem. Science 1996, 273, 1380–1383. [Google Scholar] [CrossRef] [PubMed]
- Freitas, M.; Axelsson, L.G.; Cayuela, C.; Midtvedt, T.; Trugnan, G. Indigenous microbes and their soluble factors differentially modulate intestinal glycosylation steps in vivo. Use of a “lectin assay” to survey in vivo glycosylation changes. Histochem. Cell Biol. 2005, 124, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Rubio-del-Campo, A.; Coll-Marques, J.M.; Yebra, M.J.; Buesa, J.; Perez-Martinez, G.; Monedero, V.; Rodriguez-Diaz, J. Noroviral p-particles as an in vitro model to assess the interactions of noroviruses with probiotics. PLoS ONE 2014, 9, e89586. [Google Scholar]
- Rodriguez-Diaz, J.; Garcia-Mantrana, I.; Vila-Vicent, S.; Gozalbo-Rovira, R.; Buesa, J.; Monedero, V.; Collado, M.C. Relevance of secretor status genotype and microbiota composition in susceptibility to rotavirus and norovirus infections in humans. Sci. Rep. 2017, 7, 45559. [Google Scholar] [CrossRef] [PubMed]
- Wacklin, P.; Tuimala, J.; Nikkila, J.; Sebastian, T.; Makivuokko, H.; Alakulppi, N.; Laine, P.; Rajilic-Stojanovic, M.; Paulin, L.; de Vos, W.M.; et al. Faecal microbiota composition in adults is associated with the FUT2 gene determining the secretor status. PLoS ONE 2014, 9, e94863. [Google Scholar] [CrossRef] [PubMed]
- Turpin, W.; Bedrani, L.; Espin-Garcia, O.; Xu, W.; Silverberg, M.S.; Smith, M.I.; Guttman, D.S.; Griffiths, A.; Moayyedi, P.; Panaccione, R.; et al. FUT2 genotype and secretory status are not associated with fecal microbial composition and inferred function in healthy subjects. Gut Microbes 2018, 9, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Davenport, E.R.; Goodrich, J.K.; Bell, J.T.; Spector, T.D.; Ley, R.E.; Clark, A.G. ABO antigen and secretor statuses are not associated with gut microbiota composition in 1,500 twins. BMC Genom. 2016, 17, 941. [Google Scholar] [CrossRef] [PubMed]
- Turpin, W.; Espin-Garcia, O.; Xu, W.; Silverberg, M.S.; Kevans, D.; Smith, M.I.; Guttman, D.S.; Griffiths, A.; Panaccione, R.; Otley, A.; et al. Association of host genome with intestinal microbial composition in a large healthy cohort. Nat. Genet. 2016, 48, 1413–1417. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Thingholm, L.B.; Skieceviciene, J.; Rausch, P.; Kummen, M.; Hov, J.R.; Degenhardt, F.; Heinsen, F.A.; Ruhlemann, M.C.; Szymczak, S.; et al. Genome-wide association analysis identifies variation in vitamin D receptor and other host factors influencing the gut microbiota. Nat. Genet. 2016, 48, 1396–1406. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Allele | Mutation Type | Phenotype | Population |
|---|---|---|---|
| Se302 | Missense | Non-secretor a | Thai, Bangladeshi |
| Se385 | Missense | Weak-secretor | East Asian |
| Se428 | Nonsense | Non-secretor | Caucasian, African, Meso-American, Central Asian |
| Se571 | Nonsense | Non-secretor | Filipino and Samoan |
| Se628 | Nonsense | Non-secretor | Japanese and Chinese |
| Se658 | Nonsense | Non-secretor | Chinese |
| Se357,480,778 | Frameshift | Non-secretor | African |
| Se849 | Nonsense | Non-secretor | Chinese and Filipino |
| Sedel1 | Deletion | Non-secretor | Indian and Bangladeshi |
| Sedel2 | Deletion | Non-secretor | Samoan |
| Sefus | Fusion | Weak-secretor | Japanese |
| Genotype | Country | O | A | B | AB | Secretor | Non-Secretor | Type of Study | Reference | |
|---|---|---|---|---|---|---|---|---|---|---|
| GI | ||||||||||
| GI.1 | USA | 21/28 (75%) | 10/19 (53%) | 3/7 (43%) | 0/1 (0%) | 34/55 (62%) | 0/22 (0%) | Challenge | [28] | |
| GI.1 a | USA | 25/26 (96%) | 14/18 (78%) | 3/5 (60%) | 0/2 (0%) | 42/43 (98%) | 0/8 (0%) | Challenge | [34,35,37] | |
| GI.3 | Sweden | 10/23 (43%) | 14/32 (44%) | 2/12 (17%) | 0/1 (0%) | 26/68 (38%) | 7/15 (47%) | Outbreak | [12] | |
| GI.3 | Netherlands | 11/11 (100%) | 9/9 (100%) | 0/2 (0%) | 0/0 (0%) | 20/22 (91%) | 4/7 (57%) | Outbreak | [39] | |
| GII-nonGII.4 | ||||||||||
| GII.1 | USA | N.A | N.A | N.A | N.A | + c | + | Active surveillance | [40] | |
| GII.1 | Ecuador | N.A | N.A | N.A | N.A | + | + | Birth cohort | [41] | |
| GII.2 a | USA | 4/8 (50%) | 2/4 (50%) | 1/1 (100%) | 2/2 (100%) | 8/12 (67%) | 1/3 (33%) | Challenge | [36] | |
| GII.3 b | China | 3/6 (50%) | 5/8 (63%) | 1/2 (50%) | 0/0 (0%) | 9/14 (64%) | 0/2 (0%) | Outbreak | [42] | |
| GII.3 b | China | 8/22 (36%) | 4/6 (67%) | 4/8 (50%) | 3/3 (100%) | 18/38 (47%) | 1/1 (100%) | Outbreak | [43] | |
| GII.3 b | China | N.A | N.A | N.A | N.A | 15/102 (15%) | 1/22 (5%) | Active surveillance | [44] | |
| GII.3 | Tunisia | + | + | + | + | 23/76 (30%) | 5/22 (23%) | Passive surveillance | [45] | |
| GII.3 d | Vietnam | N.A | N.A | N.A | N.A | 23/229 (10%) | 5/31 (16%) | Surveillance | [46] | |
| GII.6 | USA | N.A | N.A | N.A | N.A | + | +/− | Active surveillance | [40] | |
| GII.7 | USA | N.A | N.A | N.A | N.A | + | + | Active surveillance | [40] | |
| GII.10 | Burkina Faso | 4/71 (6%) | 1/34 (3%) | 0/55 (0%) | 0/4 (0%) | 5/164 (3%) | 0/44 (0%) | Passive surveillance | [47] | |
| GII.17 | China | 23/69 (33%) | 23/46 (50%) | 18/47 (38%) | 3/7 (43%) | 67/169 (40%) | 2/23 (9%) | Outbreak | [48] | |
| GII.23 | Ecuador | N.A | N.A | N.A | N.A | + | − | Birth cohort | [41] | |
| GII.4 Variant | Country | O | A | B | AB | Secretor | Non-Secretor | Type of Study | Reference |
|---|---|---|---|---|---|---|---|---|---|
| New Orleans 2009 | USA | N.A | N.A | N.A | N.A | + a | − | Active surveillance | [40] |
| Den Haag 2006b | USA | N.A | N.A | N.A | N.A | + | − | Active surveillance | [40] |
| Sydney 2012 | USA | N.A | N.A | N.A | N.A | + | − | Active surveillance | [40] |
| Den Haag 2006b | USA | + | + | + | + | 15/25 (60%) | 1/2 (50%) | Outbreaks | [51] |
| New Orleans 2009 | USA | + | + | + | + | 39/49 (80%) | 2/3 (66%) | Outbreaks | [51] |
| Sydney 2012 | USA | + | + | + | + | 5/9 (55%) | 0/2 (0%) | Outbreaks | [51] |
| New Orleans 2009 b | Ecuador | N.A | N.A | N.A | N.A | + | − | Birth cohort | [41] |
| Den Haag 2006b b | Ecuador | N.A | N.A | N.A | N.A | + | − | Birth cohort | [41] |
| Farmington Hills 2002 | USA | NA | NA | NA | NA | 16/23 (70%) | 1/17 (6%) | Challenge | [33] |
| Den Haag 2006b | China | N.A | N.A | N.A | N.A | 17/102 (17%) | 1/22 (5%) | Active surveillance | [44] |
| Hunter 2004 | Spain | N.A | N.A | N.A | N.A | 33/43 (77%) | 1/17 (6%) | Outbreak | [50] |
| N.A | Denmark | N.A | N.A | N.A | N.A | 29/52 (56%) | 0/9 (0%) | Outbreak | [30] |
| N.A | Sweden | N.A | N.A | N.A | N.A | 53/97 (55%) | 0/18 (0%) | Outbreaks | [49] |
| N.A | China | 7/50 (14%) | 25/52 (48%) | 9/28 (32%) | 3/5 (60%) | 41/115 (36%) | 0/15 (0%) | Outbreak | [42] |
| Den Haag 2006b c | China | 24/35 (69%) | 20/28 (71%) | 7/9 (78%) | 2/2 (100%) | 49/69 (71%) | 4/5 (80%) | Outbreak | [43] |
| Mixed d,e | Vietnam | N.A | N.A | N.A | N.A | 22/229 (10%) | 0/31 (0%) | Surveillance | [46] |
| Mixed | Burkina Faso | 1/71 (1%) | 0/34 (0%) | 7/55 (13%) | 1/4 (25%) | 9/164 (5%) | 1/44 (2%) | Passive surveillance | [47] |
| Mixed | Nicaragua | + | + | + | − f | + | − f | Passive surveillance | [53] |
| Unknown c | Israel | 16/30 (53%) | 17/34 (50%) | 7/16 (44%) | 6/11 (55%) | N.A | N.A | Outbreak | [54] |
| Unknown c | Israel | 36/66 (55%) | 28/77 (36%) | 21/45 (47%) | 7/25 (28%) | N.A | N.A | Outbreak | [54] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nordgren, J.; Svensson, L. Genetic Susceptibility to Human Norovirus Infection: An Update. Viruses 2019, 11, 226. https://doi.org/10.3390/v11030226
Nordgren J, Svensson L. Genetic Susceptibility to Human Norovirus Infection: An Update. Viruses. 2019; 11(3):226. https://doi.org/10.3390/v11030226
Chicago/Turabian StyleNordgren, Johan, and Lennart Svensson. 2019. "Genetic Susceptibility to Human Norovirus Infection: An Update" Viruses 11, no. 3: 226. https://doi.org/10.3390/v11030226
APA StyleNordgren, J., & Svensson, L. (2019). Genetic Susceptibility to Human Norovirus Infection: An Update. Viruses, 11(3), 226. https://doi.org/10.3390/v11030226