Genetic Evolution and Molecular Selection of the HE Gene of Influenza C Virus

Abstract
1. Introduction
2. Materials and Methods
2.1. Sequence Data
2.2. Phylogenetic Analysis
2.3. Selection Analysis
2.4. Nucleotide Composition Analysis
2.5. Relative Synonymous Codon Usage (RSCU)
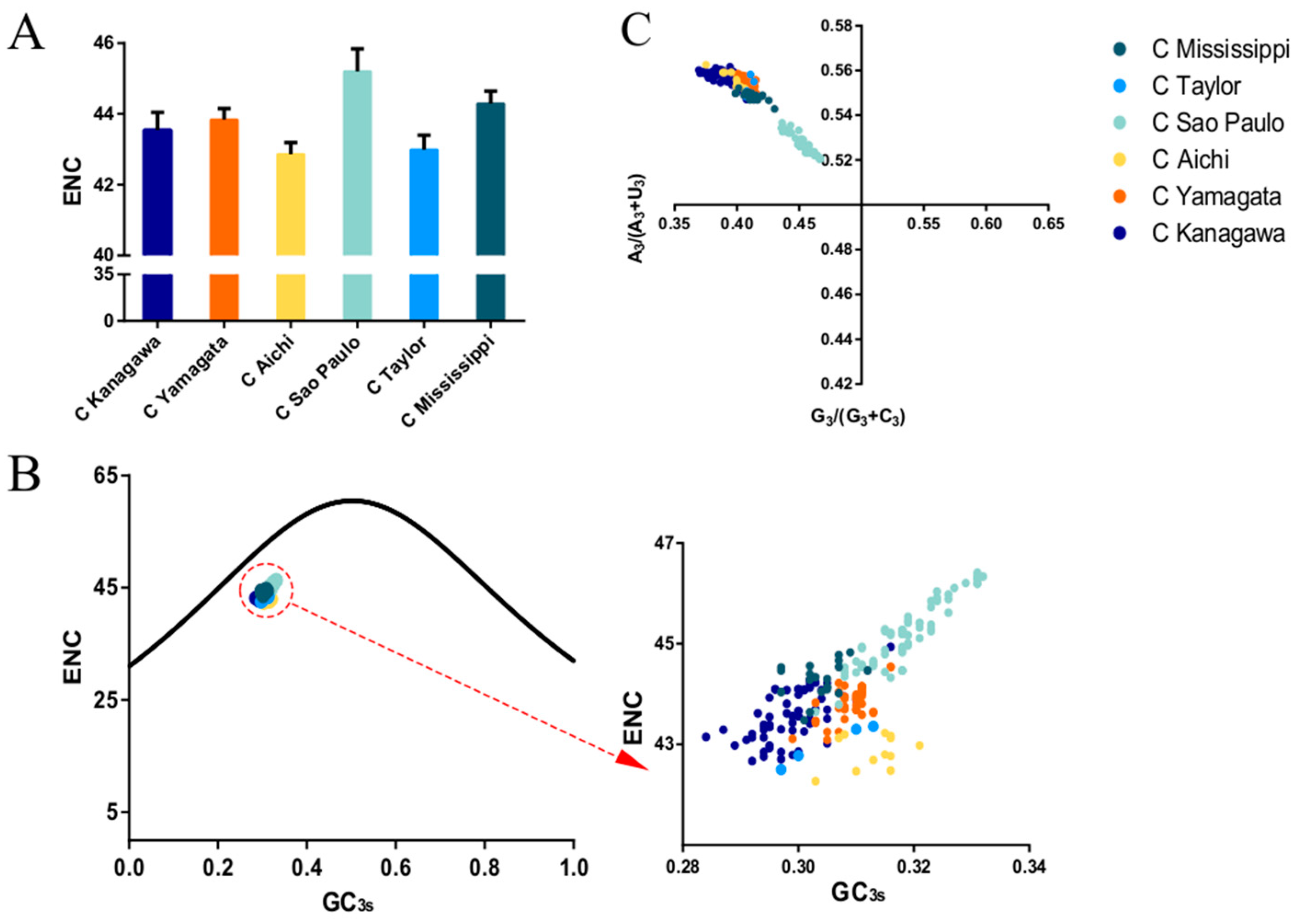
2.6. Effective Number of Codons (ENC)
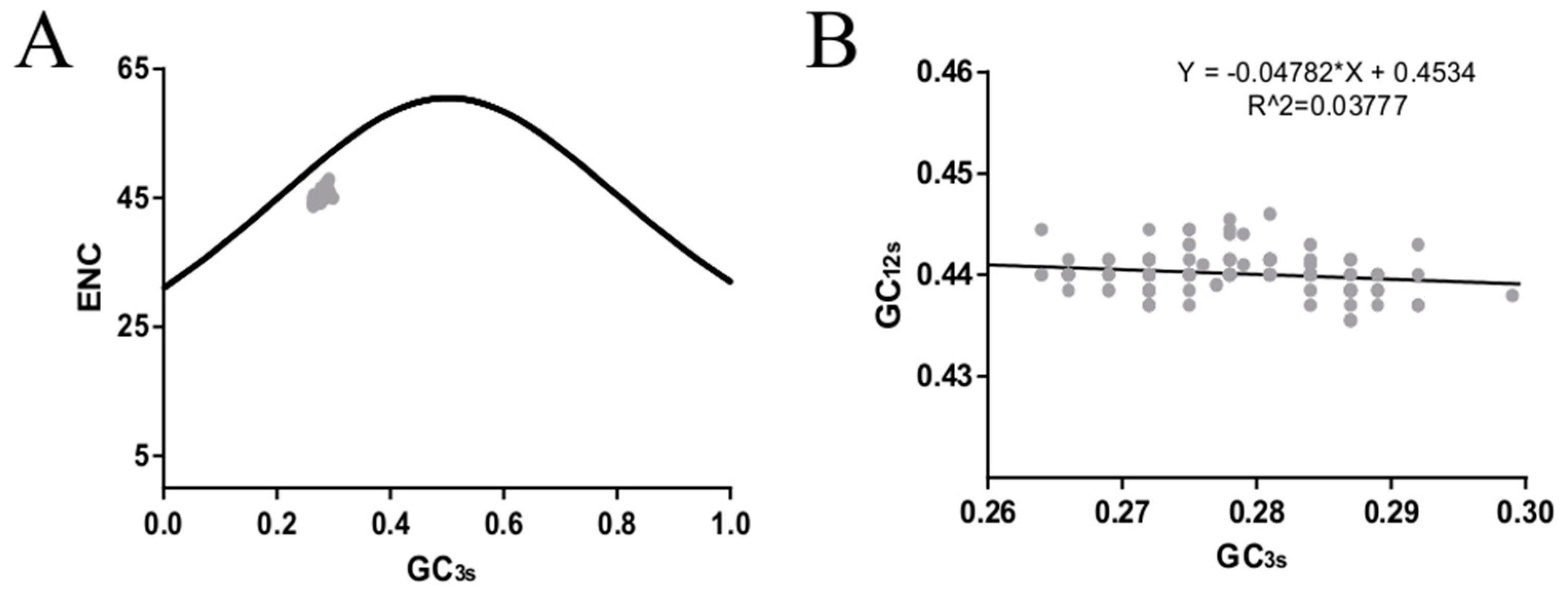
2.7. ENC Plot
2.8. Neutrality Plot Analysis
2.9. Principal Component Analysis (PCA)
2.10. Parity Rule 2 Analysis (PR2)
2.11. Codon Adaptation Index (CAI) Analysis
2.12. Relative Codon Deoptimization Index (RCDI)
2.13. Similarity Index (SiD)
2.14. Relative Dinucleotide Abundance Analysis
3. Results
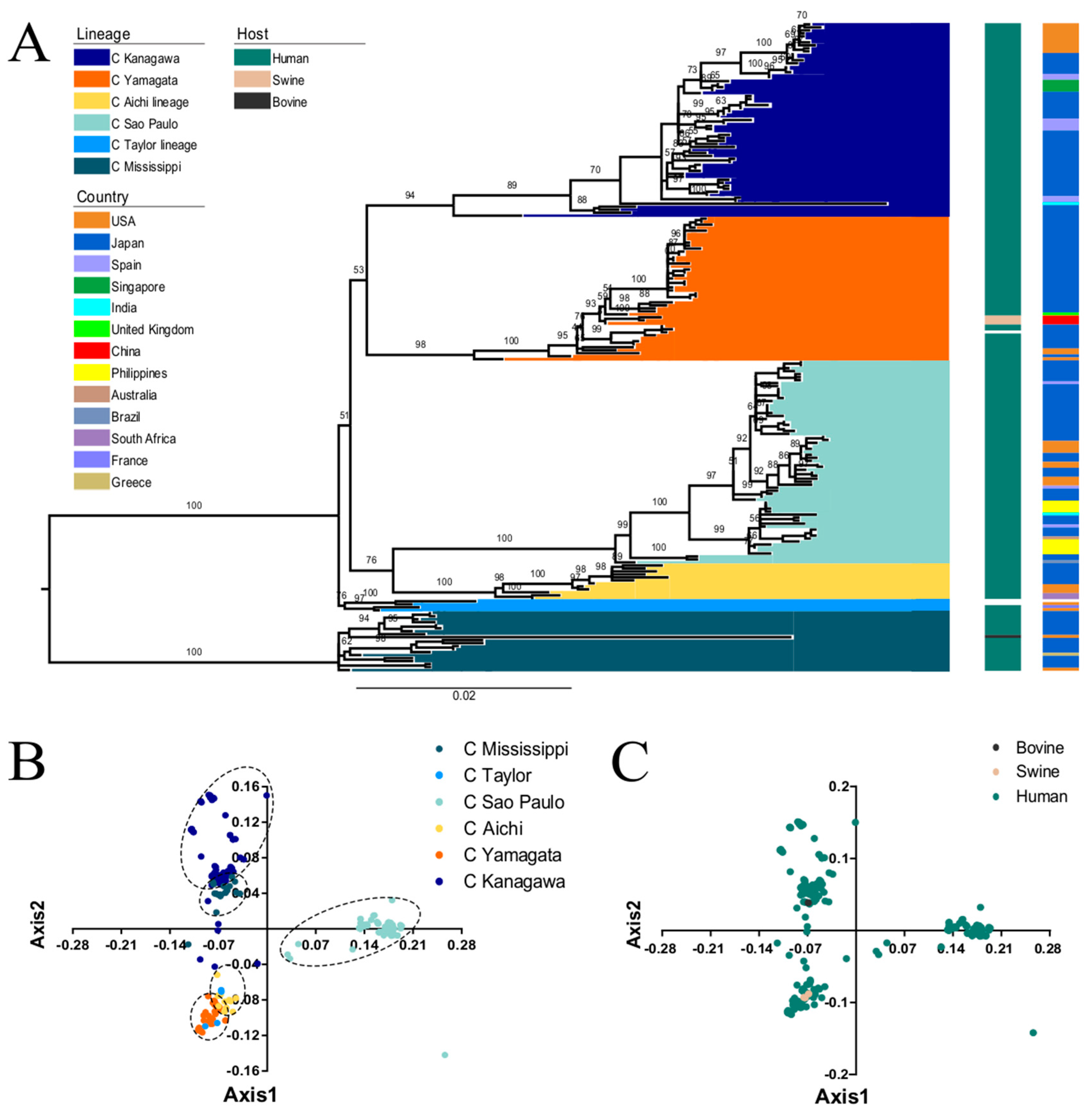
3.1. Phylogenetic Analysis of the ICV HE Gene
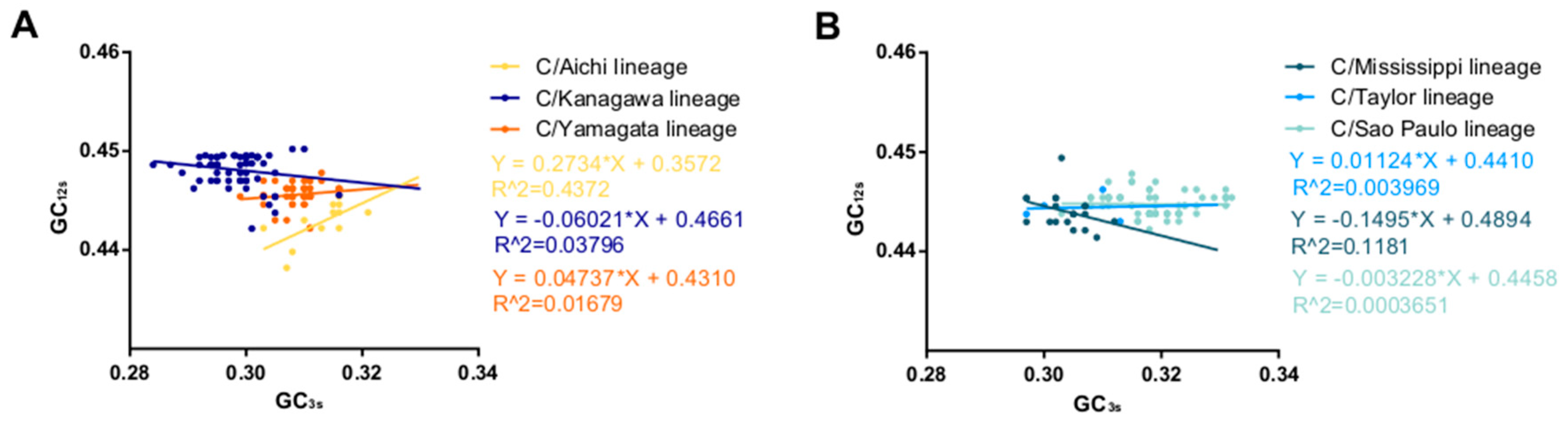
3.2. PCA Analysis of ICV HE Gene
3.3. The ICV HE Coding Regions Are Rich in A and U Nucleotides
3.4. Codon Usage Bias of the HE Coding Sequence
3.5. The HE Gene Displays a Contrasting Codon Usage Pattern with its Hosts
3.6. Natural Selection Dominates the Codon Usage Pattern of the ICV HE Gene
3.7. Dinucleotide Abundance Plays an Important Role in Shaping the Codon Usage Bias of the HE Gene
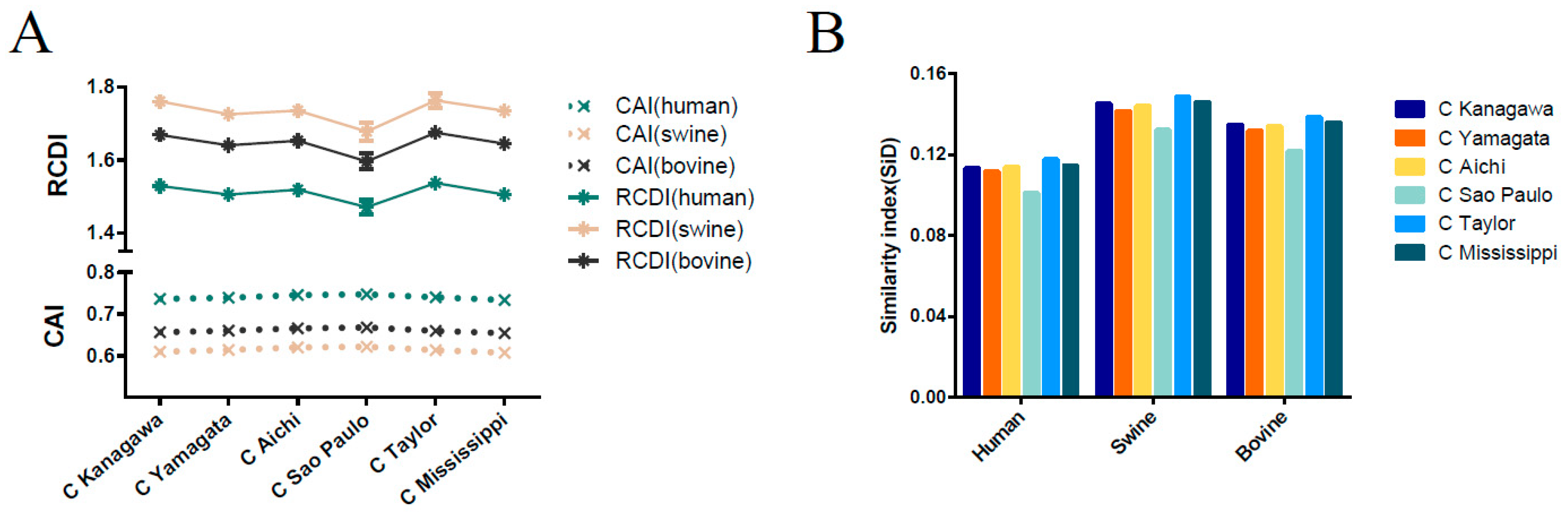
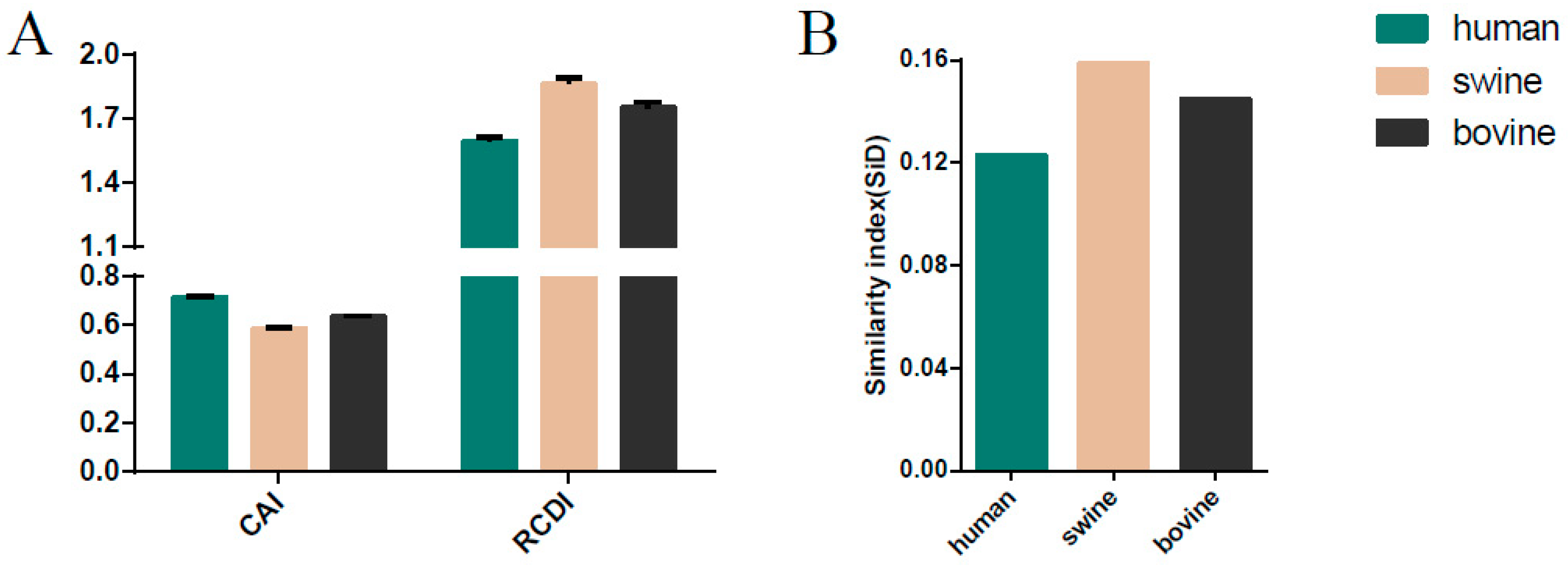
3.8. Host-Specific Codon Adaption Patterns of ICV
3.9. Sus scrofa Induced the Stronger Selection Pressure on ICV
3.10. Selection Analysis of the ICV HE Gene
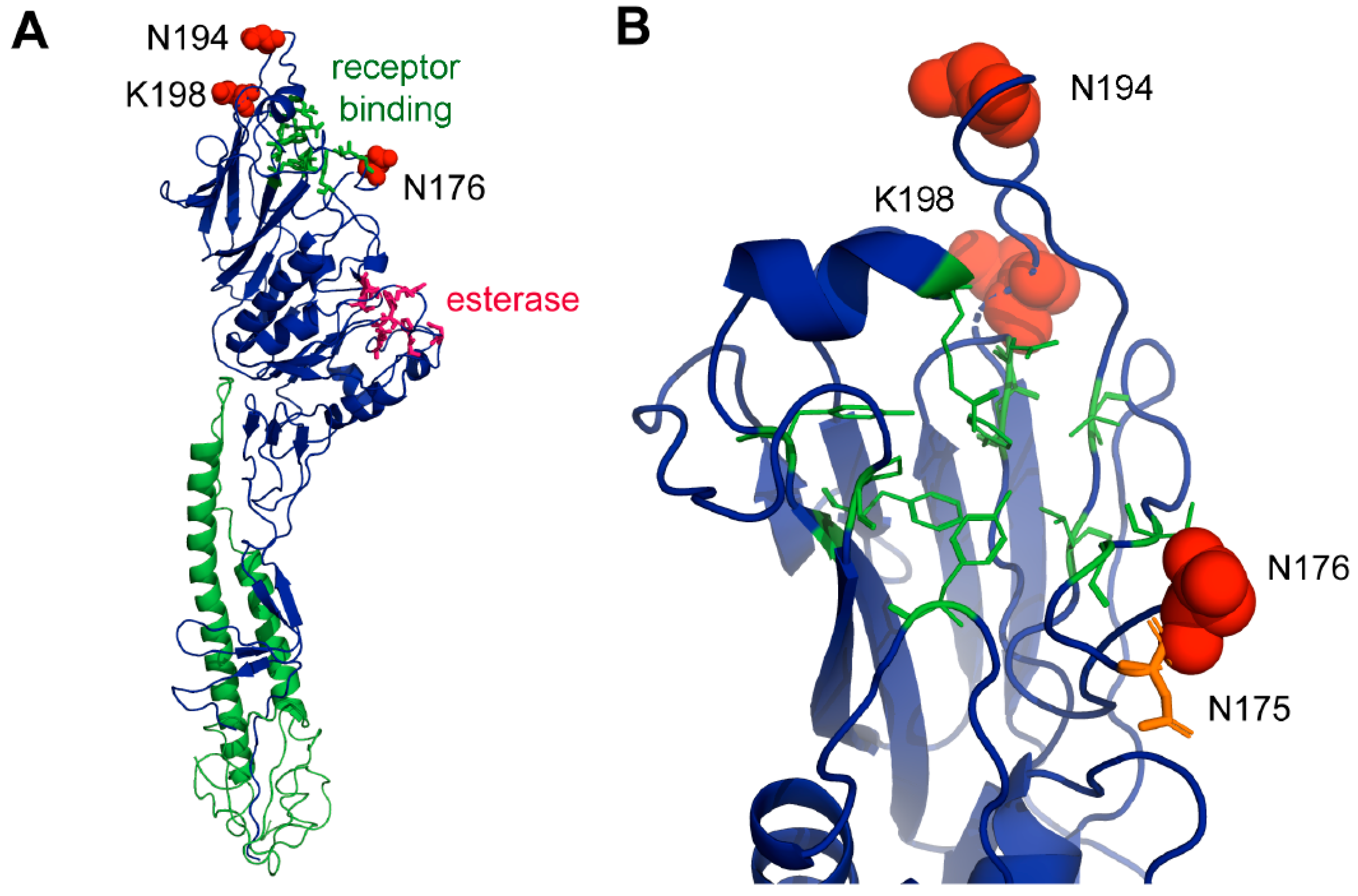
3.11. Structural Analysis of Sites under Positive Selection
4. Discussion
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Taylor, R.M. Studies on survival of influenza virus between epidemics and antigenic variants of the virus. Am. J. Public Health Nations Health 1949, 39, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Dykes, A.C.; Cherry, J.D.; Nolan, C.E. A clinical, epidemiologic, serologic, and virologic study of influenza c virus infection. Arch. Intern. Med. 1980, 140, 1295. [Google Scholar] [CrossRef] [PubMed]
- O’Callaghan, R.J.; Gohd, R.S.; Labat, D.D. Human antibody to influenza c virus: Its age-related distribution and distinction from receptor analogs. Infect. Immun. 1980, 30, 500–505. [Google Scholar] [PubMed]
- Homma, M.; Ohyama, S.; Katagiri, S. Age distribution of the antibody to type c influenza virus. Microbiol. Immunol. 1982, 26, 639–642. [Google Scholar] [CrossRef]
- Guo, Y.J.; Jin, F.G.; Wang, P.; Wang, M.; Zhu, J.M. Isolation of influenza c virus from pigs and experimental infection of pigs with influenza c virus. J. Gen. Virol. 1983, 64, 177–182. [Google Scholar] [PubMed]
- Zhang, H.; Porter, E.; Lohman, M.; Lu, N.; Peddireddi, L.; Hanzlicek, G.; Marthaler, D.; Liu, X.; Bai, J. Influenza C virus in cattle with respiratory disease, united states, 2016–2018. Emerg. Infect. Dis. 2018, 24, 1926–1929. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, S.; Ohizumi, A.; Homma, M. An outbreak of type c influenza in a children’s home. J. Infect. Dis. 1983, 148, 51–56. [Google Scholar] [CrossRef]
- Moriuchi, H.; Katsushima, N.; Nishimura, H.; Nakamura, K.; Numazaki, Y. Community-acquired influenza c virus infection in children. J. Pediatr. 1991, 118, 235–238. [Google Scholar] [CrossRef]
- Matsuzaki, Y.; Katsushima, N.; Nagai, Y.; Shoji, M.; Itagaki, T.; Sakamoto, M.; Kitaoka, S.; Mizuta, K.; Nishimura, H. Clinical features of influenza c virus infection in children. J. Infect. Dis. 2006, 193, 1229–1235. [Google Scholar] [CrossRef]
- Shimizu, Y.; Abiko, C.; Ikeda, T.; Mizuta, K.; Matsuzaki, Y. Influenza c virus and human metapneumovirus infections in hospitalized children with lower respiratory tract illness. Pediatr. Infecti. Dis. J. 2015, 34, 1273–1275. [Google Scholar] [CrossRef]
- Thielen, B.K.; Friedlander, H.; Bistodeau, S.; Shu, B.; Lynch, B.; Martin, K.; Bye, E.; Comosabetti, K.; Boxrud, D.; Strain, A.K. Detection of influenza C viruses among outpatients and patients hospitalized for severe acute respiratory infection, minnesota, 2013–2016. Clin. Infect. Dis. 2018, 66, 1092–1098. [Google Scholar] [CrossRef]
- Herrler, G.; Compans, R.W.; Meier-Ewert, H. A precursor glycoprotein in influenza c virus. Virology 1979, 99, 49–56. [Google Scholar] [CrossRef]
- Herrler, G.; Dürkop, I.; Becht, H.; Klenk, H.D. The glycoprotein of influenza c virus is the haemagglutinin, esterase and fusion factor. J. Gen. Virol. 1988, 69, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Herrler, G.; Klenk, H.D. Structure and function of the hef glycoprotein of influenza C virus. Adv. Virus Res. 1991, 40, 213–234. [Google Scholar] [PubMed]
- Sugawara, K.; Kitame, F.; Nishimura, H.; Nakamura, K. Operational and topological analyses of antigenic sites on influenza c virus glycoprotein and their dependence on glycosylation. J. Gen. Virol. 1988, 69, 537. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, M.; Sugawara, K.; Adachi, K.; Hongo, S.; Nishimura, H.; Kitame, F.; Nakamura, K. Location of neutralizing epitopes on the hemagglutinin-esterase protein of influenza C virus. Virology 1992, 189, 79–87. [Google Scholar] [CrossRef]
- Sugawara, K.; Nishimura, H.; Hongo, S.; Muraki, Y.; Kitame, F.; Nakamura, K. Construction of an antigenic map of the haemagglutinin-esterase protein of influenza C virus. J. Gen. Virol. 1993, 74, 1661. [Google Scholar] [CrossRef] [PubMed]
- Nakada, S.; Creager, R.S.; Krystal, M.; Aaronson, R.P.; Palese, P. Influenza C virus hemagglutinin: Comparison with influenza a and b virus hemagglutinins. J. Virol. 1984, 50, 118–124. [Google Scholar] [PubMed]
- Pfeifer, J.B.; Compans, R.W. Structure of the influenza C glycoprotein gene as determined from cloned DNA. Virus Res. 1984, 1, 281–296. [Google Scholar] [CrossRef]
- Matsuzaki, Y.; Sugawara, K.; Furuse, Y.; Shimotai, Y.; Hongo, S.; Oshitani, H.; Mizuta, K.; Nishimura, H. Genetic lineage and reassortment of influenza C viruses circulating between 1947 and 2014. J. Virol. 2016, 90, 8251–8265. [Google Scholar] [CrossRef]
- Stoletzki, N.; Eyre-Walker, A. Synonymous codon usage in escherichia coli: Selection for translational accuracy. Mol. Biol. Evol. 2007, 24, 374. [Google Scholar] [CrossRef] [PubMed]
- Burns, C.C.; Shaw, J.; Campagnoli, R.; Jorba, J.; Vincent, A.; Quay, J.; Kew, O. Modulation of poliovirus replicative fitness in hela cells by deoptimization of synonymous codon usage in the capsid region. J. Virol. 2006, 80, 3259–3272. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.; Papamichail, D.; Coleman, J.R.; Skiena, S.; Wimmer, E. Reduction of the rate of poliovirus protein synthesis through large-scale codon deoptimization causes attenuation of viral virulence by lowering specific infectivity. J. Virol. 2006, 80, 9687–9696. [Google Scholar] [CrossRef] [PubMed]
- Costafreda, M.I.; Pérezrodriguez, F.J.; D’Andrea, L.; Guix, S.; Ribes, E.; Bosch, A.; Pintó, R.M. Hepatitis a virus adaptation to cellular shutoff is driven by dynamic adjustments of codon usage and results in the selection of populations with altered capsids. J. Virol. 2014, 88, 5029–5041. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. Mega7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Keane, T.M.; Creevey, C.J.; Pentony, M.M.; Naughton, T.J.; McLnerney, J.O. Assessment of methods for amino acid matrix selection and their use on empirical data shows that ad hoc assumptions for choice of matrix are not justified. BMC Evol. Biol. 2006, 6, 29. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. Raxml version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Pond, S.L.K.; Scheffler, K. Fubar: A fast, unconstrained bayesian approximation for inferring selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Kosakovsky Pond, S.L. Detecting individual sites subject to episodic diversifying selection. PLOS Genetics 2012, 8, e1002764. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Frost, S.D. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef]
- Sharp, P.M.; Li, W.H. An evolutionary perspective on synonymous codon usage in unicellular organisms. J. Mol. Evol. 1986, 24, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.H.; Smith, D.K.; Rabadan, R.; Peiris, M.; Poon, L.L. Codon usage bias and the evolution of influenza A viruses. Codon usage biases of influenza virus. BMC Evol. Biol. 2010, 10, 253. [Google Scholar] [CrossRef] [PubMed]
- Wright, F. The ’effective number of codons’ used in a gene. Gene 1990, 87, 23–29. [Google Scholar] [CrossRef]
- Comeron, J.M.; Aguadé, M. An evaluation of measures of synonymous codon usage bias. J. Mol. Evol. 1998, 47, 268. [Google Scholar] [CrossRef] [PubMed]
- Sueoka, N. Directional mutation pressure and neutral molecular evolution. Proc. Natl. Acad. Sci. USA 1988, 85, 2653–2657. [Google Scholar] [CrossRef] [PubMed]
- Sueoka, N. Intrastrand parity rules of DNA base composition and usage biases of synonymous codons. J. Mol. Evol. 1995, 40, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Sueoka, N. Translation-coupled violation of parity rule 2 in human genes is not the cause of heterogeneity of the DNA G+C content of third codon position. Gene 1999, 238, 53–58. [Google Scholar] [CrossRef]
- Sharp, P.M.; Li, W. The codon adaptation index-a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef]
- Puigbò, P.; Bravo, I.G.; Garciavallve, S. Caical: A combined set of tools to assess codon usage adaptation. Biol. Direct. 2008, 3, 1–8. [Google Scholar] [CrossRef]
- Puigbò, P.; Aragonès, L.; Garcia-Vallvé, S. Rcdi/ercdi: A web-server to estimate codon usage deoptimization. BMC Res. Notes 2010, 3, 87. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, J.; Sun, D.; Ma, Q.; Chen, H.; Ma, L.; Ding, Y.; Liu, Y. The distribution of synonymous codon choice in the translation initiation region of dengue virus. PLoS ONE 2013, 8, e77239. [Google Scholar] [CrossRef] [PubMed]
- Karlin, S.; Doerfler, W.; Cardon, L.R. Why is cpg suppressed in the genomes of virtually all small eukaryotic viruses but not in those of large eukaryotic viruses? J. Virol. 1994, 68, 2889–2897. [Google Scholar] [PubMed]
- Furuse, Y.; Matsuzaki, Y.; Nishimura, H.; Oshitani, H. Analyses of evolutionary characteristics of the hemagglutinin-esterase gene of influenza c virus during a period of 68 years reveals evolutionary patterns different from influenza A and B viruses. Viruses 2016, 8, 321. [Google Scholar] [CrossRef]
- Matsuzaki, Y.; Sugawara, K.; Abiko, C.; Ikeda, T.; Aoki, Y.; Mizuta, K.; Katsushima, N.; Katsushima, F.; Katsushima, Y.; Itagaki, T.; et al. Epidemiological information regarding the periodic epidemics of influenza c virus in japan (1996–2013) and the seroprevalence of antibodies to different antigenic groups. J. Clin. Virol. 2014, 61, 87–93. [Google Scholar] [CrossRef]
- Zhang, H.; Porter, E.P.; Lohman, M.; Lu, N.; Peddireddi, L.; Hanzlicek, G.; Marthaler, D.; Liu, X.; Bai, J. Complete genome sequence of an influenza C virus strain identified from a sick calf in the united states. Microbiol. Resour. Announc. 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Nasrullah, I.; Butt, A.M.; Tahir, S.; Idrees, M.; Tong, Y. Genomic analysis of codon usage shows influence of mutation pressure, natural selection, and host features on marburg virus evolution. BMC Evol. Biol. 2015, 15, 174. [Google Scholar] [CrossRef]
- Rosenthal, P.B.; Zhang, X.; Formanowski, F.; Fitz, W.; Wong, C.H.; Meier-Ewert, H.; Skehel, J.J.; Wiley, D.C. Structure of the haemagglutinin-esterase-fusion glycoprotein of influenza C virus. Nature 1998, 396, 92–96. [Google Scholar] [CrossRef]
- Wang, M.; Veit, M. Hemagglutinin-esterase-fusion (HEF) protein of influenza C virus. Protein Cell 2016, 7, 28–45. [Google Scholar] [CrossRef]
- Matsuzaki, Y.; Sugawara, K.; Furuse, Y.; Shimotai, Y.; Hongo, S.; Mizuta, K.; Nishimura, H. Neutralizing epitopes and residues mediating the potential antigenic drift of the hemagglutinin-esterase protein of influenza C virus. Viruses 2018, 10. [Google Scholar] [CrossRef]
- Sugahara, K.; Hongo, S.; Sugawara, K.; Li, Z.N.; Tsuchiya, E.; Muraki, Y.; Matsuzaki, Y.; Nakamura, K. Role of individual oligosaccharide chains in antigenic properties, intracellular transport, and biological activities of influenza c virus hemagglutinin-esterase protein. Virology 2001, 285, 153–164. [Google Scholar] [CrossRef]
- Jenkins, G.M.; Holmes, E.C. The extent of codon usage bias in human rna viruses and its evolutionary origin. Virus Res. 2003, 92, 1–7. [Google Scholar] [CrossRef]
- Greenbaum, B.D.; Levine, A.J.; Bhanot, G.; Rabadan, R. Patterns of evolution and host gene mimicry in influenza and other rna viruses. PLoS Pathog. 2008, 4, e1000079. [Google Scholar] [CrossRef] [PubMed]
- Belalov, I.S.; Lukashev, A.N. Causes and implications of codon usage bias in rna viruses. PLoS ONE 2013, 8, e56642. [Google Scholar] [CrossRef] [PubMed]
- Bera, B.C.; Virmani, N.; Kumar, N.; Anand, T.; Pavulraj, S.; Rash, A.; Elton, D.; Rash, N.; Bhatia, S.; Sood, R. Genetic and codon usage bias analyses of polymerase genes of equine influenza virus and its relation to evolution. BMC Genomics 2017, 18, 652. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Liu, W.J.; Peng, S.W.; Sun, X.Y.; Frazer, I. Papillomavirus capsid protein expression level depends on the match between codon usage and trna availability. J. Virol. 1999, 73, 4972–4982. [Google Scholar] [PubMed]
- Hause, B.M.; Ducatez, M.; Collin, E.A.; Ran, Z.; Liu, R.; Sheng, Z.; Armien, A.; Kaplan, B.; Chakravarty, S.; Hoppe, A.D.; et al. Isolation of a novel swine influenza virus from oklahoma in 2011 which is distantly related to human influenza c viruses. PLoS Pathog. 2013, 9, e1003176. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Fu, X.; Li, G.; Kerlin, F.; Veit, M. Novel influenza D virus: Epidemiology, pathology, evolution and biological characteristics. Virulence 2017, 8, 1580–1591. [Google Scholar] [CrossRef]
- Yan, Z.; Wang, R.; Zhang, L.; Shen, B.; Wang, N.; Xu, Q.; He, W.; He, W.; Li, G.; Su, S. Evolutionary changes of the novel influenza d virus hemagglutinin-esterase fusion gene revealed by the codon usage pattern. Virulence 2019, 10, 1–9. [Google Scholar] [CrossRef]
- Hu, J.S.; Wang, Q.Q.; Zhang, J.; Chen, H.T.; Xu, Z.W.; Zhu, L.; Ding, Y.Z.; Ma, L.N.; Xu, K.; Gu, Y.X. The characteristic of codon usage pattern and its evolution of hepatitis C virus. Infect. Genetics Evol. 2011, 11, 2098–2102. [Google Scholar] [CrossRef]
- Butt, A.M.; Nasrullah, I.; Qamar, R.; Tong, Y. Evolution of codon usage in zika virus genomes is host and vector specific. Emer. Microbes Infect. 2016, 5, e107. [Google Scholar] [CrossRef]
- Anhlan, D.; Grundmann, N.; Makalowski, W.; Ludwig, S.; Scholtissek, C. Origin of the 1918 pandemic H1N1 influenza A virus as studied by codon usage patterns and phylogenetic analysis. RNA 2011, 17, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Gu, W.; Ma, J.; Sun, X.; Lu, Z. Analysis of synonymous codon usage in H5N1 virus and other influenza A viruses. Biosystems 2005, 81, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Wang, R.; Zhang, C.; Wang, S.; He, W.; Zhang, J.; Liu, J.; Cai, Y.; Zhou, J.; Su, S. Genetic and evolutionary analysis of emerging H3N2 canine influenza virus. Emerg. Microbes Infect. 2018, 7, 73. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Zinovyev, A.; Képès, F. Codon adaptation index as a measure of dominating codon bias. Bioinformatics 2003, 19, 2005–2015. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Abiko, C.; Peng, G.; Muraki, Y.; Sugawara, K.; Hongo, S.; Kitame, F.; Mizuta, K.; Numazaki, Y.; Suzuki, H. Interspecies transmission of influenza c virus between humans and pigs. Virus Res. 1997, 48, 71–79. [Google Scholar] [CrossRef]
- Muraki, Y.; Hongo, S.; Sugawara, K.; Kitame, F.; Nakamura, K. Evolution of the haemagglutinin-esterase gene of influenza c virus. J. Gen. Virol. 1996, 77, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, Y.; Mizuta, K.; Sugawara, K.; Tsuchiya, E.; Muraki, Y.; Hongo, S.; Suzuki, H.; Nishimura, H. Frequent reassortment among influenza C viruses. J. Virol. 2003, 77, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, Y.; Takao, S.; Shimada, S.; Mizuta, K.; Sugawara, K.; Takashita, E.; Muraki, Y.; Hongo, S.; Nishimura, H. Characterization of antigenically and genetically similar influenza c viruses isolated in Japan during the 1999–2000 season. Epidemiol. Infect. 2004, 132, 709. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AA | Codon | Influenza C Virus HE Gene | Hosts | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Overall | C/Aichi | C/Kanagawa | C/Mississippi | C Sao Paul | C/Taylor | C Yamagata | H.sapiens | Sus scrofa | Bos taurus | ||
| Phe | UUU(F) | 1.27 | 1.2 | 1.3 | 1.39 | 1.24 | 1.21 | 1.24 | 0.93 | 0.79 | 0.85 |
| UUC(F) | 0.73 | 0.8 | 0.7 | 0.61 | 0.76 | 0.79 | 0.76 | 1.07 | 1.21 | 1.15 | |
| Leu | UUA(L) | 1.08 | 0.99 | 1.32 | 1.26 | 0.77 | 1 | 1.16 | 0.46 | 0.32 | 0.38 |
| UUG(L) | 1.87 | 1.95 | 1.66 | 1.78 | 2.26 | 1.97 | 1.62 | 0.77 | 0.67 | 0.71 | |
| CUU(L) | 1.18 | 1.06 | 1.22 | 1.25 | 1.13 | 1.2 | 1.21 | 0.79 | 1.35 | 0.7 | |
| CUC(L) | 0.28 | 0.47 | 0.3 | 0.11 | 0.32 | 0.31 | 0.23 | 1.17 | 1.35 | 1.26 | |
| CUA(L) | 0.72 | 0.76 | 0.8 | 0.9 | 0.61 | 0.66 | 0.66 | 0.43 | 0.33 | 0.36 | |
| CUG(L) | 0.86 | 0.76 | 0.69 | 0.69 | 0.91 | 0.87 | 1.11 | 2.37 | 2.68 | 2.59 | |
| Ile | AUU(I) | 1.33 | 1.37 | 1.34 | 1.32 | 1.39 | 1.33 | 1.22 | 1.08 | 0.91 | 0.98 |
| AUC(I) | 0.86 | 0.78 | 0.9 | 0.85 | 0.83 | 0.85 | 0.89 | 1.41 | 1.67 | 1.57 | |
| AUA(I) | 0.81 | 0.86 | 0.76 | 0.83 | 0.78 | 0.82 | 0.88 | 0.51 | 0.42 | 0.45 | |
| Val | GUU(V) | 1.39 | 1.23 | 1.4 | 1.35 | 1.47 | 1.47 | 1.31 | 0.73 | 0.57 | 0.64 |
| GUC(V) | 0.66 | 0.77 | 0.73 | 0.68 | 0.49 | 0.59 | 0.77 | 0.95 | 1.07 | 1.01 | |
| GUA(V) | 0.79 | 0.6 | 0.79 | 0.91 | 0.8 | 0.81 | 0.77 | 0.47 | 0.34 | 0.4 | |
| GUG(V) | 1.16 | 1.4 | 1.08 | 1.06 | 1.25 | 1.13 | 1.15 | 1.85 | 2.03 | 1.95 | |
| Ser | UCU(S) | 1.7 | 1.55 | 1.65 | 1.71 | 1.73 | 1.66 | 1.75 | 1.13 | 0.99 | 1.04 |
| UCC(S) | 0.28 | 0.44 | 0.32 | 0.27 | 0.24 | 0.32 | 0.26 | 1.31 | 1.5 | 1.37 | |
| UCA(S) | 1.74 | 1.8 | 1.72 | 1.57 | 1.82 | 1.82 | 1.69 | 0.9 | 0.73 | 0.79 | |
| UCG(S) | 0.05 | 0 | 0.15 | 0.11 | 0 | 0 | 0 | 0.33 | 0.39 | 0.39 | |
| AGU(S) | 0.96 | 1.02 | 0.8 | 0.8 | 1.16 | 0.95 | 0.92 | 0.9 | 0.77 | 0.87 | |
| AGC(S) | 1.27 | 1.19 | 1.36 | 1.54 | 1.05 | 1.26 | 1.38 | 1.44 | 1.62 | 1.53 | |
| Pro | CCU(P) | 1.68 | 1.9 | 1.68 | 1.35 | 1.83 | 1.59 | 1.56 | 1.15 | 1.05 | 1.08 |
| CCC(P) | 0.66 | 0.42 | 0.64 | 0.78 | 0.62 | 0.72 | 0.77 | 1.29 | 1.46 | 1.39 | |
| CCA(P) | 1.59 | 1.68 | 1.68 | 1.61 | 1.42 | 1.69 | 1.67 | 1.11 | 0.94 | 1 | |
| CCG(P) | 0.07 | 0 | 0 | 0.25 | 0.14 | 0 | 0.01 | 0.45 | 0.56 | 0.53 | |
| Thr | ACU(T) | 1.75 | 1.61 | 1.79 | 1.85 | 1.71 | 1.62 | 1.77 | 0.99 | 0.83 | 0.89 |
| ACC(T) | 0.99 | 1.08 | 0.97 | 0.9 | 1.08 | 0.98 | 0.93 | 1.42 | 1.68 | 1.55 | |
| ACA(T) | 1.2 | 1.19 | 1.24 | 1.13 | 1.21 | 1.21 | 1.18 | 1.14 | 0.92 | 1.01 | |
| ACG(T) | 0.05 | 0.12 | 0.01 | 0.12 | 0 | 0.19 | 0.12 | 0.46 | 0.57 | 0.56 | |
| Ala | GCU(A) | 1.94 | 2.02 | 1.9 | 1.84 | 2.02 | 1.92 | 1.89 | 1.06 | 0.96 | 1 |
| GCC(A) | 0.54 | 0.61 | 0.51 | 0.54 | 0.5 | 0.56 | 0.6 | 1.6 | 1.8 | 1.71 | |
| GCA(A) | 1.32 | 1.38 | 1.4 | 1.51 | 1.21 | 1.36 | 1.28 | 0.91 | 0.74 | 0.8 | |
| GCG(A) | 0.2 | 0 | 0.19 | 0.11 | 0.26 | 0.17 | 0.22 | 0.42 | 0.5 | 0.48 | |
| Tyr | UAU(Y) | 1.13 | 1.1 | 1.18 | 1.23 | 1.1 | 1.11 | 1.09 | 0.89 | 0.73 | 0.79 |
| UAC(Y) | 0.87 | 0.9 | 0.82 | 0.77 | 0.9 | 0.89 | 0.91 | 1.11 | 1.27 | 1.21 | |
| His | CAU(H) | 1.61 | 1.5 | 1.73 | 1.74 | 1.57 | 1.57 | 1.46 | 0.84 | 0.7 | 0.75 |
| CAC(H) | 0.39 | 0.5 | 0.27 | 0.26 | 0.43 | 0.43 | 0.54 | 1.16 | 1.3 | 1.25 | |
| Gln | CAA(Q) | 1.74 | 1.82 | 1.79 | 1.73 | 1.68 | 1.84 | 1.73 | 0.53 | 0.44 | 0.46 |
| CAG(Q) | 0.26 | 0.18 | 0.21 | 0.27 | 0.32 | 0.16 | 0.27 | 1.47 | 1.56 | 1.54 | |
| Asn | AAU(N) | 1.35 | 1.4 | 1.3 | 1.36 | 1.37 | 1.37 | 1.37 | 1.08 | 0.79 | 0.81 |
| AAC(N) | 0.65 | 0.6 | 0.7 | 0.64 | 0.63 | 0.63 | 0.63 | 1.41 | 1.21 | 1.19 | |
| Lys | AAA(K) | 1.55 | 1.51 | 1.5 | 1.52 | 1.63 | 1.48 | 1.53 | 0.87 | 0.76 | 0.78 |
| AAG(K) | 0.45 | 0.49 | 0.5 | 0.48 | 0.37 | 0.52 | 0.47 | 1.13 | 1.24 | 1.22 | |
| Asp | GAU(D) | 1.12 | 1.04 | 1.12 | 1.01 | 1.11 | 1.14 | 1.19 | 0.93 | 0.8 | 0.84 |
| GAC(D) | 0.88 | 0.96 | 0.88 | 0.99 | 0.89 | 0.86 | 0.81 | 1.07 | 1.2 | 1.16 | |
| Glu | GAA(E) | 1.69 | 1.72 | 1.79 | 1.67 | 1.57 | 1.75 | 1.73 | 0.84 | 0.72 | 0.78 |
| GAG(E) | 0.31 | 0.28 | 0.21 | 0.33 | 0.43 | 0.25 | 0.27 | 1.16 | 1.28 | 1.22 | |
| Cys | UGU(C) | 0.79 | 0.61 | 0.77 | 0.86 | 0.84 | 0.74 | 0.75 | 0.91 | 0.79 | 0.85 |
| UGC(C) | 1.21 | 1.39 | 1.23 | 1.14 | 1.16 | 1.26 | 1.25 | 1.09 | 1.21 | 1.15 | |
| Arg | CGU(R) | 0.17 | 0.25 | 0.23 | 0.24 | 0.01 | 0.25 | 0.25 | 0.48 | 0.44 | 0.49 |
| CGC(R) | 0.07 | 0 | 0 | 0 | 0.24 | 0 | 0 | 1.1 | 1.31 | 1.17 | |
| CGA(R) | 0.19 | 0 | 0.31 | 0.23 | 0.25 | 0 | 0.01 | 0.65 | 0.6 | 0.68 | |
| CGG(R) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1.21 | 1.29 | 1.32 | |
| AGA(R) | 4.24 | 4.7 | 4.25 | 4.46 | 3.66 | 4.76 | 4.8 | 1.29 | 1.12 | 1.14 | |
| AGG(R) | 1.32 | 1.05 | 1.22 | 1.06 | 1.84 | 0.99 | 0.94 | 1.27 | 1.23 | 1.2 | |
| Gly | GGU(G) | 0.3 | 0.29 | 0.29 | 0.39 | 0.3 | 0.3 | 0.29 | 0.65 | 0.57 | 0.64 |
| GGC(G) | 0.54 | 0.57 | 0.58 | 0.58 | 0.51 | 0.48 | 0.5 | 1.35 | 1.46 | 1.43 | |
| GGA(G) | 2.51 | 2.48 | 2.62 | 2.39 | 2.42 | 2.63 | 2.52 | 1 | 0.91 | 0.95 | |
| GGG(G) | 0.65 | 0.67 | 0.51 | 0.64 | 0.76 | 0.59 | 0.68 | 1 | 1.05 | 0.99 | |
| Gene | AA | FEL | SLAC | FUBAR | MEME | ||||
|---|---|---|---|---|---|---|---|---|---|
| dN-dS | p-Value | dN-dS | p-Value | dN-dS | Post.Pro | ω + | p-Value | ||
| HE | 176 | 2.019 | 0.019 | 5.44 | 0.101 | 4.873 | 0.983 | >100 | 0.03 |
| 194 | 4.011 | 0.008 | 11.1 | 0.016 | 9.813 | 0.998 | >100 | 0.00 | |
| 198 | 3.679 | 0.020 | 8.64 | 0.022 | 8.031 | 0.992 | >100 | 0.03 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, W.; Zhang, L.; He, W.; Zhang, X.; Wen, B.; Wang, C.; Xu, Q.; Li, G.; Zhou, J.; Veit, M.; et al. Genetic Evolution and Molecular Selection of the HE Gene of Influenza C Virus. Viruses 2019, 11, 167. https://doi.org/10.3390/v11020167
Zhang W, Zhang L, He W, Zhang X, Wen B, Wang C, Xu Q, Li G, Zhou J, Veit M, et al. Genetic Evolution and Molecular Selection of the HE Gene of Influenza C Virus. Viruses. 2019; 11(2):167. https://doi.org/10.3390/v11020167
Chicago/Turabian StyleZhang, Wenyan, Letian Zhang, Wanting He, Xu Zhang, Baiqing Wen, Congcong Wang, Qiuhua Xu, Gairu Li, Jiyong Zhou, Michael Veit, and et al. 2019. "Genetic Evolution and Molecular Selection of the HE Gene of Influenza C Virus" Viruses 11, no. 2: 167. https://doi.org/10.3390/v11020167
APA StyleZhang, W., Zhang, L., He, W., Zhang, X., Wen, B., Wang, C., Xu, Q., Li, G., Zhou, J., Veit, M., & Su, S. (2019). Genetic Evolution and Molecular Selection of the HE Gene of Influenza C Virus. Viruses, 11(2), 167. https://doi.org/10.3390/v11020167