Geographic Distribution of HCV-GT3 Subtypes and Naturally Occurring Resistance Associated Substitutions

 and
and
Abstract
:1. Introduction
2. Materials and Methods
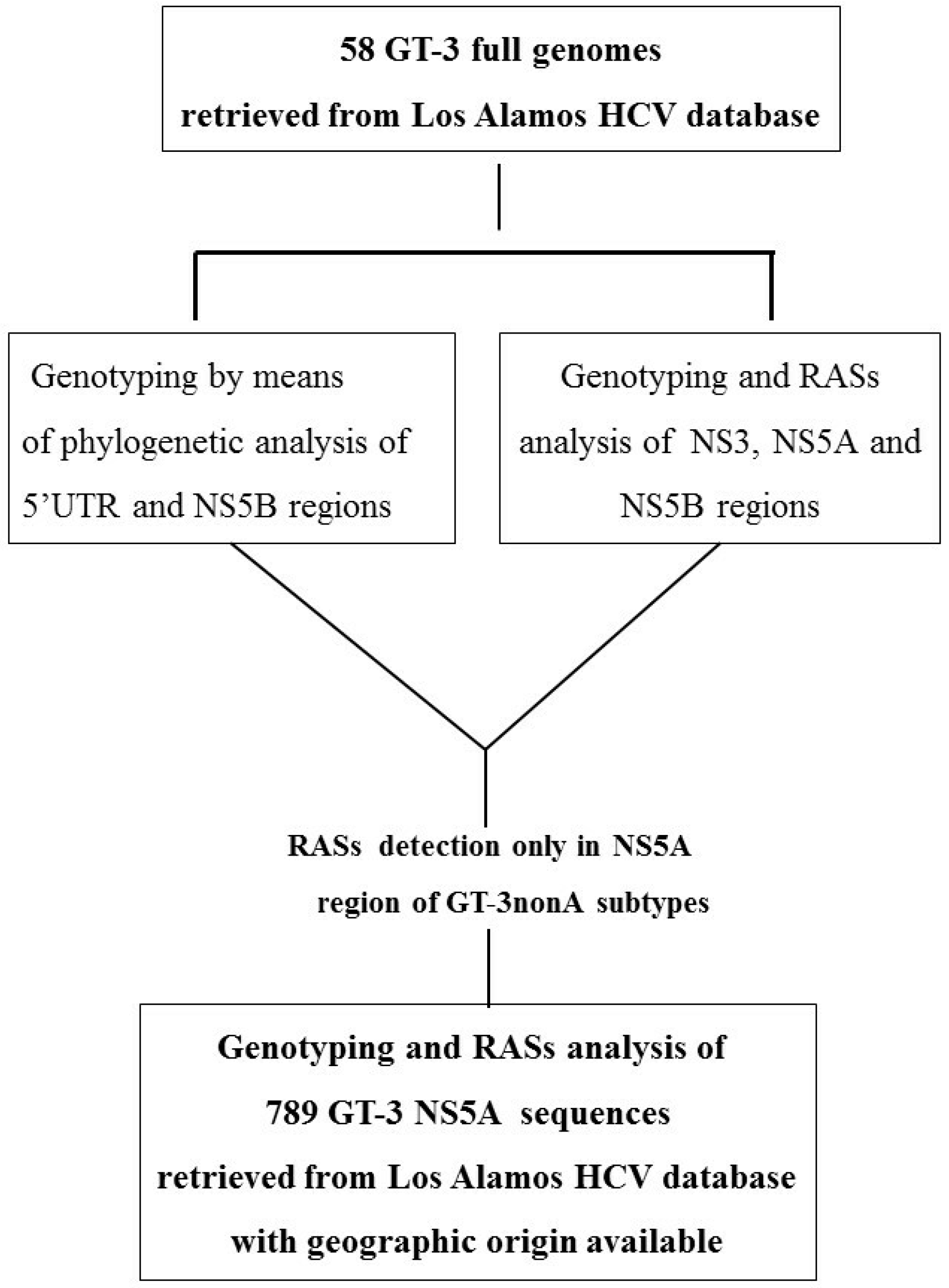
2.1. Design of the Study and Methodological Approach
2.2. Phylogenetic Analysis of 5′UTR, NS3, NS5A, and NS5B Domains
2.3. Selective Pressure Analysis on NS5A Domain 1
2.4. Statistical Analysis
3. Results
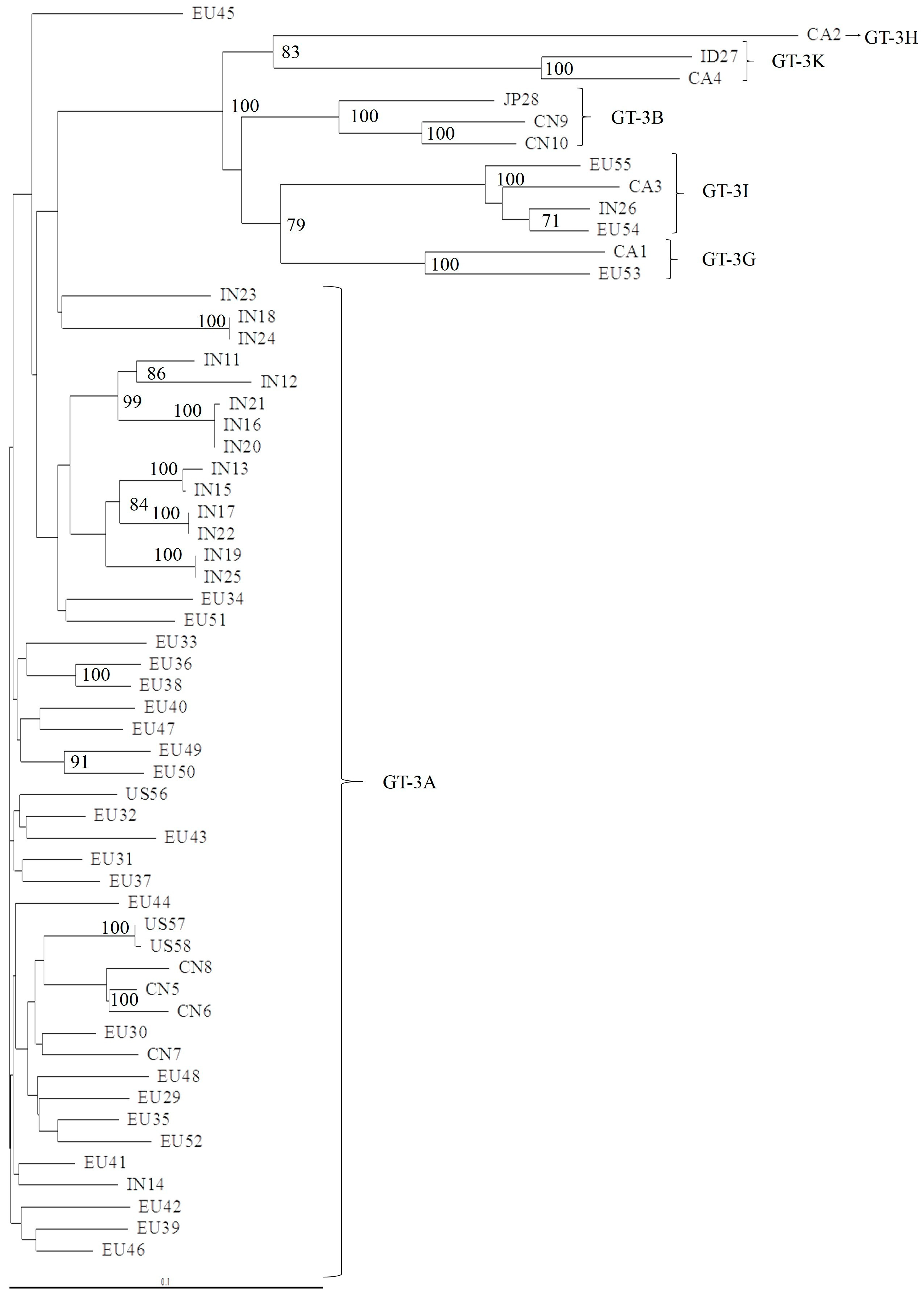
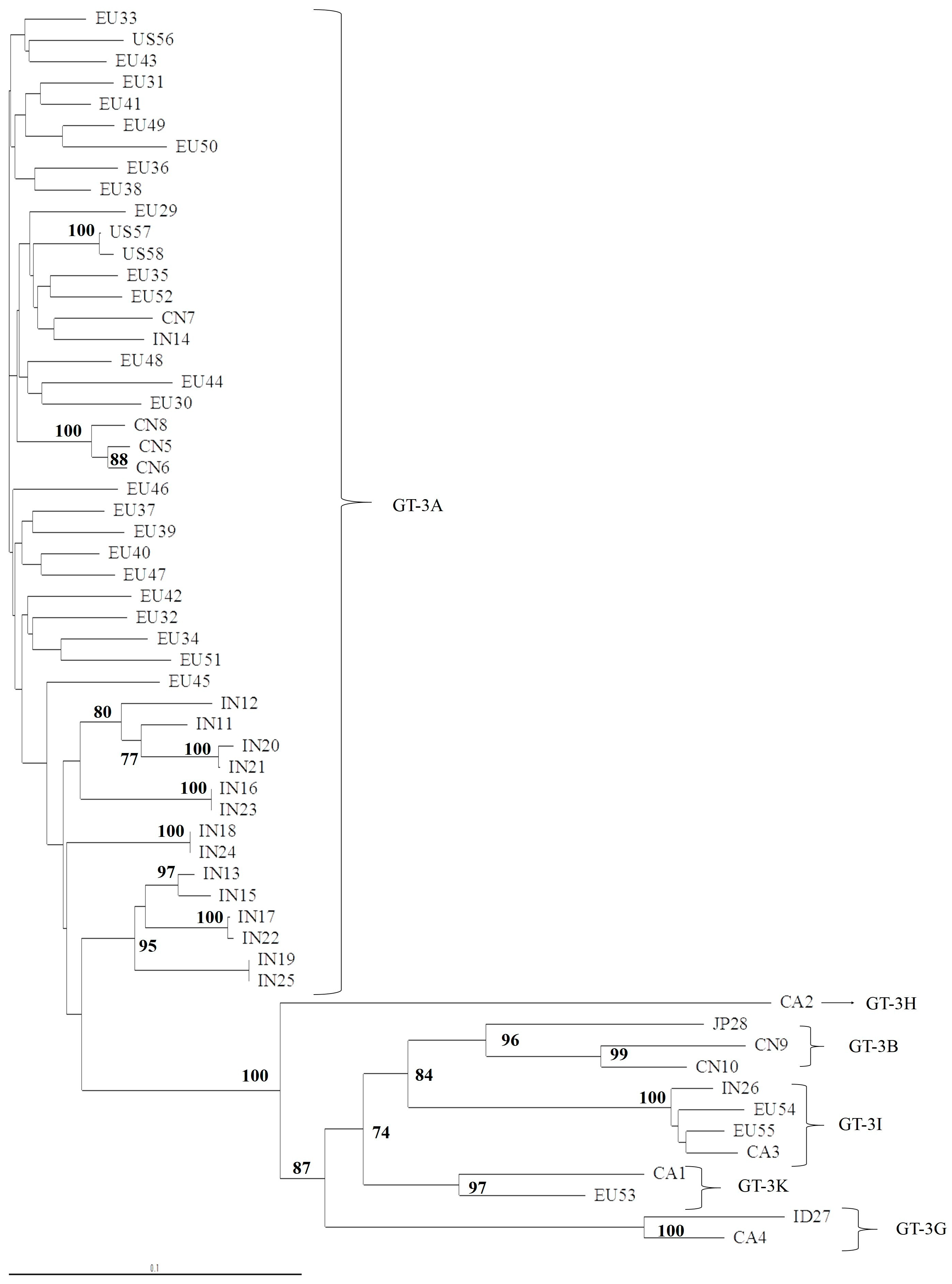
3.1. Concordance of GT-3 Subtypes Resulting from Phylogenetic Analysis of Different Regions (5′UTR, NS3, NS5A, and NS5B) in 58 Isolates Obtained from Los Alamos HCV Database
3.2. NS5A Analysis and Geographic Distribution, According to GT-3 Subtypes
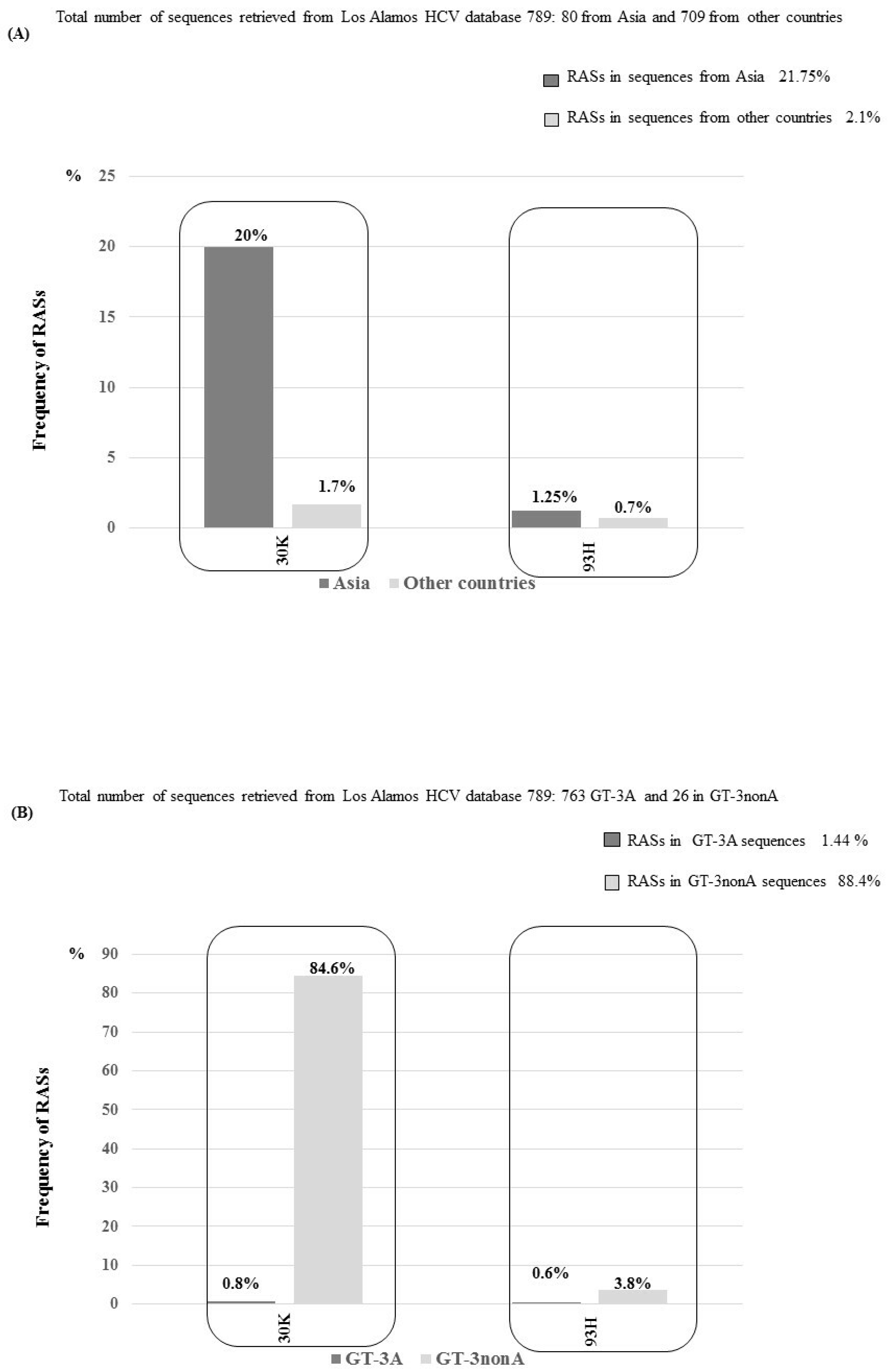
3.3. NS5A RASs According to Geographic Distribution and Subtypes
3.4. Selective Pressure on NS5A Domain 1 According to Subtype and Geographic Origin
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Conflicts of Interest
References
- Smith, D.B.; Bukh, J.; Kuiken, C.; Muerhoff, A.S.; Rice, C.M.; Stapleton, J.T.; Simmonds, P. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: updated criteria and genotype assignment web resource. Hepatology 2014, 59, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Gower, E.; Estes, C.; Blach, S.; Razavi-Shearer, K.; Razavi, H. Global epidemiology and genotype distribution of the hepatitis C virus infection. J. Hepatol. 2014, 61, S45–S57. [Google Scholar] [CrossRef] [PubMed]
- Welzel, T.M.; Bhardwaj, N.; Hedskog, C.; Chodavarapu, K.; Camus, G.; McNally, J.; Brainard, D.; Miller, M.D.; Mo, H.; Svarovskaia, E.; et al. Global epidemiology of HCV subtypes and resistance-associated substitutions evaluated by sequencing-based subtype analyses. J. Hepatol. 2017, 67, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Ceccherini Silberstein, F.; Di Maio, V.C.; Aragri, M.; Ciotti, M.; Cento, V.; Perno, C.F. Hepatitis C virus gene sequencing as a tool for precise genotyping in the era of new direct antiviral agents. Hepatology 2016, 63, 1058–1059. [Google Scholar] [CrossRef] [PubMed]
- AASLD/IDSA/IAS–USA. Recommendations for Testing, Managing, and Treating Hepatitis C. Available online: http://www.hcvguidelines.org (accessed on 17 February 2019).
- EASL. Recommendations on Treatment of Hepatitis C. Available online: http://www.easl.eu/research/our-contributions/clinical-practiceguidelines (accessed on 17 February 2019).
- Chen, Z.W.; Li, H.; Ren, H.; Hu, P. Global prevalence of pre-existing HCV variants resistant to direct-acting antiviral agents (DAAs): Mining the GenBank HCV genome data. Sci. Rep. 2016, 6, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, D.; Zhou, N.; Ueland, J.; Monikowski, A.; McPhee, F. Natural prevalence of NS5A polymorphisms in subjects infected with hepatitis C virus genotype 3 and their effects on the antiviral activity of NS5A inhibitors. J. Clin. Virol. 2013, 57, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.T.; Deming, P. Closing the gap: the challenges of treating hepatitis C virus genotype 3 infection. Pharmacotherapy 2017, 37, 735–747. [Google Scholar] [CrossRef]
- Pawlotsky, J.M. Hepatitis C Virus Resistance to Direct-Acting Antiviral Drugs in Interferon-Free Regimens. Gastroenterology 2016, 151, 70–86. [Google Scholar] [CrossRef]
- Lontok, E.; Harrington, P.; Howe, A.; Kieffer, T.; Lennerstrand, J.; Lenz, O.; McPhee, F.; Mo, H.; Parkin, N.; Pilot-Matias, T.; et al. Hepatitis C virus drug resistance-associated substitutions: state of the art summary. Hepatology 2015, 62, 1623–1632. [Google Scholar] [CrossRef]
- Sarrazin, C. The importance of resistance to direct antiviral drugs in HCV infection in clinical practice. J. Hepatol. 2016, 64, 486–504. [Google Scholar] [CrossRef]
- Carrasco, I.; Arias, A.; Benítez-Gutiérrez, L.; Lledó, G.; Requena, S.; Cuesta, M.; Cuervas-Mons, V.; de Mendoza, C. Baseline NS5A resistance associated substitutions may impair DAA response in real world hepatitis C patients. J. Med. Virol. 2018, 90, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Los Alamos HCV Database. Available online: https://hcv.lanl.gov (accessed on 17 February 2019).
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Kosakovsky Pond, S.L. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef]
- Ng, T.I.; Krishnan, P.; Pilot-Matias, T.; Kati, W.; Schnell, G.; Beyer, J.; Reisch, T.; Lu, L.; Dekhtyar, T.; Irvin, M.; et al. In Vitro Antiviral Activity and Resistance Profile of the Next-Generation Hepatitis C Virus NS5A Inhibitor Pibrentasvir. Antimicrob. Agents Chemother. 2017, 61, e02558-16. [Google Scholar] [CrossRef] [PubMed]
- Lawitz, E.J.; Dvory-Sobol, H.; Doehle, B.P.; Worth, A.S.; McNally, J.; Brainard, D.M.; Link, J.O.; Miller, M.D.; Mo, H. Clinical resistance to velpatasvir (GS-5816), a novel pan-genotypic inhibitor of the hepatitis C virus NS5A protein. Antimicrob. Agents Chemother. 2016, 60, 5368–5378. [Google Scholar] [CrossRef] [PubMed]
- Patiño-Galindo, J.Á.; Salvatierra, K.; González-Candelas, F.; López-Labrador, F.X. Comprehensive Screening for Naturally Occurring Hepatitis C Virus Resistance to Direct-Acting Antivirals in the NS3, NS5A, and NS5B Genes in Worldwide Isolates of Viral Genotypes 1 to 6. Antimicrob. Agents Chemother. 2016, 60, 2402–2416. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.R.; Cooper, J.N.; Lalezari, J.P.; Lawitz, E.; Pockros, P.J.; Gitlin, N.; Freilich, B.F.; Younes, Z.H.; Harlan, W.; Ghalib, R.; et al. All-oral 12-week treatment with daclatasvir plus sofosbuvir in patients with hepatitis C virus genotype 3 infection: ALLY-3 phase III study. Hepatology 2015, 61, 1127–1135. [Google Scholar] [CrossRef] [PubMed]
- Foster, G.R.; Afdhal, N.; Roberts, S.K.; Bräu, N.; Gane, E.J.; Pianko, S.; Lawitz, E.; Thompson, A.; Shiffman, M.L.; Cooper, C.; et al. Sofosbuvir and Velpatasvir for HCV Genotype 2 and 3 Infection. N. Engl. J. Med. 2015, 373, 2608–2617. [Google Scholar] [CrossRef] [PubMed]
- Pianko, S.; Flamm, S.L.; Shiffman, M.L.; Kumar, S.; Strasser, S.I.; Dore, G.J.; McNally, J.; Brainard, D.M.; Han, L.; Doehle, B.; et al. Sofosbuvir Plus Velpatasvir Combination Therapy for Treatment-Experienced Patients With Genotype 1 or 3 Hepatitis C Virus Infection: A Randomized Trial. Ann. Intern. Med. 2015, 163, 809–817. [Google Scholar] [CrossRef]
- Curry, M.P.; O’Leary, J.G.; Bzowej, N.; Muir, A.J.; Korenblat, K.M.; Fenkel, J.M.; Reddy, KR.; Lawitz, E.; Flamm, S.L.; Schiano, T.; et al. Sofosbuvir and Velpatasvir for HCV in Patients with Decompensated Cirrhosis. N. Engl. J. Med. 2015, 373, 2618–2628. [Google Scholar] [CrossRef]
- Bourlière, M.; Gordon, S.C.; Flamm, S.L.; Cooper, C.L.; Ramji, A.; Tong, M.; Ravendhran, N.; Vierling, J.M.; Tran, T.T.; Pianko, S.; et al. Sofosbuvir, Velpatasvir, and Voxilaprevir for Previously Treated HCV Infection. N. Engl. J. Med. 2017, 376, 2134–2146. [Google Scholar] [CrossRef]
- Kwo, P.Y.; Poordad, F.; Asatryan, A.; Wang, S.; Wyles, D.L.; Hassanein, T.; Felizarta, F.; Sulkowski, M.S.; Gane, E.; Maliakkal, B.; et al. Glecaprevir and pibrentasvir yield high response rates in patients with HCV genotype 1–6 without cirrhosis. J. Hepatol. 2017, 67, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Lambert, S.M.; Langley, D.R.; Garnett, J.A.; Angell, R.; Hedgethorne, K.; Meanwell, N.A.; Matthews, S.J. The crystal structure of NS5A domain 1 from genotype 1a reveals new clues to the mechanism of action for dimeric HCV inhibitors. Protein Sci. 2014, 23, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.H.; O’Boyle Ii, D.R.; Zhang, Y.; Wang, C.; Nower, P.; Valera, L.; Roberts, S.; Nettles, R.E.; Fridell, R.A.; Gao, M. Impact of a baseline polymorphism on the emergence of resistance to the hepatitis C virus nonstructural protein 5A replication complex inhibitor, BMS-790052. Hepatology 2012, 55, 1692–1699. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GT-3A | GT-3B ° | GT-3G | GT-3H | GT-3K °° | GT-3I | |
|---|---|---|---|---|---|---|
| Brazil (N = 597) | 597 | - | - | - | - | - |
| Australia (N = 18) | 18 | - | - | - | - | - |
| Europe (N = 86) | 83 | - | 1 | - | - | 2 |
| Pakistan (N = 25) | 25 | - | - | - | - | - |
| Thailand (N = 21) | 18 | 3 | - | - | - | - |
| China (N =16) | 4 | 12 | - | - | - | - |
| India (N = 16) | 15 | - | - | - | - | 1 |
| Other (N = 8) | 3 | - | 1 | 1 | 1 | 2 |
| TOTAL | 763 | 15 | 2 | 1 | 1 | 5 |
| RASs | Replicative Capacity | DCV ° FC [8] | VEL °° FC [17] | PIB ** FC [16] |
|---|---|---|---|---|
| A30K | 66 | 44 | 50 | - |
| Y93H A30K + Y93H | 34 | 2154 | 723 | 2.3 |
| Data Set | N | Site | alpha | beta- | p- | beta+ | p+ | LTR | p-Value |
|---|---|---|---|---|---|---|---|---|---|
| Overall | 789 | 62 | 0.78 | 0.31 | 0.76 | 8.28 | 0.24 | 14.20 | 0.00 |
| Asia | 80 | 30 | 0.65 | 0.00 | 0.85 | 7.96 | 0.15 | 9.16 | 0.00 |
| 62 | 1.37 | 0.25 | 0.73 | 10.35 | 0.27 | 6.83 | 0.01 | ||
| GT-3A | 62 | 62 | 0.98 | 0.00 | 0.72 | 12.20 | 0.28 | 6.90 | 0.01 |
| GT-non3A | 18 | 30 | 0.00 | 0.00 | 0.89 | 8.49 | 0.11 | 9.28 | 0.00 |
| Other countries | 709 | 62 | 0.63 | 0.51 | 0.91 | 13.26 | 0.09 | 5.10 | 0.04 |
| GT-3A | 701 | 62 | 0.63 | 0.57 | 0.87 | 11.03 | 0.13 | 6.40 | 0.02 |
| GT-non3A | 8 * | - | - | - | - | - | - | - | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bagaglio, S.; Messina, E.; Hasson, H.; Galli, A.; Uberti-Foppa, C.; Morsica, G. Geographic Distribution of HCV-GT3 Subtypes and Naturally Occurring Resistance Associated Substitutions. Viruses 2019, 11, 148. https://doi.org/10.3390/v11020148
Bagaglio S, Messina E, Hasson H, Galli A, Uberti-Foppa C, Morsica G. Geographic Distribution of HCV-GT3 Subtypes and Naturally Occurring Resistance Associated Substitutions. Viruses. 2019; 11(2):148. https://doi.org/10.3390/v11020148
Chicago/Turabian StyleBagaglio, Sabrina, Emanuela Messina, Hamid Hasson, Andrea Galli, Caterina Uberti-Foppa, and Giulia Morsica. 2019. "Geographic Distribution of HCV-GT3 Subtypes and Naturally Occurring Resistance Associated Substitutions" Viruses 11, no. 2: 148. https://doi.org/10.3390/v11020148
APA StyleBagaglio, S., Messina, E., Hasson, H., Galli, A., Uberti-Foppa, C., & Morsica, G. (2019). Geographic Distribution of HCV-GT3 Subtypes and Naturally Occurring Resistance Associated Substitutions. Viruses, 11(2), 148. https://doi.org/10.3390/v11020148