Detection of RNA-Dependent RNA Polymerase of Hubei Reo-Like Virus 7 by Next-Generation Sequencing in Aedes aegypti and Culex quinquefasciatus Mosquitoes from Brazil


 , ,
, ,  and
and 
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
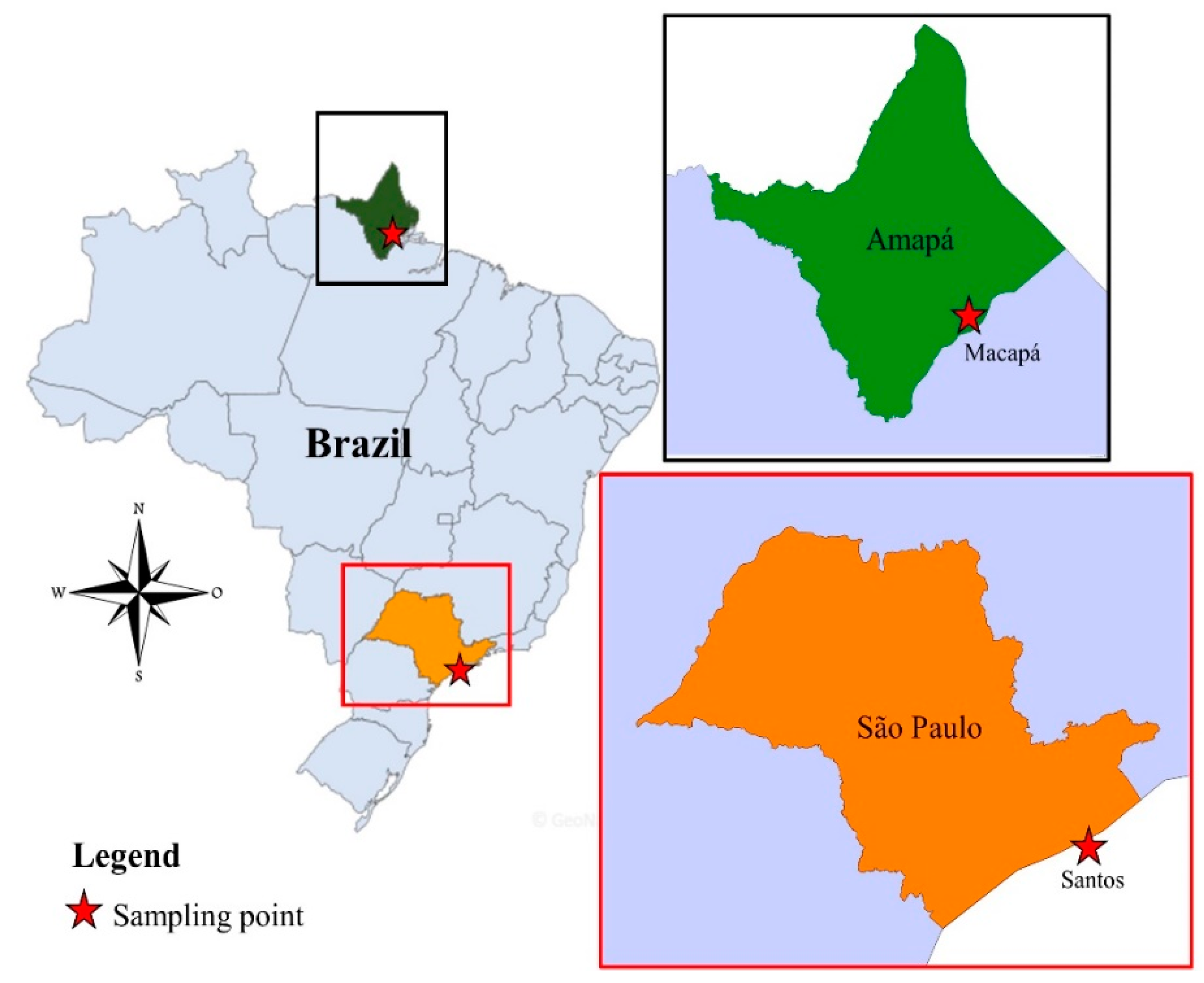
2.1. Mosquitoes Collection
2.2. Sample Processing and Next-Generation Sequencing (NGS)
2.3. Sequence Analysis
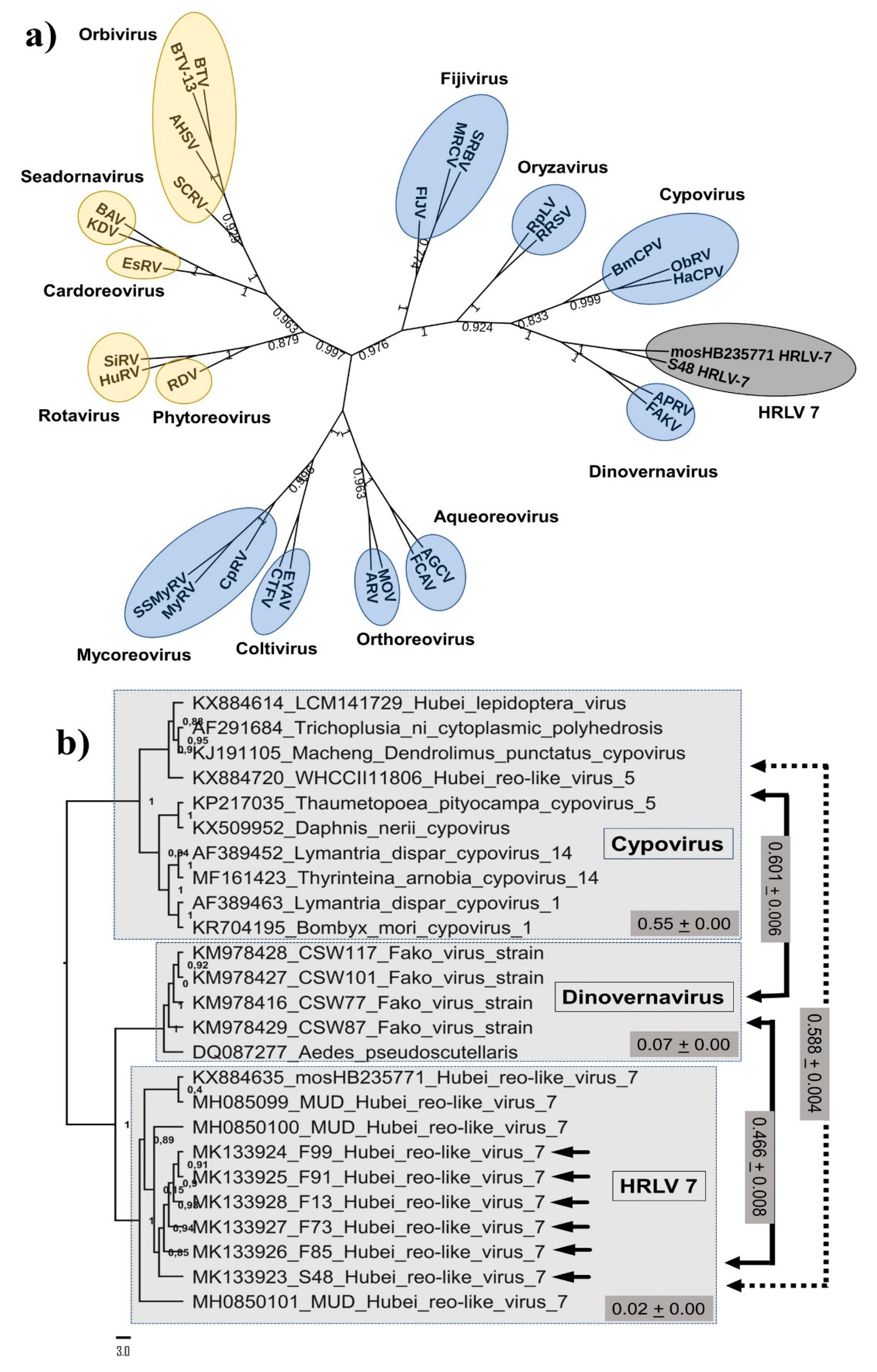
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Delwart, E.L. Viral metagenomics. Rev. Med. Virol. 2007, 17, 115–131. [Google Scholar] [CrossRef] [PubMed]
- Moormann, A.M.; Snider, C.J.; Chelimo, K. The company malaria keeps: how co-infection with Epstein-Barr virus leads to endemic Burkitt lymphoma. Curr. Opin. Infect. Dis. 2011, 24, 435. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Neville, P.; Nicholson, J.; Eden, J.-S.; Imrie, A.; Holmes, E.C. High Resolution Meta-Transcriptomics Reveals the Ecological Dynamics of Mosquito-Associated RNA Viruses in Western Australia. J. Virol. 2017, 91, e00680-17. [Google Scholar] [CrossRef] [PubMed]
- Fauver, J.R.; Grubaugh, N.D.; Krajacich, B.J.; Weger-Lucarelli, J.; Lakin, S.M.; Fakoli, L.S., III; Bolay, F.K.; Diclaro, J.W., II; Dabiré, K.R.; Foy, B.D. West African Anopheles gambiae mosquitoes harbor a taxonomically diverse virome including new insect-specific flaviviruses, mononegaviruses, and totiviruses. Virology 2016, 498, 288–299. [Google Scholar] [CrossRef]
- Sadeghi, M.; Altan, E.; Deng, X.; Barker, C.M.; Fang, Y.; Coffey, L.L.; Delwart, E. Virome of > 12 thousand Culex mosquitoes from throughout California. Virology 2018, 523, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Depoux, A.; Philibert, A.; Rabier, S.; Philippe, H.J.; Fontanet, A.; Flahault, A. A multi-faceted pandemic: A review of the state of knowledge on the Zika virus. Public Health Rev. 2018, 39, 10. [Google Scholar] [CrossRef]
- Rossetto, E.V.; Angerami, R.N.; de Albuquerque Luna, E.J. What to expect from the 2017 yellow fever outbreak in Brazil? Rev. Inst. Med. Trop. Sao Paulo 2017, 59, e17. [Google Scholar] [CrossRef] [PubMed]
- Goldani, L.Z. Yellow fever outbreak in Brazil, 2017. Braz. J. Infect. Dis. 2017, 21, 123–124. [Google Scholar] [CrossRef]
- Paixão, E.S.; Teixeira, M.G.; Rodrigues, L.C. Zika, chikungunya and dengue: the causes and threats of new and re-emerging arboviral diseases. BMJ Glob. Health 2017, 3, e000530. [Google Scholar] [CrossRef]
- Lim, E.S.; Zhou, Y.; Zhao, G.; Bauer, I.K.; Droit, L.; Ndao, I.M.; Warner, B.B.; Tarr, P.I.; Wang, D.; Holtz, L.R. Early life dynamics of the human gut virome and bacterial microbiome in infants. Nat. Med. 2015, 21, 1228–1234. [Google Scholar] [CrossRef]
- Zablocki, O.; van Zyl, L.; Adriaenssens, E.M.; Rubagotti, E.; Tuffin, M.; Cary, S.C.; Cowan, D. High-level diversity of tailed phages, eukaryote-associated viruses, and virophage-like elements in the metaviromes of antarctic soils. Appl. Environ. Microbiol. 2014, 80, 6888–6897. [Google Scholar] [CrossRef]
- Shi, M.; Lin, X.D.; Tian, J.H.; Chen, L.J.; Chen, X.; Li, C.X.; Qin, X.C.; Li, J.; Cao, J.P.; Eden, J.S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef]
- Temmam, S.; Monteil-Bouchard, S.; Robert, C.; Baudoin, J.P.; Sambou, M.; Aubadie-Ladrix, M.; Labas, N.; Raoult, D.; Mediannikov, O.; Desnues, C. Characterization of viral communities of biting midges and identification of novel thogotovirus species and rhabdovirus genus. Viruses 2016, 8, 77. [Google Scholar] [CrossRef]
- Roux, S.; Chan, L.K.; Egan, R.; Malmstrom, R.R.; McMahon, K.D.; Sullivan, M.B. Ecogenomics of virophages and their giant virus hosts assessed through time series metagenomics. Nat. Commun. 2017, 8, 858. [Google Scholar] [CrossRef]
- Li, C.X.; Shi, M.; Tian, J.H.; Lin, X.D.; Kang, Y.J.; Chen, L.J.; Qin, X.C.; Xu, J.; Holmes, E.C.; Zhang, Y.Z. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. Elife 2015, 4, e05378. [Google Scholar] [CrossRef]
- Vibin, J.; Chamings, A.; Collier, F.; Klaassen, M.; Nelson, T.M.; Alexandersen, S. Metagenomics detection and characterisation of viruses in faecal samples from Australian wild birds. Sci. Rep. 2018, 8, 8686. [Google Scholar] [CrossRef]
- Consoli, R.A.G.B.; de Oliveira, R.L. Principais mosquitos de importância sanitária no Brasil, 1st ed.; Editora Fiocruz: Rio de Janeiro, Brazil, 1994; ISBN 85-85676-03-5. [Google Scholar]
- Da Costa, A.C.; Thézé, J.; Komninakis, S.C.V.; Sanz-Duro, R.L.; Felinto, M.R.L.; Moura, L.C.C.; de Oliveira Barroso, I.M.; Santos, L.E.C.; de Lemos Nunes, M.A.; Moura, A.A. Spread of Chikungunya Virus East/Central/South African Genotype in Northeast Brazil. Emerg. Infect. Dis. 2017, 23, 1742. [Google Scholar] [CrossRef]
- Deng, X.; Naccache, S.N.; Ng, T.; Federman, S.; Li, L.; Chiu, C.Y.; Delwart, E.L. An ensemble strategy that significantly improves de novo assembly of microbial genomes from metagenomic next-generation sequencing data. Nucleic Acids Res. 2015, 43, e46. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2017. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Attoui, H.; Fang, Q.; Jaafar, F.M.; Cantaloube, J.F.; Biagini, P.; de Micco, P.; de Lamballerie, X. Common evolutionary origin of aquareoviruses and orthoreoviruses revealed by genome characterization of Golden shiner reovirus, Grass carp reovirus, Striped bass reovirus and golden ide reovirus (genus Aquareovirus, family Reoviridae). J. Gen. Virol. 2002, 33, 1941–1951. [Google Scholar] [CrossRef] [PubMed]
- Attoui, H.; Mertens, P.P.C.; Becnel, J.; Belaganahalli, S.; Bergoin, M.; Brussard, C.P. Family Reoviridae. In Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Academic Press: London, UK, 2012; pp. 541–637. ISBN 9780123846846. [Google Scholar]
- Auguste, A.J.; Kaelber, J.T.; Fokam, E.B.; Guzman, H.; Carrington, C.V.F.; Erasmus, J.H.; Kamgang, B.; Popov, V.L.; Jakana, J.; Liu, X.; et al. A Newly Isolated Reovirus Has the Simplest Genomic and Structural Organization of Any Reovirus. J. Virol. 2015, 89, 676–687. [Google Scholar] [CrossRef]
- Novella, I.S.; Presloid, J.B.; Taylor, R.T. RNA replication errors and the evolution of virus pathogenicity and virulence. Curr. Opin. Virol. 2014, 9, 143–147. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ribeiro, G.d.O.; Monteiro, F.J.C.; Rego, M.O.d.S.; Ribeiro, E.S.D.; Castro, D.F.d.; Caseiro, M.M.; Souza Marinho, R.d.S.; Komninakis, S.V.; Witkin, S.S.; Deng, X.; et al. Detection of RNA-Dependent RNA Polymerase of Hubei Reo-Like Virus 7 by Next-Generation Sequencing in Aedes aegypti and Culex quinquefasciatus Mosquitoes from Brazil. Viruses 2019, 11, 147. https://doi.org/10.3390/v11020147
Ribeiro GdO, Monteiro FJC, Rego MOdS, Ribeiro ESD, Castro DFd, Caseiro MM, Souza Marinho RdS, Komninakis SV, Witkin SS, Deng X, et al. Detection of RNA-Dependent RNA Polymerase of Hubei Reo-Like Virus 7 by Next-Generation Sequencing in Aedes aegypti and Culex quinquefasciatus Mosquitoes from Brazil. Viruses. 2019; 11(2):147. https://doi.org/10.3390/v11020147
Chicago/Turabian StyleRibeiro, Geovani de Oliveira, Fred Julio Costa Monteiro, Marlisson Octavio da S Rego, Edcelha Soares D’Athaide Ribeiro, Daniela Funayama de Castro, Marcos Montani Caseiro, Robson dos Santos Souza Marinho, Shirley Vasconcelos Komninakis, Steven S. Witkin, Xutao Deng, and et al. 2019. "Detection of RNA-Dependent RNA Polymerase of Hubei Reo-Like Virus 7 by Next-Generation Sequencing in Aedes aegypti and Culex quinquefasciatus Mosquitoes from Brazil" Viruses 11, no. 2: 147. https://doi.org/10.3390/v11020147
APA StyleRibeiro, G. d. O., Monteiro, F. J. C., Rego, M. O. d. S., Ribeiro, E. S. D., Castro, D. F. d., Caseiro, M. M., Souza Marinho, R. d. S., Komninakis, S. V., Witkin, S. S., Deng, X., Delwart, E., Sabino, E. C., da Costa, A. C., & Leal, É. (2019). Detection of RNA-Dependent RNA Polymerase of Hubei Reo-Like Virus 7 by Next-Generation Sequencing in Aedes aegypti and Culex quinquefasciatus Mosquitoes from Brazil. Viruses, 11(2), 147. https://doi.org/10.3390/v11020147