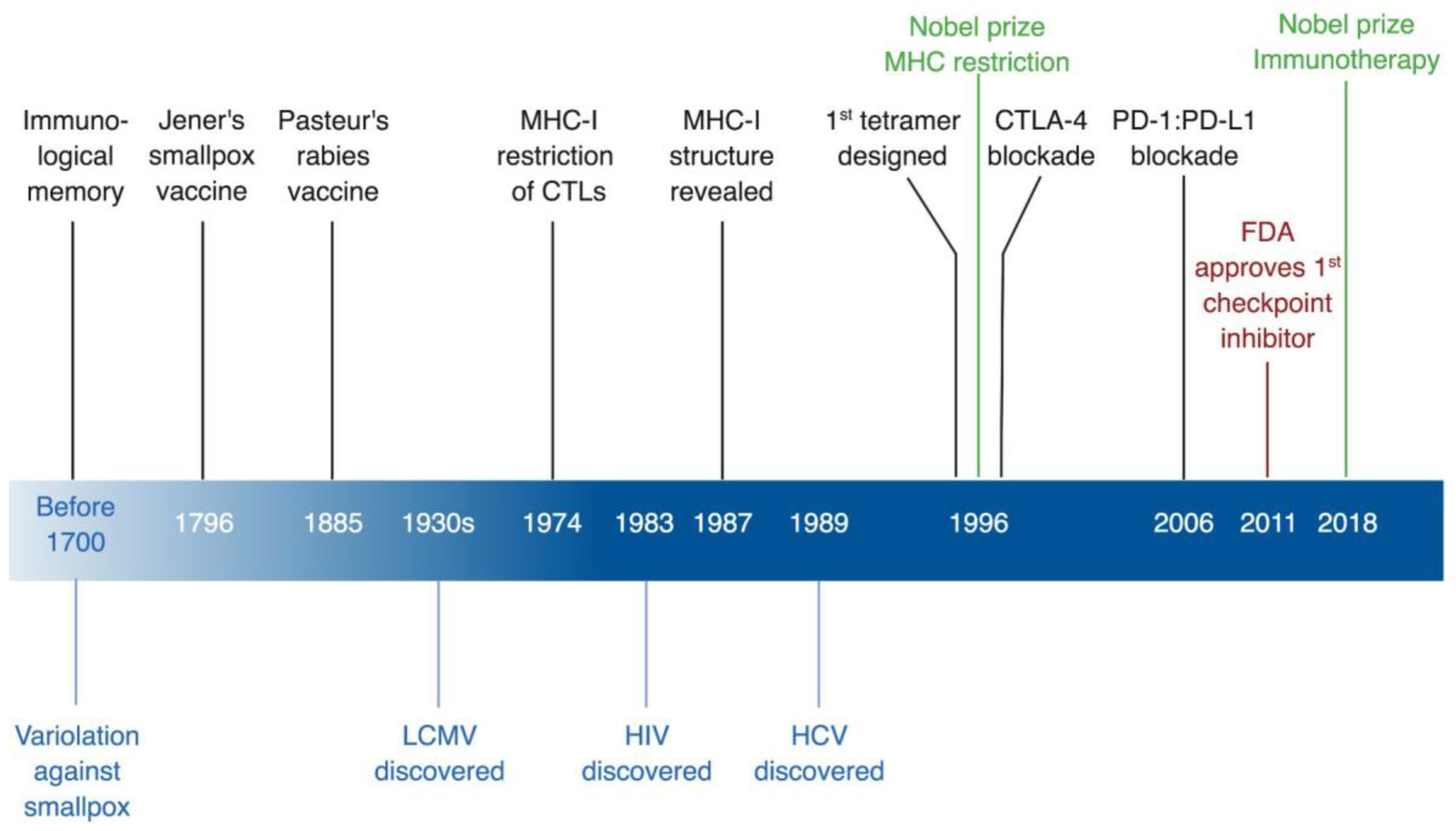
Viruses Teaching Immunology: Role of LCMV Model and Human Viral Infections in Immunological Discoveries

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
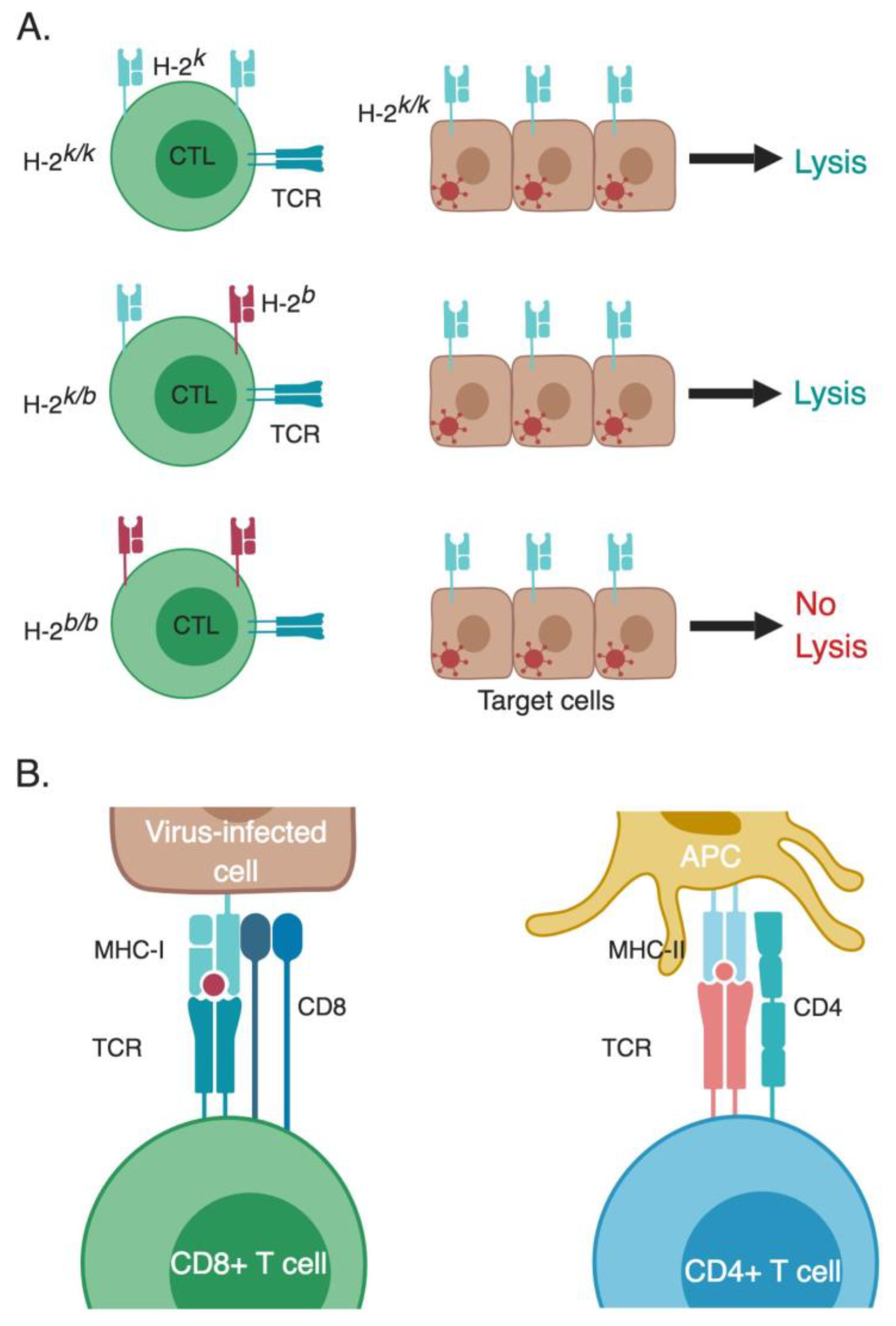
2. MHC Restriction
3. Tetramer Development and Related Discoveries
3.1. Tetramers
3.2. Escape Mutation
3.3. Protective HLA Alleles
3.4. LCMV; Armstrong versus Clone-13
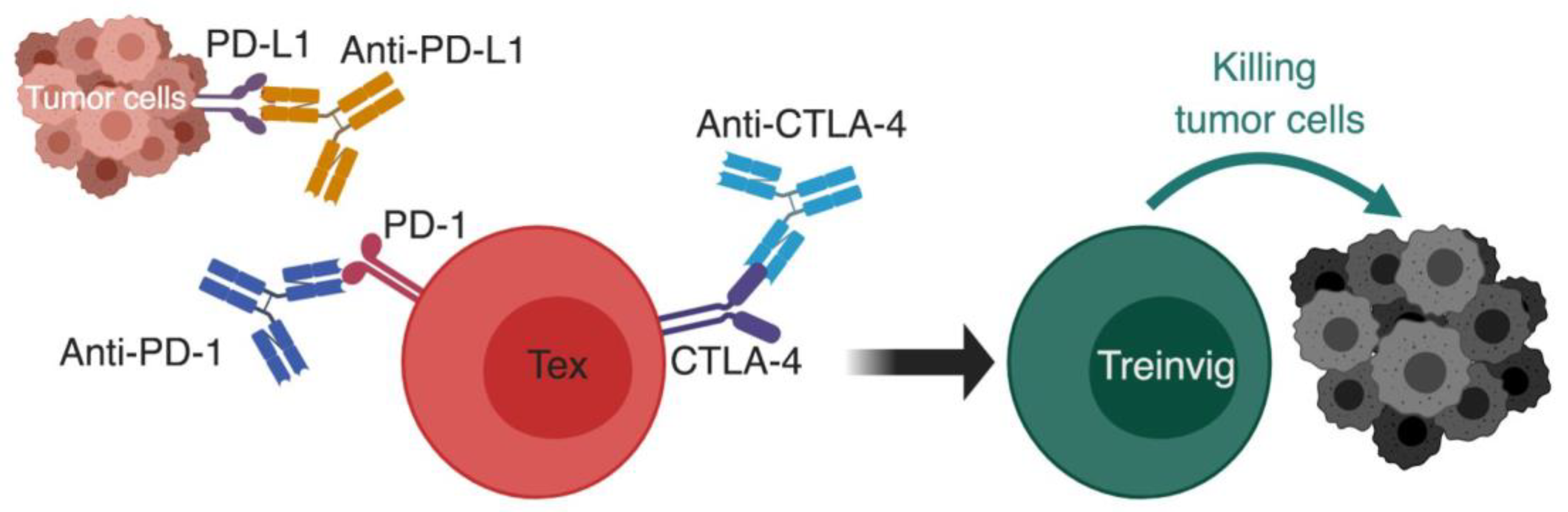
4. Immunotherapy and T-Cell Exhaustion
5. Conclusions
Funding
Conflicts of Interest
References
- Riedel, S. Edward jenner and the history of smallpox and vaccination. Proc. (Bayl. Univ. Med. Cent.) 2005, 18, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.A. Louis pasteur, the father of immunology? Front. Immunol. 2012, 3, 68. [Google Scholar] [CrossRef] [PubMed]
- Pircher, H.; Moskophidis, D.; Rohrer, U.; Burki, K.; Hengartner, H.; Zinkernagel, R.M. Viral escape by selection of cytotoxic t cell-resistant virus variants in vivo. Nature 1990, 346, 629–633. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.M.; Altfeld, M.; Geer, S.C.; Kalife, E.T.; Moore, C.; O’Sullivan K, M.; Desouza, I.; Feeney, M.E.; Eldridge, R.L.; Maier, E.L.; et al. Selective escape from CD8+ T-cell responses represents a major driving force of human immunodeficiency virus type 1 (HIV-1) sequence diversity and reveals constraints on HIV-1 evolution. J. Virol. 2005, 79, 13239–13249. [Google Scholar] [CrossRef] [PubMed]
- Timm, J.; Lauer, G.M.; Kavanagh, D.G.; Sheridan, I.; Kim, A.Y.; Lucas, M.; Pillay, T.; Ouchi, K.; Reyor, L.L.; Schulze zur Wiesch, J.; et al. CD8 epitope escape and reversion in acute HCV infection. J. Exp. Med. 2004, 200, 1593–1604. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Gelderblom, H.R. Structure and classification of viruses. In Medical Microbiology, 4th ed.; Baron, S., Ed.; University of Texas Medical Branch at Galveston: Galveston, TX, USA, 1996. [Google Scholar]
- Oldstone, M.B. An odyssey to viral pathogenesis. Annu. Rev. Pathol. 2016, 11, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Ramachandran, S.; Mann, M.; Popkin, D.L. Role of lymphocytic choriomeningitis virus (LCMV) in understanding viral immunology: Past, present and future. Viruses 2012, 4, 2650–2669. [Google Scholar] [CrossRef] [PubMed]
- Zinkernagel, R.M.; Doherty, P.C. Restriction of in vitro T cell-mediated cytotoxicity in lymphocytic choriomeningitis within a syngeneic or semiallogeneic system. Nature 1974, 248, 701–702. [Google Scholar] [CrossRef] [PubMed]
- Doherty, P.C.; Zinkernagel, R.M. H-2 compatibility is required for T-cell-mediated lysis of target cells infected with lymphocytic choriomeningitis virus. J. Exp. Med. 1975, 141, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Altman, J.D.; Moss, P.A.; Goulder, P.J.; Barouch, D.H.; McHeyzer-Williams, M.G.; Bell, J.I.; McMichael, A.J.; Davis, M.M. Phenotypic analysis of antigen-specific T lymphocytes. Science 1996, 274, 94–96. [Google Scholar] [CrossRef] [PubMed]
- Migueles, S.A.; Sabbaghian, M.S.; Shupert, W.L.; Bettinotti, M.P.; Marincola, F.M.; Martino, L.; Hallahan, C.W.; Selig, S.M.; Schwartz, D.; Sullivan, J.; et al. HLA B*5701 is highly associated with restriction of virus replication in a subgroup of HIV-infected long term nonprogressors. Proc. Natl. Acad. Sci. USA 2000, 97, 2709–2714. [Google Scholar] [CrossRef] [PubMed]
- Den Uyl, D.; van der Horst-Bruinsma, I.E.; van Agtmael, M. Progression of HIV to AIDS: A protective role for hla-b27? AIDS Rev. 2004, 6, 89–96. [Google Scholar] [PubMed]
- Neumann-Haefelin, C.; McKiernan, S.; Ward, S.; Viazov, S.; Spangenberg, H.C.; Killinger, T.; Baumert, T.F.; Nazarova, N.; Sheridan, I.; Pybus, O.; et al. Dominant influence of an HLA-B27 restricted CD8+ T cell response in mediating HCV clearance and evolution. Hepatology 2006, 43, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.Y.; Kuntzen, T.; Timm, J.; Nolan, B.E.; Baca, M.A.; Reyor, L.L.; Berical, A.C.; Feller, A.J.; Johnson, K.L.; Schulze zur Wiesch, J.; et al. Spontaneous control of HCV is associated with expression of HLA-B 57 and preservation of targeted epitopes. Gastroenterology 2011, 140, 686–696.e1. [Google Scholar] [CrossRef] [PubMed]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [PubMed]
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006, 439, 682–687. [Google Scholar] [CrossRef]
- Tang, J.; Pearce, L.; O’Donnell-Tormey, J.; Hubbard-Lucey, V.M. Trends in the global immuno-oncology landscape. Nat. Rev. Drug Discov. 2018, 17, 783–784. [Google Scholar] [CrossRef]
- Tang, J.; Yu, J.X.; Hubbard-Lucey, V.M.; Neftelinov, S.T.; Hodge, J.P.; Lin, Y. Trial watch: The clinical trial landscape for PD1/PDL1 immune checkpoint inhibitors. Nat. Rev. Drug Discov. 2018, 17, 854–855. [Google Scholar] [CrossRef]
- Wherry, E.J.; Blattman, J.N.; Murali-Krishna, K.; van der Most, R.; Ahmed, R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J. Virol. 2003, 77, 4911–4927. [Google Scholar] [CrossRef]
- Wherry, E.J.; Ha, S.J.; Kaech, S.M.; Haining, W.N.; Sarkar, S.; Kalia, V.; Subramaniam, S.; Blattman, J.N.; Barber, D.L.; Ahmed, R. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 2007, 27, 670–684. [Google Scholar] [CrossRef] [PubMed]
- Charles, A.; Janeway, J. Paul Travers, Mark Walport, and Mark J Shlomchik. In Immunobiology: The Immune System in Health and Disease, 5th ed.; Garland Science: New York, NY, USA, 2001. [Google Scholar]
- J.Moticka, E. (Ed.) Activation of t lymphocytes and mhc restriction. In A Historical Perspective on Evidence-Based Immunology; Elsevier: New York, NY, USA, 2015; pp. 169–179. [Google Scholar]
- Vyas, J.M.; van der Veen, A.G.; Ploegh, H.L. The known unknowns of antigen processing and presentation. Nat. Rev. Immunol. 2008, 8, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Neefjes, J.; Jongsma, M.L.; Paul, P.; Bakke, O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol. 2011, 11, 823–836. [Google Scholar] [CrossRef] [PubMed]
- Muckenfuss, R.S.A.C.; Webster, L. Etiology of the 1933 epidemic of encephalitis. JAMA 1934, 103, 731–733. [Google Scholar] [CrossRef]
- Traub, E. An epidemic in a mouse colony due to the virus of acute lymphocytic choriomeningitis. J. Exp. Med. 1936, 63, 533–546. [Google Scholar] [CrossRef]
- Michael, J.; Buchmeier, M.D.B.; Peters, C.J. Arenaviridae: The viruses and their replication. In Fields Virology, 4th ed.; David, M., Knipe, P.M.H., Eds.; Lippincot Williams & Wilkins: Philadelphia, PA, USA, 2001; Volume 2, pp. 1635–1668. [Google Scholar]
- Moller, G. Demonstration of mouse isoantigens at the cellular level by the fluorescent antibody technique. J. Exp. Med. 1961, 114, 415–434. [Google Scholar] [CrossRef]
- Davie, J.M.; Paul, W.E. Receptors on immunocompetent cells. Ii. Specificity and nature of receptors on dinitrophenylated guinea pig albumin- 125 I-binding lymphocytes of normal guinea pigs. J. Exp. Med. 1971, 134, 495–516. [Google Scholar] [CrossRef]
- Raff, M.C.; Feldmann, M.; de Petris, S. Monospecificity of bone marrow-derived lymphocytes. J. Exp. Med. 1973, 137, 1024–1030. [Google Scholar] [CrossRef]
- Bankhurst, A.D.; Warner, N.L.; Sprent, J. Surface immunoglobulins on thymus and thymus-derived lymphoid cells. J. Exp. Med. 1971, 134, 1005–1015. [Google Scholar] [CrossRef]
- Lesley, J.F.; Kettman, J.R.; Dutton, R.W. Immunoglobulins on the surface of thymus-derived cells engaged in the initiation of a humoral immune response. J. Exp. Med. 1971, 134, 618–629. [Google Scholar] [CrossRef]
- Leclerc, J.C.; Gomard, E.; Levy, J.P. Cell-mediated reaction against tumors induced by oncornaviruses. I. Kinetics and specificity of the immune response in murine sarcoma virus (MSV)-induced tumors and transplanted lymphomas. Int. J. Cancer 1972, 10, 589–601. [Google Scholar] [CrossRef] [PubMed]
- Lavrin, D.H.; Herberman, R.B.; Nunn, M.; Soares, N. In vitro cytotoxicity studies of murine sarcoma virus-induced immunity in mice. J. Natl. Cancer Inst. 1973, 51, 1497–1508. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, A.S.; Shevach, E.M. Function of macrophages in antigen recognition by guinea pig T lymphocytes. I. Requirement for histocompatible macrophages and lymphocytes. J. Exp. Med. 1973, 138, 1194–1212. [Google Scholar] [CrossRef] [PubMed]
- Shevach, E.M.; Rosenthal, A.S. Function of macrophages in antigen recognition by guinea pig T lymphocytes. II. Role of the macrophage in the regulation of genetic control of the immune response. J. Exp. Med. 1973, 138, 1213–1229. [Google Scholar] [CrossRef] [PubMed]
- Shearer, G.M. Cell-mediated cytotoxicity to trinitrophenyl-modified syngeneic lymphocytes. Eur. J. Immunol. 1974, 4, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Doherty, P.C.; Zinkernagel, R.M.; Ramshaw, I.A. Specificity and development of cytotoxic thymus-derived lymphocytes in lymphocytic choriomeningitis. J. Immunol. 1974, 112, 1548–1552. [Google Scholar]
- Zinkernagel, R.M.; Doherty, P.C. Immunological surveillance against altered self components by sensitised T lymphocytes in lymphocytic choriomeningitis. Nature 1974, 251, 547–548. [Google Scholar] [CrossRef]
- Zinkernagel, R.M.; Doherty, P.C. H-2 compatability requirement for T-cell-mediated lysis of target cells infected with lymphocytic choriomeningitis virus. Different cytotoxic T-cell specificities are associated with structures coded for in H-2K or H-2D. J. Exp. Med. 1975, 141, 1427–1436. [Google Scholar] [CrossRef]
- Gorer, P.; Lyman, S.; Snell, G.D. Studies on the genetic and antigenic basis of tumour transplantation. Linkage between a histocompatibility gene and ‘fused’ in mice. Proc. R. Soc. Lond. 1948, 135, 499–505. [Google Scholar] [CrossRef]
- Oldstone, M.B.; Dixon, F.J.; Mitchell, G.F.; McDevitt, H.O. Histocompatibility-linked genetic control of disease susceptibility. Murine lymphocytic choriomeningitis virus infection. J. Exp. Med. 1973, 137, 1201–1212. [Google Scholar] [CrossRef]
- Zinkernagel, R.M.; Doherty, P.C. Cytotoxic thymus-derived lymphocytes in cerebrospinal fluid of mice with lymphocytic choriomeningitis. J. Exp. Med. 1973, 138, 1266–1269. [Google Scholar] [CrossRef] [PubMed]
- Doherty, P.C.; Zinkernagel, R.M. T-cell-mediated immunopathology in viral infections. Transplant. Rev. 1974, 19, 89–120. [Google Scholar] [CrossRef] [PubMed]
- Zinkernagel, R.M.; Doherty, P.C. The discovery of mhc restriction. Immunol. Today 1997, 18, 14–17. [Google Scholar] [CrossRef]
- Kindred, B.; Shreffler, D.C. H-2 dependence of co-operation between T and B cells in vivo. J. Immunol. 1972, 109, 940–943. [Google Scholar] [PubMed]
- Katz, D.H.; Hamaoka, T.; Benacerraf, B. Cell interactions between histoincompatible T and B lymphocytes. II. Failure of physiologic cooperative interactions between T and B lymphocytes from allogeneic donor strains in humoral response to hapten-protein conjugates. J. Exp. Med. 1973, 137, 1405–1418. [Google Scholar] [CrossRef] [PubMed]
- Katz, D.H.; Hamaoka, T.; Dorf, M.E.; Benacerraf, B. Cell interactions between histoincompatible T and B lymphocytes. The H-2 gene complex determines successful physiologic lymphocyte interactions. Proc. Natl. Acad. Sci. USA 1973, 70, 2624–2628. [Google Scholar] [CrossRef]
- Hotchin, J.; Benson, L. The pathogenesis of lymphocytic choriomeningitis in mice: The effects of different inoculation routes and the footpad response. J. Immunol. 1963, 91, 460–468. [Google Scholar]
- Seamer, J.; Barlow, J.L.; Gledhill, A.W.; Hotchin, J. Increased susceptibility of mice to lymphocytic choriomeningitis virus after peripheral inoculation. Virology 1963, 21, 309–316. [Google Scholar] [CrossRef]
- Lehmann-Grube, F. Dose-response relationships of lymphocytic choriomeningitis viruses in mice and L cell tube cultures. J. Hyg. (Lond.) 1969, 67, 269–278. [Google Scholar] [CrossRef]
- Lehmann-Grube, F. Lymphocytic choriomeningitis in the mouse. 3. Comparative titrations of virus strains in inbred mice. Arch. Gesamte. Virusforsch. 1969, 28, 303–307. [Google Scholar] [CrossRef]
- Lehmann-Grube, F.; Slenczka, W.; Tees, R. A persistent and inapparent infection of l cells with the virus of lymphocytic choriomeningitis. J. Gen. Virol. 1969, 5, 63–81. [Google Scholar] [CrossRef] [PubMed]
- Bjorkman, P.J.; Saper, M.A.; Samraoui, B.; Bennett, W.S.; Strominger, J.L.; Wiley, D.C. Structure of the human class I histocompatibility antigen, HLA-A2. Nature 1987, 329, 506–512. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.H.; Jardetzky, T.S.; Gorga, J.C.; Stern, L.J.; Urban, R.G.; Strominger, J.L.; Wiley, D.C. Three-dimensional structure of the human class II histocompatibility antigen HLA-DR1. Nature 1993, 364, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Allison, J.P.; McIntyre, B.W.; Bloch, D. Tumor-specific antigen of murine T-lymphoma defined with monoclonal antibody. J. Immunol. 1982, 129, 2293–2300. [Google Scholar] [PubMed]
- Meuer, S.C.; Fitzgerald, K.A.; Hussey, R.E.; Hodgdon, J.C.; Schlossman, S.F.; Reinherz, E.L. Clonotypic structures involved in antigen-specific human T cell function. Relationship to the t3 molecular complex. J. Exp. Med. 1983, 157, 705–719. [Google Scholar] [CrossRef] [PubMed]
- Haskins, K.; Kubo, R.; White, J.; Pigeon, M.; Kappler, J.; Marrack, P. The major histocompatibility complex-restricted antigen receptor on T cells. I. Isolation with a monoclonal antibody. J. Exp. Med. 1983, 157, 1149–1169. [Google Scholar] [CrossRef] [PubMed]
- Hedrick, S.M.; Cohen, D.I.; Nielsen, E.A.; Davis, M.M. Isolation of cDNA clones encoding T cell-specific membrane-associated proteins. Nature 1984, 308, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Yanagi, Y.; Yoshikai, Y.; Leggett, K.; Clark, S.P.; Aleksander, I.; Mak, T.W. A human T cell-specific cDNA clone encodes a protein having extensive homology to immunoglobulin chains. Nature 1984, 308, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Saito, H.; Kranz, D.M.; Takagaki, Y.; Hayday, A.C.; Eisen, H.N.; Tonegawa, S. Complete primary structure of a heterodimeric T-cell receptor deduced from cDNA sequences. Nature 1984, 309, 757–762. [Google Scholar] [CrossRef] [PubMed]
- Chapman, S.J.; Hill, A.V. Human genetic susceptibility to infectious disease. Nat. Rev. Genet. 2012, 13, 175–188. [Google Scholar] [CrossRef]
- Newell, E.W.; Sigal, N.; Nair, N.; Kidd, B.A.; Greenberg, H.B.; Davis, M.M. Combinatorial tetramer staining and mass cytometry analysis facilitate T-cell epitope mapping and characterization. Nat. Biotechnol. 2013, 31, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Dolton, G.; Lissina, A.; Skowera, A.; Ladell, K.; Tungatt, K.; Jones, E.; Kronenberg-Versteeg, D.; Akpovwa, H.; Pentier, J.M.; Holland, C.J.; et al. Comparison of peptide-major histocompatibility complex tetramers and dextramers for the identification of antigen-specific T cells. Clin. Exp. Immunol. 2014, 177, 47–63. [Google Scholar] [CrossRef] [PubMed]
- Bukh, J.; Miller, R.H.; Purcell, R.H. Genetic heterogeneity of hepatitis c virus: Quasispecies and genotypes. Semin. Liver Dis. 1995, 15, 41–63. [Google Scholar] [CrossRef] [PubMed]
- Thimme, R.; Lohmann, V.; Weber, F. A target on the move: Innate and adaptive immune escape strategies of hepatitis C virus. Antiviral Res. 2006, 69, 129–141. [Google Scholar] [CrossRef]
- Cox, A.L.; Mosbruger, T.; Mao, Q.; Liu, Z.; Wang, X.H.; Yang, H.C.; Sidney, J.; Sette, A.; Pardoll, D.; Thomas, D.L.; et al. Cellular immune selection with hepatitis C virus persistence in humans. J. Exp. Med. 2005, 201, 1741–1752. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.M.; O’Connor, D.H.; Jing, P.; Dzuris, J.L.; Mothe, B.R.; Vogel, T.U.; Dunphy, E.; Liebl, M.E.; Emerson, C.; Wilson, N.; et al. Tat-specific cytotoxic T lymphocytes select for SIV escape variants during resolution of primary viraemia. Nature 2000, 407, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.M.; Altfeld, M.; Yu, X.G.; O’Sullivan, K.M.; Lichterfeld, M.; Le Gall, S.; John, M.; Mothe, B.R.; Lee, P.K.; Kalife, E.T.; et al. Selection, transmission, and reversion of an antigen-processing cytotoxic T-lymphocyte escape mutation in human immunodeficiency virus type 1 infection. J. Virol. 2004, 78, 7069–7078. [Google Scholar] [CrossRef]
- Frankel, A.D.; Young, J.A. HIV-1: Fifteen proteins and an RNA. Annu. Rev. Biochem. 1998, 67, 1–25. [Google Scholar] [CrossRef]
- Abdel-Hakeem, M.S.; Shoukry, N.H. Protective immunity against hepatitis C: Many shades of gray. Front. Immunol. 2014, 5, 274. [Google Scholar] [CrossRef]
- Kulpa, D.A.; Chomont, N. HIV persistence in the setting of antiretroviral therapy: When, where and how does HIV hide? J. Virus Erad. 2015, 1, 59–66. [Google Scholar]
- Gaudieri, S.; Rauch, A.; Park, L.P.; Freitas, E.; Herrmann, S.; Jeffrey, G.; Cheng, W.; Pfafferott, K.; Naidoo, K.; Chapman, R.; et al. Evidence of viral adaptation to HLA class I-restricted immune pressure in chronic hepatitis C virus infection. J. Virol. 2006, 80, 11094–11104. [Google Scholar] [CrossRef]
- Soderholm, J.; Ahlen, G.; Kaul, A.; Frelin, L.; Alheim, M.; Barnfield, C.; Liljestrom, P.; Weiland, O.; Milich, D.R.; Bartenschlager, R.; et al. Relation between viral fitness and immune escape within the hepatitis C virus protease. Gut 2006, 55, 266–274. [Google Scholar] [CrossRef]
- Kuntzen, T.; Timm, J.; Berical, A.; Lewis-Ximenez, L.L.; Jones, A.; Nolan, B.; Schulze zur Wiesch, J.; Li, B.; Schneidewind, A.; Kim, A.Y.; et al. Viral sequence evolution in acute hepatitis C virus infection. J. Virol 2007, 81, 11658–11668. [Google Scholar] [CrossRef]
- Jones, N.A.; Wei, X.; Flower, D.R.; Wong, M.; Michor, F.; Saag, M.S.; Hahn, B.H.; Nowak, M.A.; Shaw, G.M.; Borrow, P. Determinants of human immunodeficiency virus type 1 escape from the primary CD8+ cytotoxic T lymphocyte response. J. Exp. Med. 2004, 200, 1243–1256. [Google Scholar] [CrossRef]
- Tester, I.; Smyk-Pearson, S.; Wang, P.; Wertheimer, A.; Yao, E.; Lewinsohn, D.M.; Tavis, J.E.; Rosen, H.R. Immune evasion versus recovery after acute hepatitis C virus infection from a shared source. J. Exp. Med. 2005, 201, 1725–1731. [Google Scholar] [CrossRef] [PubMed]
- Badr, G.; Bedard, N.; Abdel-Hakeem, M.S.; Trautmann, L.; Willems, B.; Villeneuve, J.P.; Haddad, E.K.; Sekaly, R.P.; Bruneau, J.; Shoukry, N.H. Early interferon therapy for hepatitis C virus infection rescues polyfunctional, long-lived cd8+ memory t cells. J. Virol. 2008, 82, 10017–10031. [Google Scholar] [CrossRef]
- Abdel-Hakeem, M.S.; Bedard, N.; Badr, G.; Ostrowski, M.; Sekaly, R.P.; Bruneau, J.; Willems, B.; Heathcote, E.J.; Shoukry, N.H. Comparison of immune restoration in early versus late alpha interferon therapy against hepatitis C virus. J. Virol. 2010, 84, 10429–10435. [Google Scholar] [CrossRef]
- Kelleher, A.D.; Long, C.; Holmes, E.C.; Allen, R.L.; Wilson, J.; Conlon, C.; Workman, C.; Shaunak, S.; Olson, K.; Goulder, P.; et al. Clustered mutations in HIV-1 gag are consistently required for escape from HLA-b27-restricted cytotoxic T lymphocyte responses. J. Exp. Med. 2001, 193, 375–386. [Google Scholar] [CrossRef]
- Altfeld, M.; Addo, M.M.; Rosenberg, E.S.; Hecht, F.M.; Lee, P.K.; Vogel, M.; Yu, X.G.; Draenert, R.; Johnston, M.N.; Strick, D.; et al. Influence of HLA-B57 on clinical presentation and viral control during acute HIV-1 infection. AIDS 2003, 17, 2581–2591. [Google Scholar] [CrossRef] [PubMed]
- Neumann-Haefelin, C.; Timm, J.; Schmidt, J.; Kersting, N.; Fitzmaurice, K.; Oniangue-Ndza, C.; Kemper, M.N.; Humphreys, I.; McKiernan, S.; Kelleher, D.; et al. Protective effect of human leukocyte antigen B27 in hepatitis C virus infection requires the presence of a genotype-specific immunodominant CD8+ T-cell epitope. Hepatology 2010, 51, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Neumann-Haefelin, C.; Oniangue-Ndza, C.; Kuntzen, T.; Schmidt, J.; Nitschke, K.; Sidney, J.; Caillet-Saguy, C.; Binder, M.; Kersting, N.; Kemper, M.W.; et al. Human leukocyte antigen B27 selects for rare escape mutations that significantly impair hepatitis C virus replication and require compensatory mutations. Hepatology 2011, 54, 1157–1166. [Google Scholar] [CrossRef]
- Ahmed, R.; Salmi, A.; Butler, L.D.; Chiller, J.M.; Oldstone, M.B. Selection of genetic variants of lymphocytic choriomeningitis virus in spleens of persistently infected mice. Role in suppression of cytotoxic T lymphocyte response and viral persistence. J. Exp. Med. 1984, 160, 521–540. [Google Scholar] [CrossRef]
- Matloubian, M.; Concepcion, R.J.; Ahmed, R. Cd4+ T cells are required to sustain CD8+ cytotoxic T-cell responses during chronic viral infection. J. Virol. 1994, 68, 8056–8063. [Google Scholar]
- Salvato, M.; Shimomaye, E.; Southern, P.; Oldstone, M.B. Virus-lymphocyte interactions. Iv. Molecular characterization of LCMV armstrong (CTL+) small genomic segment and that of its variant, Clone 13 (CTL-). Virology 1988, 164, 517–522. [Google Scholar] [CrossRef]
- Matloubian, M.; Somasundaram, T.; Kolhekar, S.R.; Selvakumar, R.; Ahmed, R. Genetic basis of viral persistence: Single amino acid change in the viral glycoprotein affects ability of lymphocytic choriomeningitis virus to persist in adult mice. J. Exp. Med. 1990, 172, 1043–1048. [Google Scholar] [CrossRef]
- Sullivan, B.M.; Emonet, S.F.; Welch, M.J.; Lee, A.M.; Campbell, K.P.; de la Torre, J.C.; Oldstone, M.B. Point mutation in the glycoprotein of lymphocytic choriomeningitis virus is necessary for receptor binding, dendritic cell infection, and long-term persistence. Proc. Natl. Acad. Sci. USA 2011, 108, 2969–2974. [Google Scholar] [CrossRef]
- Wherry, E.J.; Barber, D.L.; Kaech, S.M.; Blattman, J.N.; Ahmed, R. Antigen-independent memory CD8 T cells do not develop during chronic viral infection. Proc. Natl. Acad. Sci. USA 2004, 101, 16004–16009. [Google Scholar] [CrossRef]
- Blackburn, S.D.; Shin, H.; Haining, W.N.; Zou, T.; Workman, C.J.; Polley, A.; Betts, M.R.; Freeman, G.J.; Vignali, D.A.; Wherry, E.J. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat. Immunol. 2009, 10, 29–37. [Google Scholar] [CrossRef]
- Doering, T.A.; Crawford, A.; Angelosanto, J.M.; Paley, M.A.; Ziegler, C.G.; Wherry, E.J. Network analysis reveals centrally connected genes and pathways involved in CD8+ T cell exhaustion versus memory. Immunity 2012, 37, 1130–1144. [Google Scholar] [CrossRef]
- Pauken, K.E.; Sammons, M.A.; Odorizzi, P.M.; Manne, S.; Godec, J.; Khan, O.; Drake, A.M.; Chen, Z.; Sen, D.R.; Kurachi, M.; et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 2016, 354, 1160–1165. [Google Scholar] [CrossRef]
- Zajac, A.J.; Blattman, J.N.; Murali-Krishna, K.; Sourdive, D.J.; Suresh, M.; Altman, J.D.; Ahmed, R. Viral immune evasion due to persistence of activated T cells without effector function. J. Exp. Med. 1998, 188, 2205–2213. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. Car T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [PubMed]
- Twumasi-Boateng, K.; Pettigrew, J.L.; Kwok, Y.Y.E.; Bell, J.C.; Nelson, B.H. Oncolytic viruses as engineering platforms for combination immunotherapy. Nat. Rev. Cancer 2018, 18, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Scheper, W.; Kvistborg, P. Cancer neoantigens. Annu. Rev. Immunol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.P.; Simon, P.; Lower, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrors, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Aurisicchio, L.; Pallocca, M.; Ciliberto, G.; Palombo, F. The perfect personalized cancer therapy: Cancer vaccines against neoantigens. J. Exp. Clin. Cancer Res. 2018, 37, 86. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.A.; Reed, K.; et al. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef]
- Attanasio, J.; Wherry, E.J. Costimulatory and coinhibitory receptor pathways in infectious disease. Immunity 2016, 44, 1052–1068. [Google Scholar] [CrossRef]
- Brunet, J.F.; Denizot, F.; Luciani, M.F.; Roux-Dosseto, M.; Suzan, M.; Mattei, M.G.; Golstein, P. A new member of the immunoglobulin superfamily--CTLA-4. Nature 1987, 328, 267–270. [Google Scholar] [CrossRef] [PubMed]
- Krummel, M.F.; Allison, J.P. CD28 and CTLA-4 have opposing effects on the response of t cells to stimulation. J. Exp. Med. 1995, 182, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Fuller, M.J.; Zajac, A.J. Ablation of CD8 and CD4 T cell responses by high viral loads. J. Immunol. 2003, 170, 477–486. [Google Scholar] [CrossRef]
- Shin, H.; Blackburn, S.D.; Blattman, J.N.; Wherry, E.J. Viral antigen and extensive division maintain virus-specific CD8 t cells during chronic infection. J. Exp. Med. 2007, 204, 941–949. [Google Scholar] [CrossRef] [PubMed]
- Sen, D.R.; Kaminski, J.; Barnitz, R.A.; Kurachi, M.; Gerdemann, U.; Yates, K.B.; Tsao, H.W.; Godec, J.; LaFleur, M.W.; Brown, F.D.; et al. The epigenetic landscape of T cell exhaustion. Science 2016, 354, 1165–1169. [Google Scholar] [CrossRef] [PubMed]
- Pauken, K.E.; Wherry, E.J. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015, 36, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Hui, E.; Cheung, J.; Zhu, J.; Su, X.; Taylor, M.J.; Wallweber, H.A.; Sasmal, D.K.; Huang, J.; Kim, J.M.; Mellman, I.; et al. T cell costimulatory receptor CD28 is a primary target for pd-1-mediated inhibition. Science 2017, 355, 1428–1433. [Google Scholar] [CrossRef]
- Kamphorst, A.O.; Wieland, A.; Nasti, T.; Yang, S.; Zhang, R.; Barber, D.L.; Konieczny, B.T.; Daugherty, C.Z.; Koenig, L.; Yu, K.; et al. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is cd28-dependent. Science 2017, 355, 1423–1427. [Google Scholar] [CrossRef]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Mescher, M.F.; Curtsinger, J.M.; Agarwal, P.; Casey, K.A.; Gerner, M.; Hammerbeck, C.D.; Popescu, F.; Xiao, Z. Signals required for programming effector and memory development by CD8+ T cells. Immunol. Rev. 2006, 211, 81–92. [Google Scholar] [CrossRef]
- Curtsinger, J.M.; Mescher, M.F. Inflammatory cytokines as a third signal for T cell activation. Curr. Opin. Immunol. 2010, 22, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Harding, F.A.; McArthur, J.G.; Gross, J.A.; Raulet, D.H.; Allison, J.P. Cd28-mediated signalling co-stimulates murine t cells and prevents induction of anergy in T-cell clones. Nature 1992, 356, 607–609. [Google Scholar] [CrossRef] [PubMed]
- Gimmi, C.D.; Freeman, G.J.; Gribben, J.G.; Gray, G.; Nadler, L.M. Human t-cell clonal anergy is induced by antigen presentation in the absence of B7 costimulation. Proc. Natl. Acad. Sci. USA 1993, 90, 6586–6590. [Google Scholar] [CrossRef]
- Teijaro, J.R.; Ng, C.; Lee, A.M.; Sullivan, B.M.; Sheehan, K.C.; Welch, M.; Schreiber, R.D.; de la Torre, J.C.; Oldstone, M.B. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 2013, 340, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Wilson, E.B.; Yamada, D.H.; Elsaesser, H.; Herskovitz, J.; Deng, J.; Cheng, G.; Aronow, B.J.; Karp, C.L.; Brooks, D.G. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 2013, 340, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Tinoco, R.; Alcalde, V.; Yang, Y.; Sauer, K.; Zuniga, E.I. Cell-intrinsic transforming growth factor-beta signaling mediates virus-specific CD8+ T cell deletion and viral persistence in vivo. Immunity 2009, 31, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Wilson, E.B.; Brooks, D.G. The role of Il-10 in regulating immunity to persistent viral infections. Curr. Top. Microbiol. Immunol. 2011, 350, 39–65. [Google Scholar]
- Velu, V.; Titanji, K.; Zhu, B.; Husain, S.; Pladevega, A.; Lai, L.; Vanderford, T.H.; Chennareddi, L.; Silvestri, G.; Freeman, G.J.; et al. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature 2009, 458, 206–210. [Google Scholar] [CrossRef]
- Palmer, B.E.; Neff, C.P.; Lecureux, J.; Ehler, A.; Dsouza, M.; Remling-Mulder, L.; Korman, A.J.; Fontenot, A.P.; Akkina, R. In vivo blockade of the pd-1 receptor suppresses HIV-1 viral loads and improves CD4+ T cell levels in humanized mice. J. Immunol. 2013, 190, 211–219. [Google Scholar] [CrossRef]
- Day, C.L.; Kaufmann, D.E.; Kiepiela, P.; Brown, J.A.; Moodley, E.S.; Reddy, S.; Mackey, E.W.; Miller, J.D.; Leslie, A.J.; DePierres, C.; et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 2006, 443, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Boni, C.; Fisicaro, P.; Valdatta, C.; Amadei, B.; Di Vincenzo, P.; Giuberti, T.; Laccabue, D.; Zerbini, A.; Cavalli, A.; Missale, G.; et al. Characterization of hepatitis B virus (HBV)-specific T-cell dysfunction in chronic HBV infection. J. Virol. 2007, 81, 4215–4225. [Google Scholar] [CrossRef] [PubMed]
- Bengsch, B.; Seigel, B.; Ruhl, M.; Timm, J.; Kuntz, M.; Blum, H.E.; Pircher, H.; Thimme, R. Coexpression of pd-1, 2b4, cd160 and klrg1 on exhausted HCV-specific CD8+ t cells is linked to antigen recognition and T cell differentiation. PLoS Pathog. 2010, 6, e1000947. [Google Scholar] [CrossRef] [PubMed]
- Mumprecht, S.; Schurch, C.; Schwaller, J.; Solenthaler, M.; Ochsenbein, A.F. Programmed death 1 signaling on chronic myeloid leukemia-specific T cells results in T-cell exhaustion and disease progression. Blood 2009, 114, 1528–1536. [Google Scholar] [CrossRef] [PubMed]
- Schietinger, A.; Philip, M.; Krisnawan, V.E.; Chiu, E.Y.; Delrow, J.J.; Basom, R.S.; Lauer, P.; Brockstedt, D.G.; Knoblaugh, S.E.; Hammerling, G.J.; et al. Tumor-specific T cell dysfunction is a dynamic antigen-driven differentiation program initiated early during tumorigenesis. Immunity 2016, 45, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Ahmadzadeh, M.; Johnson, L.A.; Heemskerk, B.; Wunderlich, J.R.; Dudley, M.E.; White, D.E.; Rosenberg, S.A. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009, 114, 1537–1544. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huang, S.; Gong, D.; Qin, Y.; Shen, Q. Programmed death-1 upregulation is correlated with dysfunction of tumor-infiltrating CD8+ T lymphocytes in human non-small cell lung cancer. Cell Mol. Immunol. 2010, 7, 389–395. [Google Scholar] [CrossRef]
- Gandhi, M.K.; Lambley, E.; Duraiswamy, J.; Dua, U.; Smith, C.; Elliott, S.; Gill, D.; Marlton, P.; Seymour, J.; Khanna, R. Expression of LAG-3 by tumor-infiltrating lymphocytes is coincident with the suppression of latent membrane antigen-specific cd8+ t-cell function in hodgkin lymphoma patients. Blood 2006, 108, 2280–2289. [Google Scholar] [CrossRef]
- Matsuzaki, J.; Gnjatic, S.; Mhawech-Fauceglia, P.; Beck, A.; Miller, A.; Tsuji, T.; Eppolito, C.; Qian, F.; Lele, S.; Shrikant, P.; et al. Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 7875–7880. [Google Scholar] [CrossRef]
- Riches, J.C.; Davies, J.K.; McClanahan, F.; Fatah, R.; Iqbal, S.; Agrawal, S.; Ramsay, A.G.; Gribben, J.G. T cells from CLL patients exhibit features of T-cell exhaustion but retain capacity for cytokine production. Blood 2013, 121, 1612–1621. [Google Scholar] [CrossRef]





© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdel-Hakeem, M.S. Viruses Teaching Immunology: Role of LCMV Model and Human Viral Infections in Immunological Discoveries. Viruses 2019, 11, 106. https://doi.org/10.3390/v11020106
Abdel-Hakeem MS. Viruses Teaching Immunology: Role of LCMV Model and Human Viral Infections in Immunological Discoveries. Viruses. 2019; 11(2):106. https://doi.org/10.3390/v11020106
Chicago/Turabian StyleAbdel-Hakeem, Mohamed S. 2019. "Viruses Teaching Immunology: Role of LCMV Model and Human Viral Infections in Immunological Discoveries" Viruses 11, no. 2: 106. https://doi.org/10.3390/v11020106
APA StyleAbdel-Hakeem, M. S. (2019). Viruses Teaching Immunology: Role of LCMV Model and Human Viral Infections in Immunological Discoveries. Viruses, 11(2), 106. https://doi.org/10.3390/v11020106