Ecology of West Nile Virus in the Danube Delta, Romania: Phylogeography, Xenosurveillance and Mosquito Host-Feeding Patterns

 , , ,
, , , 
 ,
, 
 ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. Mosquitoes and WNV in the Danube Delta
3.2. Genome Characterization of WNV in the DDBR
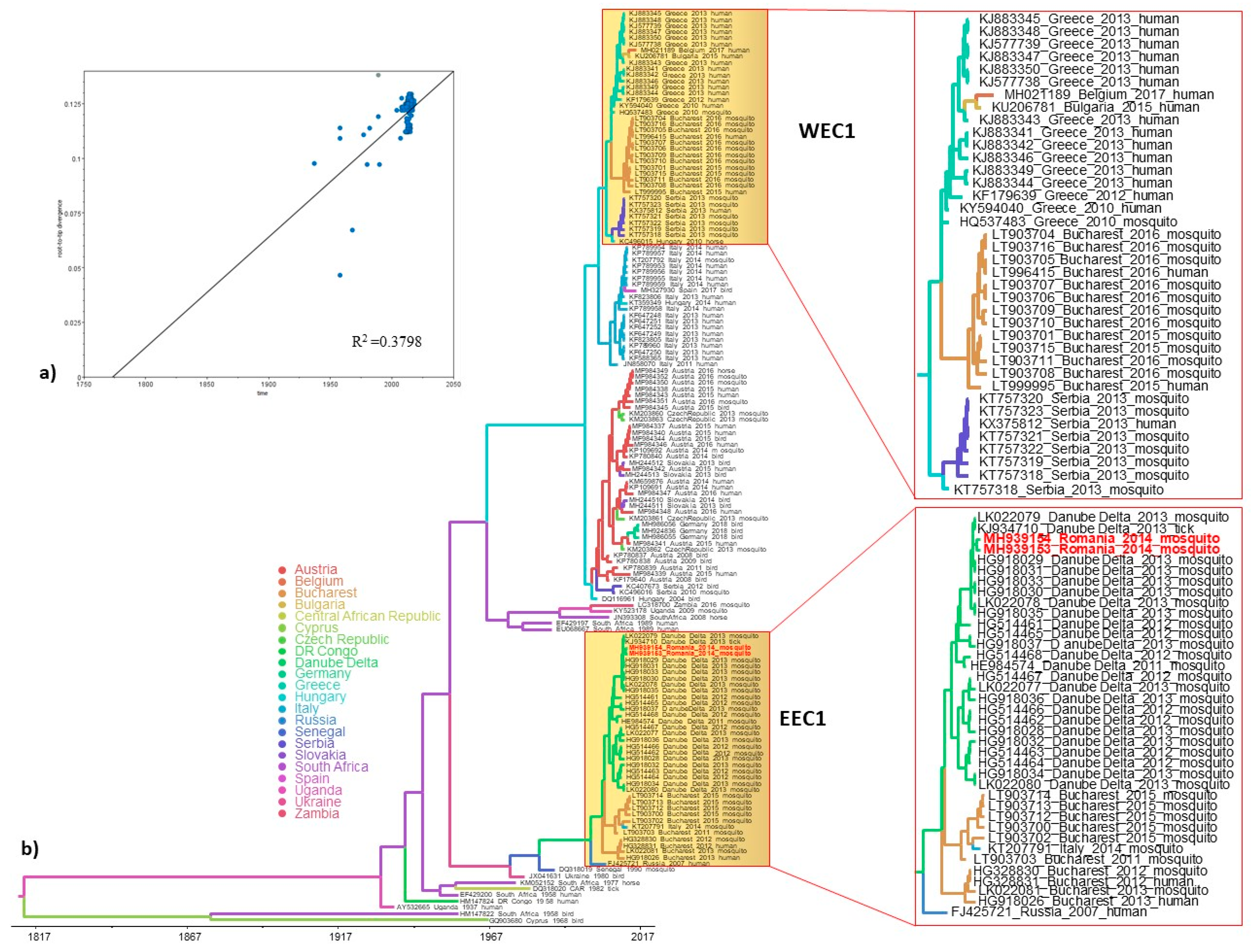
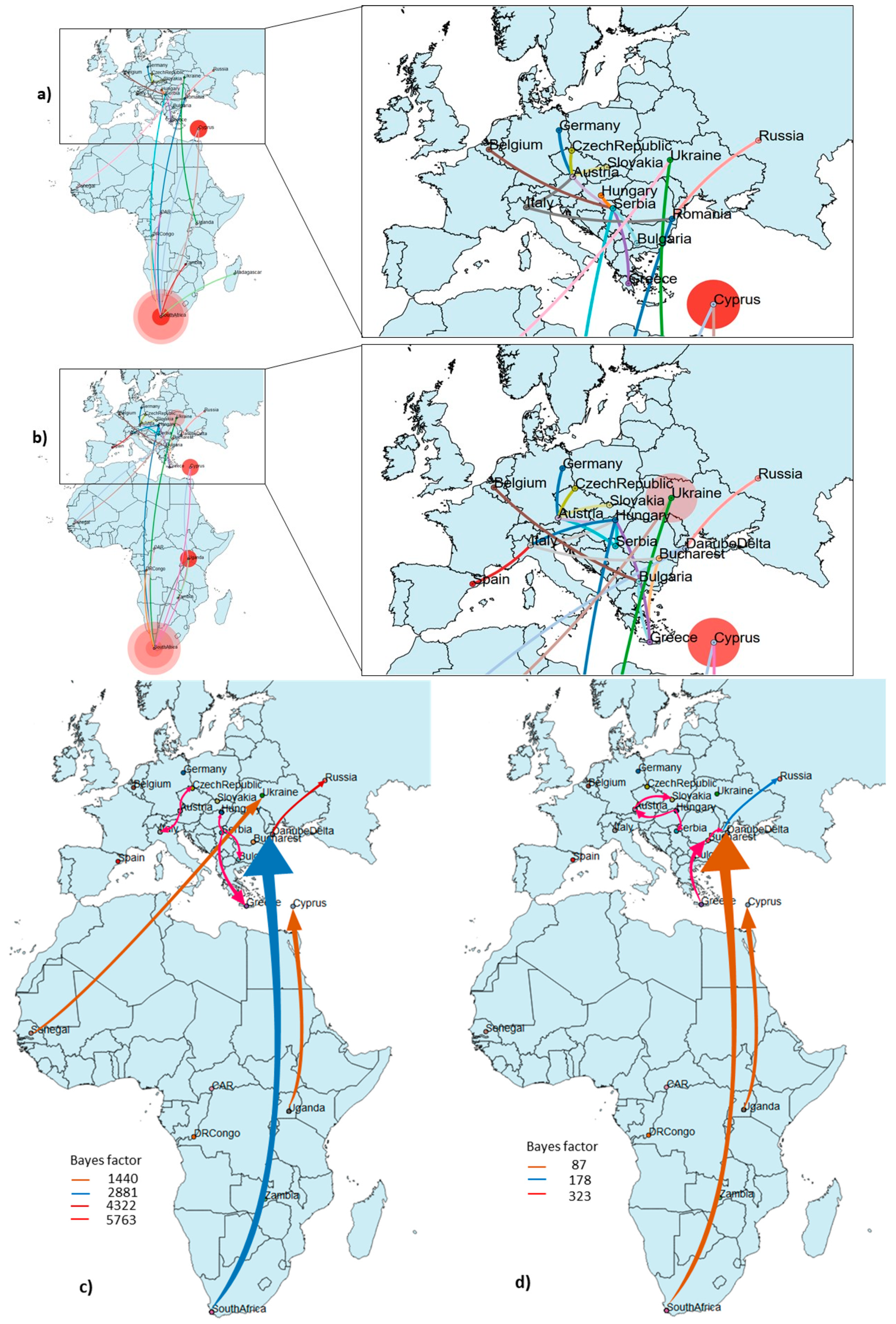
3.3. Phylogeography and Spatio-Temporal Dispersal Pattern of WNV
3.4. Screening for WNV-Specific IgG Antibodies
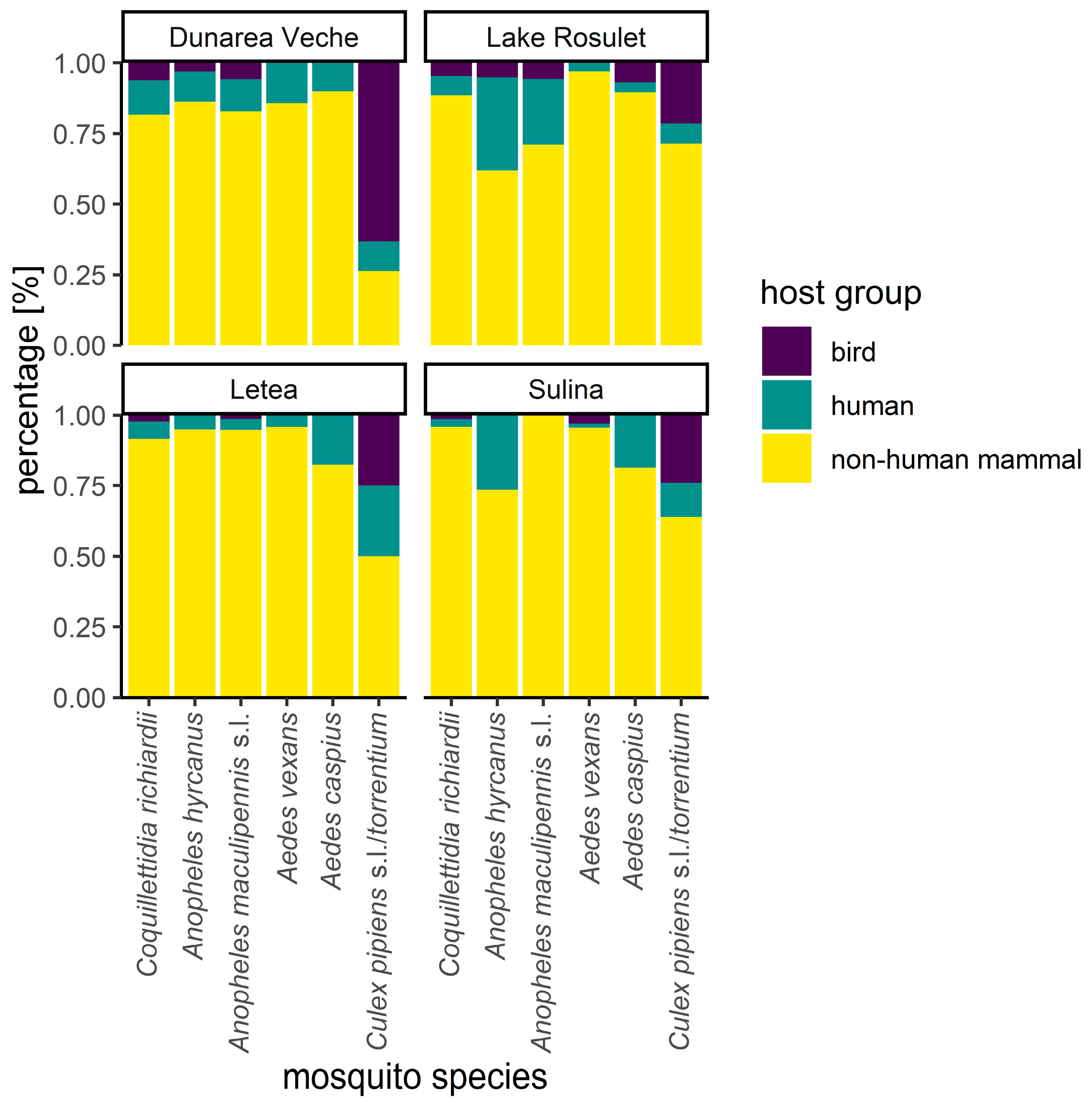
3.5. Host-Feeding Patterns
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zeller, H.; Marrama, L.; Sudre, B.; Bortel, W.V.; Warns-Petit, E. Mosquito-borne disease surveillance by the European Centre for Disease Prevention and Control. Clin. Microbiol. Infect. 2013, 19, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Hubálek, Z. Mosquito-borne viruses in Europe. Parasitol. Res. 2008, 103, 29–43. [Google Scholar] [CrossRef] [PubMed]
- Chancey, C.; Grinev, A.; Volkova, E.; Rios, M. The global ecology and epidemiology of West Nile virus. BioMed Res. Int. 2015, 2015, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Gould, E.; Pettersson, J.; Higgs, S.; Charrel, R.; de Lamballerie, X. Emerging arboviruses: Why today? One Health 2017, 4, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Weaver, S.C.; Reisen, W.K. Present and future arboviral threats. Antivir. Res. 2010, 85, 328–345. [Google Scholar] [CrossRef] [PubMed]
- Tsai, T.; Popovici, F.; Cernescu, C.; Campbell, G.; Nedelcu, N. West Nile encephalitis epidemic in southeastern Romania. Lancet 1998, 352, 767–771. [Google Scholar] [CrossRef]
- Ziegler, U.; Lühken, R.; Keller, M.; Cadar, D.; van der Grinten, E.; Michel, F.; Albrecht, K.; Eiden, M.; Rinder, M.; Lachmann, L.; et al. West Nile virus epizootic in Germany, 2018. Antivir. Res. 2019, 162, 39–43. [Google Scholar] [CrossRef]
- European Centre for Disease Prevention and Control. Historical Data by Year—West Nile Fever Seasonal Surveillance. 2019. Available online: https://www.ecdc.europa.eu/en/west-nile-fever/surveillance-and-disease-data/historical (accessed on 5 November 2019).
- Török, E.; Tomazatos, A.; Cadar, D.; Horváth, C.; Keresztes, L.; Jansen, S.; Becker, N.; Kaiser, A.; Popescu, O.; Schmidt-Chanasit, J.; et al. Pilot longitudinal mosquito surveillance study in the Danube Delta Biosphere Reserve and the first reports of Anopheles algeriensis Theobald, 1903 and Aedes hungaricus Mihályi, 1955 for Romania. Parasit. Vectors 2016, 9, 196. [Google Scholar] [CrossRef]
- Han, L.L.; Popovici, F.; Alexander, J.P., Jr.; Laurentia, V.; Tengelsen, L.A.; Cernescu, C.; Gary, H.E., Jr.; Ion-Nedelcu, N.; Campbell, G.L.; Tsai, T.F. Risk factors for West Nile virus infection and meningoencephalitis, Romania, 1996. J. Infect. Dis. 1999, 179, 230–233. [Google Scholar] [CrossRef]
- Savage, H.M.; Ceianu, C.; Nicolescu, G.; Karabatsos, N.; Lanciotti, R.; Vladimirescu, A.; Laiv, L.; Ungureanu, A.; Romanca, C.; Tsai, T.F. Entomologic and avian investigations of an epidemic of West Nile fever in Romania in 1996, with serologic and molecular characterization of a virus isolate from mosquitoes. Am. J. Trop. Med. Hyg. 1999, 61, 600–611. [Google Scholar] [CrossRef]
- Sirbu, A.; Ceianu, C.S.; Panculescu-Gatej, R.; Vasquez, A.; Tenorio, A.; Rebreanu, R.; Niedrig, M.; Nicolescu, G.; Pistol, A. Outbreak of West Nile virus infection in humans, Romania, July to October 2010. Euro Surveill. 2010, 16, 19762. [Google Scholar]
- Dinu, S.; Cotar, A.; Pănculescu-Gătej, I.; Fălcuţă, E.; Prioteasa, F.; Sîrbu, A.; Oprişan, G.; Bădescu, D.; Reiter, P.; Ceianu, C. West Nile virus circulation in south-eastern Romania, 2011 to 2013. Euro Surveill. 2015, 20, 21130. [Google Scholar] [CrossRef] [PubMed]
- Popescu, C.P.; Florescu, S.A.; Cotar, A.I.; Badescu, D.; Ceianu, C.S.; Zaharia, M.; Tardei, G.; Codreanu, D.; Ceausu, E.; Ruta, S.M. Re-emergence of severe West Nile virus neuroinvasive disease in humans in Romania, 2012 to 2017–implications for travel medicine. Travel Med. Infect. Dis. 2018, 22, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Petersen, L.R.; Brault, A.C.; Nasci, R.S. West Nile virus: Review of the literature. JAMA 2013, 310, 308–315. [Google Scholar] [CrossRef]
- Fall, G.; Di Paola, N.; Faye, M.; Dia, M.; de Melo Freire, C.C.; Loucoubar, C.; de Andrade Zanotto, P.M.; Faye, O. Biological and phylogenetic characteristics of West African lineages of West Nile virus. PLoS Negl. Trop. Dis. 2017, 11, e0006078. [Google Scholar] [CrossRef]
- May, F.J.; Davis, C.T.; Tesh, R.B.; Barrett, A.D. Phylogeography of West Nile virus. J. Virol. 2011, 85, 2964–2974. [Google Scholar] [CrossRef]
- Sambri, V.; Capobianchi, M.; Charrel, R.; Fyodorova, M.; Gaibani, P.; Gould, E.; Niedrig, M.; Papa, A.; Pierro, A.; Rossini, G.; et al. West Nile virus in Europe: Emergence, epidemiology, diagnosis, treatment, and prevention. Clin. Microbiol. Infect. 2013, 19, 699–704. [Google Scholar] [CrossRef]
- Nash, D.; Mostashari, F.; Fine, A.; Miller, J.; O’Leary, D.; Murray, K.; Huang, A.; Rosenberg, A.; Greenberg, A.; Sherman, M.; et al. The outbreak of West Nile virus infection in the New York City area in 1999. N. Engl. J. Med. 2001, 344, 1807–1814. [Google Scholar] [CrossRef]
- Lindsey, N.P.; Staples, J.E.; Lehman, J.A.; Fischer, M. Surveillance for human West Nile virus disease. MMWR Surveill. Summ. 2010, 59, 1–17. [Google Scholar]
- Petersen, L.R.; Hayes, E.B. West Nile virus in the Americas. Med. Clin. North Am. 2008, 92, 1307–1322. [Google Scholar] [CrossRef]
- Hanganu, J.; Dubyna, D.; Zhmud, E.; Grigoraş, I.; Menke, U.; Drost, H.; Ştefan, N.; Sârbu, I. Vegetation of the Biosphere Reserve “Danube Delta”—With Transboundary Vegetation Map on a 1:150,000 Scale; Danube Delta National Institute, Romania, M.G. Kholodny Institute of Botany & Danube Delta Biosphere Reserve, Ukraine and RIZA, Eds.; RIZA Rapport: Lelystad, The Netherlands, 2002. [Google Scholar]
- Prioteasa, F.L.; Falcuta, E. An annotated checklist of the mosquitoes (Diptera: Culicidae) of the Danube Delta Biosphere Reserve. Eur. Mosq. Bull. 2010, 28, 240–245. [Google Scholar]
- Cotar, A.I.; Falcuta, E.; Prioteasa, L.F.; Dinu, S.; Ceianu, C.S.; Paz, S. Transmission dynamics of the West Nile virus in mosquito vector populations under the influence of weather factors in the Danube Delta, Romania. EcoHealth 2016, 13, 796–807. [Google Scholar] [CrossRef]
- Gossner, C.M.; Marrama, L.; Carson, M.; Allerberger, F.; Calistri, P.; Dilaveris, D.; Lecollinet, S.; Morgan, D.; Nowotny, N.; Paty, M.C.; et al. West Nile virus surveillance in Europe: Moving towards an integrated animal-human-vector approach. Euro Surveill. 2017, 22, 30526. [Google Scholar] [CrossRef]
- Leighton, B.J.; Roitberg, B.D.; Belton, P.; Lowenberger, C.A. Host antibodies in mosquito bloodmeals: A potential tool to detect and monitor infectious diseases in wildlife. J. Med. Entomol. 2008, 45, 470–475. [Google Scholar] [CrossRef]
- Barbazan, P.; Palabodeewat, S.; Nitatpattana, N.; Gonzalez, J.P. Detection of host virus-reactive antibodies in blood meals of naturally engorged mosquitoes. Vector Borne Zoonotic Dis. 2009, 9, 103–108. [Google Scholar] [CrossRef]
- Kurucz, K.; Kepner, A.; Krtinic, B.; Hederics, D.; Foldes, F.; Brigetta, Z.; Jakab, F.; Kemenesi, G. Blood-meal analysis and avian malaria screening of mosquitoes collected from human-inhabited areas in Hungary and Serbia. J. Eur. Mosq. Control Assoc. 2018, 36, 3–13. [Google Scholar]
- Balenghien, T.; Fouque, F.; Sabatier, P.; Bicout, D.J. Horse-, Bird-, and Human-Seeking Behavior and Seasonal Abundance of Mosquitoes in a West Nile Virus Focus of Southern France. J. Med. Entomol. 2006, 43, 936–946. [Google Scholar] [CrossRef]
- Gomes, B.; Sousa, C.A.; Vicente, J.L.; Pinho, L.; Calderón, I.; Arez, E.; Almeida, A.P.; Donnelly, M.J.; Pinto, J. Feeding patterns of molestus and pipiens forms of Culex pipiens (Diptera: Culicidae) in a region of high hybridization. Parasit. Vectors 2013, 6, 93. [Google Scholar] [CrossRef]
- Muñoz, J.; Ruiz, S.; Soriguer, R.; Alcaide, M.; Viana, D.S.; Roiz, D.; Vázquez, A.; Figuerola, J. Feeding patterns of potential West Nile virus vectors in south-west Spain. PLoS ONE 2012, 7, e39549. [Google Scholar] [CrossRef]
- Rizzoli, A.; Bolzoni, L.; Chadwick, E.A.; Capelli, G.; Montarsi, F.; Grisenti, M.; de la Puente, J.M.; Muñoz, J.; Figuerola, J.; Soriguer, R.; et al. Understanding West Nile virus ecology in Europe: Culex pipiens host feeding preference in a hotspot of virus emergence. Parasit. Vectors 2015, 8, 213. [Google Scholar] [CrossRef]
- Martínez-de la Puente, J.; Ferraguti, M.; Ruiz, S.; Roiz, D.; Soriguer, R.C.; Figuerola, J. Culex pipiens forms and urbanization: Effects on blood feeding sources and transmission of avian Plasmodium. Malar. J. 2016, 15, 589. [Google Scholar] [CrossRef] [PubMed]
- Fălcuţă, E.; Toty, C.; Prioteasa, F.L.; Nicolescu, G.; Teodorescu, I.; Purcarea-Ciulacu, V.; Fontenille, D. Blood-meal preferences for Anopheles maculipennis (Diptera: Cuicidae) complex species in Comana, Giurgiu county (Romania). Rom. J. Biol. Zool. 2010, 55, 1–110. [Google Scholar]
- Martínez-de la Puente, J.; Muñoz, J.; Capelli, G.; Montarsi, F.; Soriguer, R.; Arnoldi, D.; Rizzoli, A.; Figuerola, J. Avian malaria parasites in the last supper: Identifying encounters between parasites and the invasive Asian mosquito tiger and native mosquito species in Italy. Malar. J. 2015, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Triana, L.M.; Brugman, V.A.; Prosser, S.W.J.; Weland, C.; Nikolova, N.; Thorne, L.; Marco, M.F.D.; Fooks, A.R.; Johnson, N. Molecular approaches for blood meal analysis and species identification of mosquitoes (Insecta: Diptera: Culicidae) in rural locations in southern England, United Kingdom. Zootaxa 2017, 4250, 67. [Google Scholar] [CrossRef] [PubMed]
- Börstler, J.; Jöst, H.; Garms, R.; Krüger, A.; Tannich, E.; Becker, N.; Schmidt-Chanasit, J.; Lühken, R. Host-feeding patterns of mosquito species in Germany. Parasit. Vectors 2016, 9, 318. [Google Scholar] [CrossRef] [PubMed]
- Schönenberger, A.C.; Wagner, S.; Tuten, H.C.; Schaffner, F.; Torgerson, P.; Furrer, S.; Mathis, A.; Silaghi, C. Host preferences in host-seeking and blood-fed mosquitoes in Switzerland: Host preferences in mosquitoes. Med. Vet. Entomol. 2015, 30, 39–52. [Google Scholar] [CrossRef]
- Becker, N.; Petric, D.; Zgomba, M.; Boase, C.; Madon, M.; Dahl, C.; Kaiser, A. Mosquitoes and Their Control, 2nd ed.; Springer: Berlin/Heidelberg, Germany, 2010; ISBN 978-3-540-92873-7. [Google Scholar]
- Rudolf, M.; Czajka, C.; Börstler, J.; Melaun, C.; Jöst, H.; von Thien, H.; Badusche, M.; Becker, N.; Schmidt-Chanasit, J.; Krüger, A.; et al. First nationwide surveillance of Culex pipiens complex and Culex torrentium mosquitoes demonstrated the presence of Culex pipiens biotype pipiens/molestus hybrids in Germany. PLoS ONE 2013, 8, e71832. [Google Scholar] [CrossRef]
- Chao, D.Y.; Davis, B.S.; Chang, G.J.J. Development of multiplex real-time reverse transcriptase PCR assays for detecting eight medically important flaviviruses in mosquitoes. J. Clin. Microbiol. 2007, 45, 584–589. [Google Scholar] [CrossRef]
- Becker, N.; Jöst, H.; Ziegler, U.; Eiden, M.; Höper, D.; Emmerich, P.; Fichet-Calvet, E.; Ehichioya, D.U.; Czajka, C.; Gabriel, M.; et al. Epizootic emergence of Usutu virus in wild and captive birds in Germany. PLoS ONE 2012, 7, e32604. [Google Scholar] [CrossRef]
- Jungbauer, C.; Hourfar, M.K.; Stiasny, K.; Aberle, S.W.; Cadar, D.; Schmidt-Chanasit, J.; Mayr, W.R. West Nile virus lineage 2 infection in a blood donor from Vienna, Austria, August 2014. J. Clin. Virol. 2015, 64, 16–19. [Google Scholar] [CrossRef]
- Burkett-Cadena, N.D.; Graham, S.P.; Hassan, H.K.; Guyer, C.; Eubanks, M.D.; Katholi, C.R.; Unnasch, T.R. Blood feeding patterns of potential arbovirus vectors of the genus culex targeting ectothermic hosts. Am. J. Trop. Med. Hyg. 2008, 79, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Kitano, T.; Umetsu, K.; Tian, W.; Osawa, M. Two universal primer sets for species identification among vertebrates. Int. J. Leg. Med. 2007, 121, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Kocher, T.D.; Thomas, W.K.; Meyer, A.; Edwards, S.V.; Paabo, S.; Villablanca, F.X.; Wilson, A.C. Dynamics of mitochondrial DNA evolution in animals: Amplification and sequencing with conserved primers. Proc. Natl. Acad. Sci. USA 1989, 86, 6196–6200. [Google Scholar] [CrossRef] [PubMed]
- R Core Team R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013.
- Wickham, H. The split-apply-combine strategy for data analysis. J. Stat. Softw. 2011, 40, 1–29. [Google Scholar] [CrossRef]
- Wickham, H.; François, R.; Henry, L.; Müller, K. Dplyr: A Grammar of Data Manipulation. 2019. Available online: https://rdrr.io/cran/dplyr/ (accessed on 5 November 2019).
- Bache, S.M.; Wickham, H. Magrittr: A Forward-Pipe Operator for R. 2014. [Google Scholar]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009; ISBN 978-0-387-98140-6. [Google Scholar]
- Tappe, D.; Schmidt-Chanasit, J.; Ries, A.; Ziegler, U.; Müller, A.; Stich, A. Ross River virus infection in a traveller returning from northern Australia. Med. Microbiol. Immunol. (Berl.) 2009, 198, 271–273. [Google Scholar] [CrossRef]
- Crivei, A.L.; Rățoi (Anton), I.; Răileanu, C.; Porea, D.; Aniţa, D.; Savuța, G.; Oșlobanu, L. First record of West Nile virus specific seroconversion in dogs from Eastern Romania. Bull. Univ. Agric. Sci. Vet. Med. Cluj Napoca Vet. Med. 2018, 75, 163. [Google Scholar] [CrossRef]
- Durand, B.; Haskouri, H.; Lowenski, S.; Vachiery, N.; Beck, C.; Lecollinet, S. Seroprevalence of West Nile and Usutu viruses in military working horses and dogs, Morocco, 2012: Dog as an alternative WNV sentinel species? Epidemiol. Infect. 2016, 144, 1857–1864. [Google Scholar] [CrossRef]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef]
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian phylogeography finds its roots. PLoS Comput. Biol. 2009, 5, e1000520. [Google Scholar] [CrossRef] [PubMed]
- Jourdain, E.; Toussaint, Y.; Leblond, A.; Bicout, D.J.; Sabatier, P.; Gauthier-Clerc, M. Bird species potentially involved in introduction, amplification, and spread of West Nile virus in a mediterranean wetland, the Camargue (Southern France). Vector Borne Zoonotic Dis. 2007, 7, 15–33. [Google Scholar] [CrossRef] [PubMed]
- Hubálek, Z.; Halouzka, J. a West Nile fever - a reemerging mosquito-borne viral disease in Europe. Emerg. Infect. Dis. 1999, 5, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Malkinson, M. Introduction of West Nile virus in the Middle East by Migrating White Storks. Emerg. Infect. Dis. 2002, 8, 392–397. [Google Scholar] [CrossRef]
- Ciccozzi, M.; Peletto, S.; Cella, E.; Giovanetti, M.; Lai, A.; Gabanelli, E.; Acutis, P.L.; Modesto, P.; Rezza, G.; Platonov, A.E.; et al. Epidemiological history and phylogeography of West Nile virus lineage 2. Infect. Genet. Evol. 2013, 17, 46–50. [Google Scholar] [CrossRef]
- Di Giallonardo, F.; Geoghegan, J.L.; Docherty, D.E.; McLean, R.G.; Zody, M.C.; Qu, J.; Yang, X.; Birren, B.W.; Malboeuf, C.M.; Newman, R.M.; et al. Fluid spatial dynamics of West Nile virus in the United States: Rapid spread in a permissive host environment. J. Virol. 2016, 90, 862–872. [Google Scholar] [CrossRef]
- Liu, W.J.; Chen, H.B.; Wang, X.J.; Huang, H.; Khromykh, A.A. Analysis of adaptive mutations in Kunjin virus replicon RNA reveals a novel role for the flavivirus nonstructural protein NS2A in inhibition of beta interferon promoter-driven transcription. J. Virol. 2004, 78, 12225–12235. [Google Scholar] [CrossRef]
- Armstrong, P.M.; Vossbrinck, C.R.; Andreadis, T.G.; Anderson, J.F.; Pesko, K.N.; Newman, R.M.; Lennon, N.J.; Birren, B.W.; Ebel, G.D.; Henn, M.R. Molecular evolution of West Nile virus in a northern temperate region: Connecticut, USA 1999–2008. Virology 2011, 417, 203–210. [Google Scholar] [CrossRef]
- Martínez-de la Puente, J.; Méndez, M.; Ruiz, S.; Godoy, J.A.; Soriguer, R.C.; Figuerola, J. Individual identification of endangered species using mosquito blood meals: A proof-of-concept study in Iberian lynx. Parasitol. Res. 2015, 114, 1607–1610. [Google Scholar] [CrossRef]
- García-Bocanegra, I.; Jurado-Tarifa, E.; Cano-Terriza, D.; Martínez, R.; Pérez-Marín, J.E.; Lecollinet, S. Exposure to West Nile virus and tick-borne encephalitis virus in dogs in Spain. Transbound. Emerg. Dis. 2018, 65, 765–772. [Google Scholar] [CrossRef]
- García-Bocanegra, I.; Arenas-Montes, A.; Napp, S.; Jaén-Téllez, J.A.; Fernández-Morente, M.; Fernández-Molera, V.; Arenas, A. Seroprevalence and risk factors associated to West Nile virus in horses from Andalusia, Southern Spain. Vet. Microbiol. 2012, 160, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Maquart, M.; Dahmani, M.; Marié, J.L.; Gravier, P.; Leparc-Goffart, I.; Davoust, B. First serological evidence of West Nile virus in horses and dogs from Corsica Island, France. Vector Borne Zoonotic Dis. 2017, 17, 275–277. [Google Scholar] [CrossRef] [PubMed]
- Popescu, D.; Necșulescu, M.; Alexse, A.; Purcărea-Ciulacu, V. West Nile virus infection of horses in anthropic ecosystems in Romania. Rev. Rom. Med. Vet. 2008, 18, 39–46. [Google Scholar]
- Port, G.R.; Boreham, P.F.L.; Bryan, J.H. The relationship of host size to feeding by mosquitoes of the Anopheles gambiae Giles complex (Diptera: Culicidae). Bull. Entomol. Res. 1980, 70, 133–144. [Google Scholar] [CrossRef]
- Hubalek, Z.; Rudolf, I.; Nowotny, N. Arboviruses pathogenic for domestic and wild animals. Adv. Virus Res. 2014, 89, 201–275. [Google Scholar] [PubMed]
- Kilpatrick, A.M.; Kramer, L.D.; Jones, M.J.; Marra, P.P.; Daszak, P. West Nile virus epidemics in North America are driven by shifts in mosquito feeding behavior. PLoS Biol. 2006, 4, e82. [Google Scholar] [CrossRef]
- Omondi, D.; Masiga, D.K.; Ajamma, Y.U.; Fielding, B.C.; Njoroge, L.; Villinger, J. Unraveling host-vector-arbovirus interactions by two-gene high resolution melting mosquito bloodmeal analysis in a Kenyan wildlife-livestock interface. PLoS ONE 2015, 10, e0134375. [Google Scholar] [CrossRef]
- Shahhosseini, N.; Friedrich, J.; Moosa-Kazemi, S.H.; Sedaghat, M.M.; Kayedi, M.H.; Tannich, E.; Schmidt-Chanasit, J.; Lühken, R. Host-feeding patterns of Culex mosquitoes in Iran. Parasit. Vectors 2018, 11, 669. [Google Scholar] [CrossRef]
- Apperson, C.S.; Hassan, H.K.; Harrison, B.A.; Savage, H.M.; Aspen, S.E.; Farajollahi, A.; Crans, W.; Daniels, T.J.; Falco, R.C.; Benedict, M.; et al. Host feeding patterns of established and potential mosquito vectors of West Nile virus in the eastern United States. Vector Borne Zoonotic Dis. 2004, 4, 71–82. [Google Scholar] [CrossRef]
- Prioteasa, F.L. Evaluating the Vectorial Potential of Danube Delta Culicids (Diptera: Insecta) for West Nile Virus (Flaviviridae). Ph.D. Thesis, Faculty of Biology, University of Bucharest, Bucharest, Wallachia, 2011. [Google Scholar]
- L’Ambert, G.; Ferré, J.B.; Schaffner, F.; Fontenille, D. Comparison of different trapping methods for surveillance of mosquito vectors of West Nile virus in Rhône Delta, France. J. Vector Ecol. 2012, 37, 269–275. [Google Scholar] [CrossRef]
- Lühken, R.; Pfitzner, W.; Börstler, J.; Garms, R.; Huber, K.; Schork, N.; Steinke, S.; Kiel, E.; Becker, N.; Tannich, E.; et al. Field evaluation of four widely used mosquito traps in Central Europe. Parasit. Vectors 2014, 7, 268. [Google Scholar] [CrossRef] [PubMed]
- Ceianu, C.S.; Ungureanu, A.; Nicolescu, G.; Cernescu, C.; Nitescu, L.; Tardei, G.; Petrescu, A.; Pitigoi, D.; Martin, D.; Ciulacu-Purcarea, V.; et al. West Nile virus surveillance in Romania: 1997–2000. Viral Immunol. 2001, 14, 251–262. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Host-Species | Mosquito Species | Dunărea Veche | Lake Roșuleț | Letea | Sulina | Sum |
|---|---|---|---|---|---|---|
| dog | Aedes caspius | 1 (1) | 0 (0) | 0 (2) | 0 (1) | 1 (4) |
| Aedes vexans | 0 (1) | 0 (3) | 0 (1) | 0 (2) | 0 (7) | |
| Anopheles hyrcanus | 0 (2) | 1 (2) | 1 (5) | 1 (1) 1 | 3 (10) | |
| Anopheles maculipennis s.l. | 0 (3) | 1 (13) | 0 (2) | 0 (1) | 1 (19) | |
| Coquillettidia richiardii | 0 (5) | 1 (40) | 0 (7) | 1 (10) | 2 (62) | |
| Culex modestus | 0 (0) | 0 (1) | 0 (0) | 0 (0) | 0 (1) | |
| Culex pipiens s.l./torrentium | 0 (1) | 0 (5) | 0 (0) | 0 (2) | 0 (8) | |
| horse | Aedes caspius | 0 (2) | 0 (6) | 0 (10) | 0 (7) | 0 (25) |
| Aedes cinereus | 0 (0) | 0 (0) | 0 (2) | 0 (0) | 0 (2) | |
| Aedes detritus | 0 (0) | 0 (0) | 0 (0) | 0 (1) | 0 (1) | |
| Aedes vexans | 0 (0) | 1 (20) | 0 (92) | 0 (8) | 1 (120) | |
| Anopheles algeriensis | 0 (0) | 0 (0) | 0 (1) | 0 (1) | 0 (2) | |
| Anopheles hyrcanus | 0 (1) | 3 (6) | 0 (65) | 0 (6) | 3 (78) | |
| Anopheles maculipennis s.l. | 0 (0) | 0 (1) | 1 (12) | 1 (8) | 2 (21) | |
| Coquillettidia richiardii | 1 (2) | 1 (32) | 3 (90) | 1 (16) | 6 (140) | |
| Culex pipiens s.l./torrentium | 0 (0) | 0 (0) | 0 (1) | 0 (1) | 0 (2) | |
| human | Aedes caspius | 0 (1) | 0 (1) | 0 (7) | 0 (5) | 0 (14) |
| Aedes flavescens | 0 (1) | 0 (1) | 0 (0) | 0 (0) | 0 (2) | |
| Aedes vexans | 0 (1) | 0 (1) | 0 (5) | 0 (1) | 0 (8) | |
| Anopheles algeriensis | 0 (0) | 0 (1) | 0 (0) | 0 (8) | 0 (9) | |
| Anopheles hyrcanus | 0 (7) | 0 (96) | 0 (20) | 0 (9) | 0 (132) | |
| Anopheles maculipennis s.l. | 0 (4) | 0 (33) | 0 (3) | 0 (0) | 0 (40) | |
| Coquillettidia richiardii | 0 (12) | 0 (28) | 0 (11) | 0 (4) | 0 (55) | |
| Culex modestus | 0 (0) | 0 (1) | 0 (0) | 0 (2) | 0 (3) | |
| Culex pipiens s.l./torrentium | 0 (2) | 0 (1) | 0 (1) | 0 (3) | 0 (7) | |
| Uranotaenia unguiculata | 0 (1) | 0 (0) | 0 (0) | 0 (0) | 0 (1) | |
| bird | Aedes caspius | 0 (0) | 0 (2) | 0 (0) | 0 (0) | 0 (2) |
| Aedes vexans | 0 (0) | 0 (0) | 0 (0) | 0 (2) | 0 (2) | |
| Anopheles hyrcanus | 0 (2) | 0 (15) | 0 (0) | 0 (0) | 0 (17) | |
| Anopheles maculipennis s.l. | 0 (2) | 0 (8) | 0 (1) | 0 (0) | 0 (11) | |
| Coquillettidia richiardii | 0 (6) | 0 (19) | 0 (4) | 0 (2) | 0 (31) | |
| Culex pipiens s.l./torrentium | 0 (12) | 0 (3) | 0 (1) | 0 (6) | 0 (22) | |
| Sum | 2 (69) | 8 (339) | 5 (343) | 4 (107) | 19 (858) |
| Coquillettidia richiardii | Anopheles hyrcanus | Anopheles maculipennis s.l. | Aedes vexans | Aedes caspius | Culex pipiens s.l./torrentium | Sum | |
|---|---|---|---|---|---|---|---|
| Anas platyrhynchos | 3 (0.4) | 1 (0.4) | 1 (0.9) | 5 (0.2) | |||
| Anatidae | 1 (0.1) | 12 (1.5) | 4 (1.4) | 1 (0.9) | 18 (0.8) | ||
| Ardea purpurea | 10 (1.2) | 1 (0.4) | 11 (0.5) | ||||
| Circus aeroginosus | 1 (0.4) | 1 (0) | |||||
| Corvus corone | 1 (0.1) | 1 (0) | |||||
| Corvus fragilegus | 1 (0.4) | 1 (0) | |||||
| Cyanistes caeruleus | 5 (0.6) | 3 (5.4) | 8 (0.3) | ||||
| Cygnus olor | 1 (0.1) | 1 (0) | |||||
| Dendrocopos syriacus | 1 (0.1) | 1 (0) | |||||
| Egretta garzetta | 1 (0.1) | 1 (0) | |||||
| Falco tinnunculus | 1 (1.8) | 1 (0) | |||||
| Gallus gallus | 1 (0.1) | 1 (0.4) | 2 (0.1) | ||||
| Hirundo rustica | 2 (0.2) | 2 (0.1) | |||||
| Ixobrychus minutus | 3 (5.4) | 3 (0.1) | |||||
| Locustella luscinoides | 3 (5.4) | 3 (0.1) | |||||
| Motacilla alba | 1 (0.1) | 1 (0) | |||||
| Netta rufina | 1 (0.1) | 1 (0) | |||||
| Nycticorax nycticorax | 4 (0.5) | 2 (0.3) | 2 (0.7) | 7 (12.5) | 15 (0.6) | ||
| Parus major | 2 (3.6) | 2 (0.1) | |||||
| Pelecanus onocrotalus | 1 (0.1) | 1 (0.4) | 2 (0.1) | ||||
| Phalacrocorax carbo | 1 (0.1) | 1 (0) | |||||
| Streptopelia orientalis | 1 (0.4) | 1 (0) | |||||
| Strix aluco | 2 (3.6) | 2 (0.1) | |||||
| Upupa epops | 1 (1.8) | 1 (0) | |||||
| Homo sapiens | 55 (6.7) | 132 (16.7) | 40 (14.3) | 8 (3.5) | 14 (13.2) | 7 (12.5) | 271 (11.5) |
| Bos taurus | 185 (22.4) | 515 (65.1) | 157 (56.1) | 78 (33.9) | 46 (43.4) | 17 (30.4) | 1009 (43) |
| Bovidae | 2 (0.2) | 2 (0.1) | |||||
| Canis aureus | 1 (0.1) | 1 (0.1) | 1 (0.9) | 3 (0.1) | |||
| Canis lupus | 62 (7.5) | 10 (1.3) | 19 (6.8) | 7 (3) | 4 (3.8) | 8 (14.3) | 111 (4.7) |
| Capra hircus | 1 (0.1) | 1 (0.1) | 2 (0.1) | ||||
| Capreolus capreolus | 1 (0.1) | 1 (0.4) | 2 (0.1) | ||||
| Chiroptera | 2 (0.2) | 1 (0.1) | 1 (0.4) | 4 (0.2) | |||
| Equus caballus | 140 (16.9) | 78 (9.9) | 21 (7.5) | 120 (52.2) | 25 (23.6) | 2 (3.6) | 391 (16.7) |
| Erinaceus europaeus | 1 (0.1) | 1 (0) | |||||
| Felis catus | 34 (4.1) | 3 (0.4) | 7 (2.5) | 1 (1.8) | 47 (2) | ||
| Lepus europaeus | 3 (0.4) | 1 (0.4) | 1 (0.9) | 1 (1.8) | 6 (0.3) | ||
| Lutra lutra | 2 (0.2) | 1 (0.1) | 3 (0.1) | ||||
| Microtus levis | 1 (1.8) | 1 (0) | |||||
| Mustela lutreola | 1 (0.1) | 1 (0) | |||||
| Mustela nivalis | 1 (0.4) | 1 (0) | |||||
| Nyctereutes procyonoides | 1 (0.1) | 1 (0) | |||||
| Ovis aries | 8 (1) | 4 (0.5) | 2 (0.9) | 2 (1.9) | 16 (0.7) | ||
| Pipistrellus kuhlii | 1 (0.1) | 1 (0) | |||||
| Rattus norvegicus | 4 (0.5) | 1 (1.8) | 7 (0.3) | ||||
| Rhinolophus hipposideros | 1 (0.1) | 1 (0) | |||||
| Sus scrofa | 299 (36.2) | 28 (3.5) | 25 (8.9) | 12 (5.2) | 11 (10.4) | 2 (3.6) | 382 (16.3) |
| blood-fed specimens | 1054 | 1454 | 568 | 343 | 234 | 88 | 3741 |
| succesful analyzed specimens 1 | 827 | 791 | 280 | 230 | 106 | 56 | 2290 |
| identified hosts per mosquito species 1 | 834 | 792 | 283 | 230 | 106 | 62 | 2307 |
| identified host taxa | 30 | 13 | 15 | 9 | 9 | 17 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tomazatos, A.; Jansen, S.; Pfister, S.; Török, E.; Maranda, I.; Horváth, C.; Keresztes, L.; Spînu, M.; Tannich, E.; Jöst, H.; et al. Ecology of West Nile Virus in the Danube Delta, Romania: Phylogeography, Xenosurveillance and Mosquito Host-Feeding Patterns. Viruses 2019, 11, 1159. https://doi.org/10.3390/v11121159
Tomazatos A, Jansen S, Pfister S, Török E, Maranda I, Horváth C, Keresztes L, Spînu M, Tannich E, Jöst H, et al. Ecology of West Nile Virus in the Danube Delta, Romania: Phylogeography, Xenosurveillance and Mosquito Host-Feeding Patterns. Viruses. 2019; 11(12):1159. https://doi.org/10.3390/v11121159
Chicago/Turabian StyleTomazatos, Alexandru, Stephanie Jansen, Stefan Pfister, Edina Török, Iulia Maranda, Cintia Horváth, Lujza Keresztes, Marina Spînu, Egbert Tannich, Hanna Jöst, and et al. 2019. "Ecology of West Nile Virus in the Danube Delta, Romania: Phylogeography, Xenosurveillance and Mosquito Host-Feeding Patterns" Viruses 11, no. 12: 1159. https://doi.org/10.3390/v11121159
APA StyleTomazatos, A., Jansen, S., Pfister, S., Török, E., Maranda, I., Horváth, C., Keresztes, L., Spînu, M., Tannich, E., Jöst, H., Schmidt-Chanasit, J., Cadar, D., & Lühken, R. (2019). Ecology of West Nile Virus in the Danube Delta, Romania: Phylogeography, Xenosurveillance and Mosquito Host-Feeding Patterns. Viruses, 11(12), 1159. https://doi.org/10.3390/v11121159