Crocodilepox Virus Evolutionary Genomics Supports Observed Poxvirus Infection Dynamics on Saltwater Crocodile (Crocodylus porosus)

 , ,
, ,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Sampling to Study Poxvirus Infection Dynamics
2.2. Extraction of DNA and PCR Screening for Poxvirus
2.3. Statistical Analyses
2.4. Virus Genome Sequencing and Analyses
2.5. Phylogenetic Analyses
2.6. Recombination Analyses
3. Results
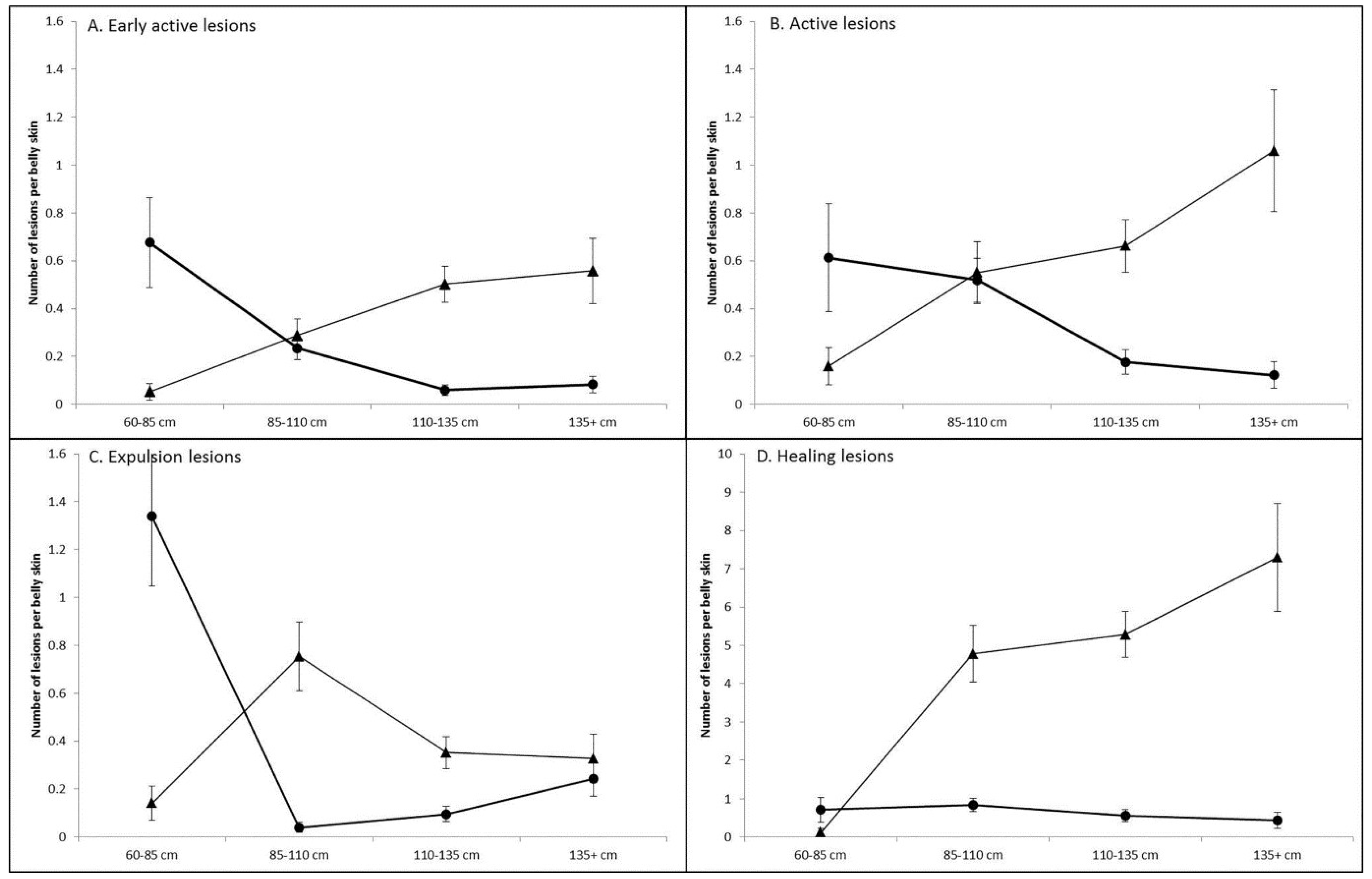
3.1. Prevalence and Infection Dynamics of Poxvirus
3.2. Characteristics of the SwCRV Genome Sequences
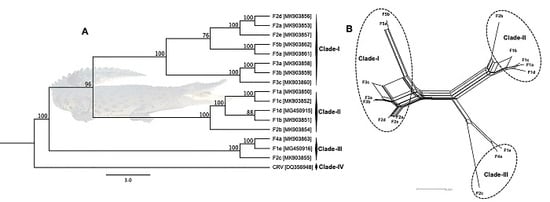
3.3. Phylogenetic Cluster Definition and Sequence Similarities
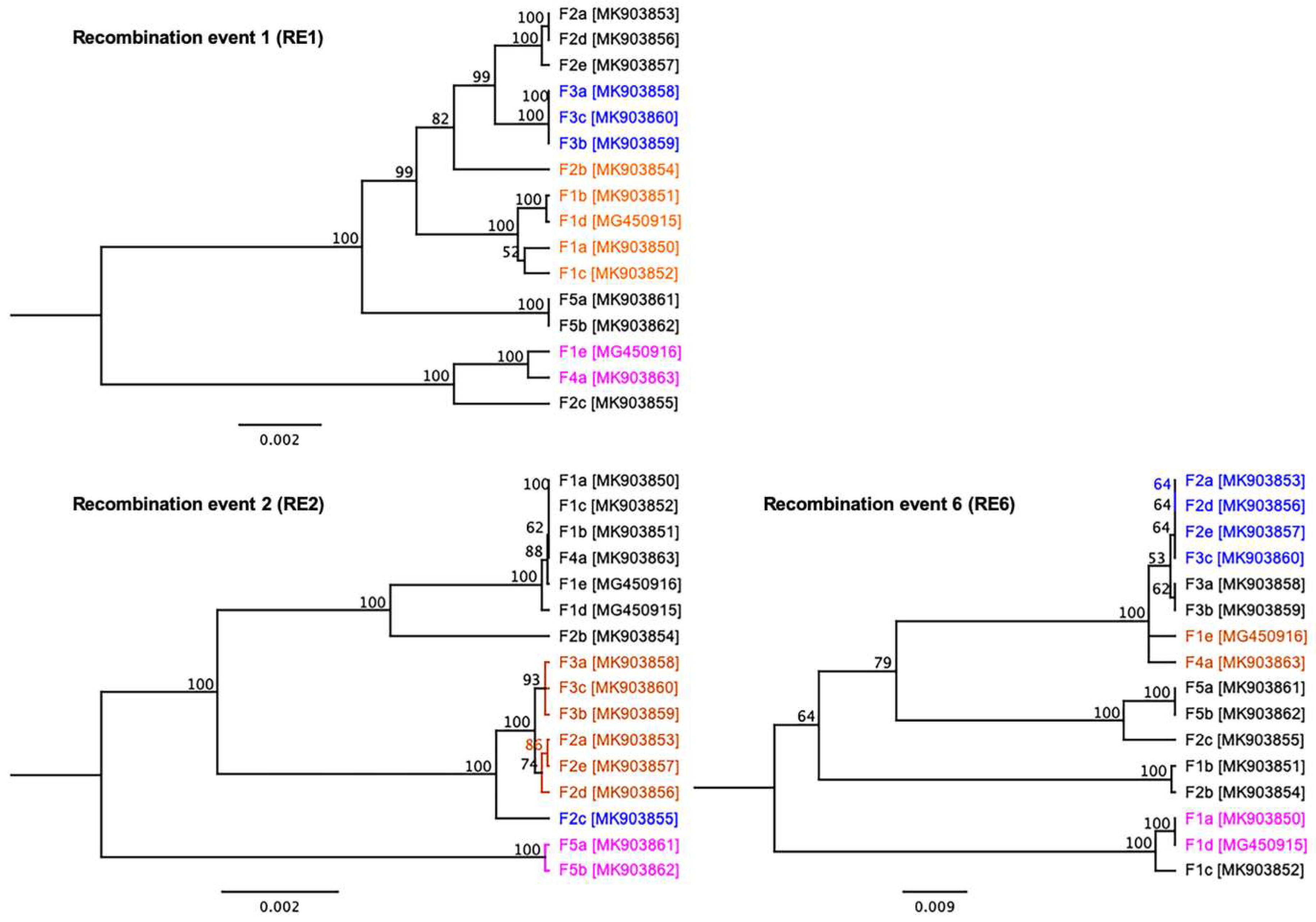
3.4. Evidence of Inter-Farm Genetic Recombination among SwCRV
4. Discussion
Supplementary Materials
Author Contributions
Data Availability
Funding
Conflicts of Interest
References
- Hughes, A.L.; Friedman, R. Poxvirus genome evolution by gene gain and loss. Mol. Phylogenet. Evol. 2005, 35, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Bratke, K.A.; McLysaght, A. Identification of multiple independent horizontal gene transfers into poxviruses using a comparative genomics approach. BMC Evol. Biol. 2008, 8, 67. [Google Scholar] [CrossRef] [PubMed]
- Sarker, S.; Isberg, S.R.; Milic, N.L.; Lock, P.; Helbig, K.J. Molecular characterization of the first saltwater crocodilepox virus genome sequences from the world’s largest living member of the Crocodylia. Sci. Rep. 2018, 8, 5623. [Google Scholar] [CrossRef] [PubMed]
- Moore, R.L.; Isberg, S.R.; Shilton, C.M.; Milic, N.L. Impact of poxvirus lesions on saltwater crocodile (Crocodylus porosus) skins. Vet. Microbiol. 2017, 211, 29–35. [Google Scholar] [CrossRef]
- ICTV. Virus Taxonomy: Classification and Nomenclature of Viruses: Ninth Report of the International Committee on Taxonomy of Viruses; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier: San Diego, CA, USA, 2012. [Google Scholar]
- Afonso, C.L.; Tulman, E.R.; Delhon, G.; Lu, Z.; Viljoen, G.J.; Wallace, D.B.; Kutish, G.F.; Rock, D.L. Genome of Crocodilepox Virus. J. Virol. 2006, 80, 4978–4991. [Google Scholar] [CrossRef]
- Pandey, G.S.; Inoue, N.; Ohshima, K.; Okada, K.; Chihaya, Y.; Fujimoto, Y. Poxvirus infection in Nile crocodiles (Crocodylus niloticus). Res. Vet. Sci. 1990, 49, 171–176. [Google Scholar] [CrossRef]
- Gerdes, G.H. Morphology of poxviruses from reptiles. Vet. Rec. 1991, 128, 452. [Google Scholar] [CrossRef]
- Horner, R.F. Poxvirus in farmed Nile crocodiles. Vet. Rec. 1988, 122, 459–462. [Google Scholar] [CrossRef]
- Marschang, R.E. Viruses Infecting Reptiles. Viruses 2011, 3, 2087–2126. [Google Scholar] [CrossRef]
- Lott, M.J.; Moore, R.L.; Milic, N.L.; Power, M.; Shilton, C.M.; Isberg, S.R. Dermatological conditions of farmed Crocodilians: A review of pathogenic agents and their proposed impact on skin quality. Vet. Microbiol. 2018, 225, 89–100. [Google Scholar] [CrossRef]
- Shilton, C.M.; Jerrett, I.V.; Davis, S.; Walsh, S.; Benedict, S.; Isberg, S.R.; Webb, G.J.; Manolis, C.; Hyndman, T.H.; Phalen, D.; et al. Diagnostic investigation of new disease syndromes in farmed Australian saltwater crocodiles (Crocodylus porosus) reveals associations with herpesviral infection. J. Vet. Diagn. Investig. 2016, 28, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Franklin, C.E.; Davis, B.M.; Peucker, S.K.; Stephenson, H.; Mayer, R.; Whittier, J.; Lever, J.; Grigg, G.C. Comparison of stress induced by manual restraint and immobilisation in the estuarine crocodile, Crocodylus porosus. J. Exp. Zool. Part A Comp. Exp. Boil. 2003, 298, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Isberg, S.R.; Gibb, K.; Chiri, E.; Kaestli, M.; Moran, J.; De Araujo, R.; Elliott, N.; Christian, K.; Melville, L.; Milic, N. Possible Chinks in the Crocodile Armour: Defining Skin Microflora; AgriFutures Australia: Canberra, Australia, 2019. [Google Scholar]
- Isberg, S.R.; Moran, J.L.; De Araujo, R.; Elliott, N.; Davis, S.S.; Melville, L. First evidence of Kunjin strain of West Nile virus associated with saltwater crocodile (Crocodylus porosus) skin lesions. Aust. Vet. J. 2019, 97, 390–393. [Google Scholar] [CrossRef] [PubMed]
- Sarker, S.; Das, S.; Lavers, J.L.; Hutton, I.; Helbig, K.; Imbery, J.; Upton, C.; Raidal, S.R. Genomic characterization of two novel pathogenic avipoxviruses isolated from pacific shearwaters (Ardenna spp.). BMC Genom. 2017, 18, 298. [Google Scholar] [CrossRef]
- Sarker, S.; Das, S.; Helbig, K.; Peters, A.; Raidal, S.R. Genome sequence of an Australian strain of canid alphaherpesvirus 1. Aust. Vet. J. 2018, 96, 24–27. [Google Scholar] [CrossRef]
- Sarker, S.; Roberts, H.K.; Tidd, N.; Ault, S.; Ladmore, G.; Peters, A.; Forwood, J.K.; Helbig, K.; Raidal, S.R. Molecular and microscopic characterization of a novel Eastern grey kangaroopox virus genome directly from a clinical sample. Sci. Rep. 2017, 7, 16472. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef]
- Martin, D.; Rybicki, E. RDP: Detection of recombination amongst aligned sequences. Bioinformatics 2000, 16, 562–563. [Google Scholar] [CrossRef]
- Padidam, M.; Sawyer, S.; Fauquet, C.M. Possible emergence of new geminiviruses by frequent recombination. Virology 1999, 265, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Posada, D.; Crandall, K.A.; Williamson, C. A modified bootscan algorithm for automated identification of recombinant sequences and recombination breakpoints. Aids Res. Hum. Retrovir. 2005, 21, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Smith, J. Analyzing the mosaic structure of genes. J. Mol. Evol. 1992, 34, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Posada, D.; Crandall, K.A. Evaluation of methods for detecting recombination from DNA sequences: Computer simulations. Proc. Natl. Acad. Sci. USA 2001, 98, 13757–13762. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, M.J.; Armstrong, J.S.; Gibbs, A.J. Sister-Scanning: A Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics 2000, 16, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Boni, M.F.; Posada, D.; Feldman, M.W. An exact nonparametric method for inferring mosaic structure in sequence triplets. Genetics 2007, 176, 1035–1047. [Google Scholar] [CrossRef]
- Martin, D.P.; Lemey, P.; Lott, M.; Moulton, V.; Posada, D.; Lefeuvre, P. RDP3: A flexible and fast computer program for analyzing recombination. Bioinformatics 2010, 26, 2462–2463. [Google Scholar] [CrossRef]
- Bruen, T.C.; Philippe, H.; Bryant, D. A Simple and Robust Statistical Test for Detecting the Presence of Recombination. Genetics 2006, 172, 2665. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef]
- Buenviaje, G.N.; Ladds, P.W.; Melville, L. Poxvirus infection in two crocodiles. Aust. Vet. J. 1992, 69, 15–16. [Google Scholar] [CrossRef]
- McKelvey, T.A.; Andrews, S.C.; Miller, S.E.; Ray, C.A.; Pickup, D.J. Identification of the Orthopoxvirus p4c Gene, Which Encodes a Structural Protein That Directs Intracellular Mature Virus Particles into A-Type Inclusions. J. Virol. 2002, 76, 11216. [Google Scholar] [PubMed]
- Okeke, M.I.; Adekoya, O.A.; Moens, U.; Tryland, M.; Traavik, T.; Nilssen, O. Comparative sequence analysis of A-type inclusion (ATI) and P4c proteins of orthopoxviruses that produce typical and atypical ATI phenotypes. Virus Genes 2009, 39, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Kastenmayer, R.J.; Maruri-Avidal, L.; Americo, J.L.; Earl, P.L.; Weisberg, A.S.; Moss, B. Elimination of A-type inclusion formation enhances cowpox virus replication in mice: Implications for orthopoxvirus evolution. Virology 2014, 452–453, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Awadalla, P. The evolutionary genomics of pathogen recombination. Nat. Rev. Genet. 2003, 4, 50–60. [Google Scholar] [CrossRef]
- Pérez-Losada, M.; Arenas, M.; Galán, J.C.; Palero, F.; González-Candelas, F. Recombination in viruses: Mechanisms, methods of study, and evolutionary consequences. Infect. Genet. Evol. 2015, 30, 296–307. [Google Scholar] [CrossRef]
- Sarker, S.; Patterson, E.I.; Peters, A.; Baker, B.G.; Forwood, J.K.; Ghorashi, S.A.; Holdsworth, M.; Baker, R.; Murray, N.; Raidal, S.R. Mutability dynamics of an emergent single stranded DNA virus in a naïve host. PLoS ONE 2014, 9, e85370. [Google Scholar] [CrossRef]
- Sarker, S.; Ghorashi, S.A.; Forwood, J.K.; Bent, J.S.; Peters, A.; Raidal, S.R. Phylogeny of beak and feather disease virus in cockatoos demonstrates host generalism and multiple-variant infections within Psittaciformes. Virology 2014, 460–461, 72–82. [Google Scholar] [CrossRef]
- Lopez-Bueno, A.; Parras-Molto, M.; Lopez-Barrantes, O.; Belda, S.; Alejo, A. Recombination events and variability among full-length genomes of co-circulating molluscum contagiosum virus subtypes 1 and 2. J. Gen. Virol. 2017, 98, 1073–1079. [Google Scholar] [CrossRef]
- Zorec, T.M.; Kutnjak, D.; Hosnjak, L.; Kusar, B.; Trcko, K.; Kocjan, B.J.; Li, Y.; Krizmaric, M.; Miljkovic, J.; Ravnikar, M.; et al. New Insights into the Evolutionary and Genomic Landscape of Molluscum Contagiosum Virus (MCV) based on Nine MCV1 and Six MCV2 Complete Genome Sequences. Viruses 2018, 10, 586. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Farm 1 | Farm 2 | |
|---|---|---|
| Sampling 1: August–October 2016 | ||
| Hatchling | 30 | 40 |
| Grower | 90 | 80 |
| Finishing pen | 20 | 20 |
| Sampling 2: January 2017 | ||
| Hatchling | 35 | 20 |
| Grower | 100 | 60 |
| Finishing pen | 20 | 20 |
| Sampling 3: August–November 2017 | ||
| Hatchling | 40 | 20 |
| Grower | 100 | 80 |
| Finishing pen | 20 | 20 |
| Sampling 4: February-–March 2018 | ||
| Hatchling | 0 | 20 |
| Grower | 100 | 40 |
| Finishing pen | 20 | 20 |
| Total | 575 | 440 |
| Sampling | Early Active | Active | Expulsion | Total Pox |
|---|---|---|---|---|
| Farm 1 | ||||
| 1 | 24 | 25 | 7 | 56 |
| 2 | 1 | 14 | 2 | 17 |
| 3 | 10 | 14 | 0 | 24 |
| 4 | 3 | 4 | 2 | 9 |
| Farm 2 | ||||
| 1 | 10 | 20 | 8 | 38 |
| 2 | 15 | 13 | 2 | 30 |
| 3 | 6 | 1 | 0 | 7 |
| 4 | 4 | 2 | 0 | 6 |
| Totals | 73 | 93 | 21 | 187 |
| % correct assignment | 67% | 84% | 100% | 82% |
| Response Variate | |||||
|---|---|---|---|---|---|
| Early Active | Active | Expulsion | Healing | ||
| Explanatory variate | Early active | 1.5 ± 0.03 *** | 1.37 ± 0.04 *** | n.s. | |
| Active | 1.27 ± 0.01 *** (0.61) | 1.08 ± 0.02 *** | 1.13 ± 0.02 *** | ||
| Expulsion | 1.11 ± 0.01 *** (0.33) | n.s. (0.27) | n.s. | ||
| Healing | 1.02 ± 0.004 *** (0.17) | 1.01 ± 0.005 ** (0.18) | n.s. (0.08) | ||
| Farm ID | Sample ID | Total Reads | Total Nucleotides | Mean Read Length | Coverage | Genome Size (bp) | GC Content (%) | ITRs in the SwCRV Genome | GenBank Accession Number | Number of Annotated Genes | References | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Position in Sense-Strand | Position in Antisense-Strand | Length | |||||||||||
| Farm 1 | F1a | 2,182,778 | 464,144,061 | 229.47 | 171.75 | 187,468 | 62.00 | 1-1140 | 187,468-186,329 | 1140 | MK903850 | 216 | This study |
| F1b | 625,406 | 171,737,498 | 289.27 | 367.73 | 187,223 | 62.00 | 1-1622 | 187,223-185,602 | 1622 | MK903851 | 218 | This study | |
| F1c | 2,929,464 | 562,334,027 | 218.46 | 77.96 | 186,383 | 62.00 | 1-902 | 186,383-185,482 | 902 | MK903852 | 213 | This study | |
| F1d | 2,263,362 | 580,836,186 | 256.63 | 1905.90 | 187,976 | 61..90 | 1-1700 | 187,976-186,277 | 1700 | MG450915 | 218 | Sarker et al. 2018 | |
| F1e | 770,348 | 260,017,678 | 267.43 | 476.58 | 184,894 | 62.20 | 1-1254 | 184,894-183,641 | 1254 | MG450916 | 215 | Sarker et al. 2018 | |
| Farm 2 | F2a | 755,168 | 180,551,171 | 260.08 | 268.82 | 187,295 | 62.00 | 1-945 | 187,295-186,351 | 945 | MK903853 | 215 | This study |
| F2b | 684,886 | 183,346,358 | 286.82 | 508.91 | 187,334 | 62.00 | 1-1655 | 187,334-185,680 | 1655 | MK903854 | 217 | This study | |
| F2c | 676,468 | 185,563,388 | 287.19 | 663.98 | 184,469 | 62.30 | 1-1617 | 184,469-182,853 | 1617 | MK903855 | 214 | This study | |
| F2d | 732,772 | 206,557,607 | 286.75 | 981.20 | 187,619 | 62.00 | 1-1291 | 187,619-186,329 | 1291 | MK903856 | 213 | This study | |
| F2e | 2,665,954 | 556,343,609 | 222.95 | 101.11 | 185,923 | 62.00 | 1-882 | 185,923-185,042 | 882 | MK903857 | 211 | This study | |
| Farm 3 | F3a | 1,199,052 | 312,997,159 | 272.30 | 1019.39 | 187,648 | 62.00 | 1-906 | 187,648-186,743 | 906 | MK903858 | 215 | This study |
| F3b | 721,906 | 201,313,232 | 287.37 | 938.56 | 187,549 | 62.00 | 1-1633 | 187,549-185,917 | 1633 | MK903859 | 216 | This study | |
| F3c | 774,274 | 213,662,759 | 286.23 | 699.52 | 187,293 | 62.00 | 1-926 | 187,293-186,368 | 926 | MK903860 | 215 | This study | |
| Farm 4 | F4a | 1,613,140 | 303,995,638 | 197.83 | 1130 | 185,168 | 62.20 | 1-1682 | 185,168-183,487 | 1682 | MK903863 | 215 | This study |
| Farm 5 | F5a | 631,050 | 173,473,167 | 286.76 | 560.34 | 186,462 | 62.10 | 1-877 | 186,462-185,586 | 877 | MK903861 | 212 | This study |
| F5b | 1,877,842 | 352,661,729 | 199.85 | 467.89 | 186,876 | 62.00 | 1-932 | 186,870-185,939 | 932 | MK903862 | 213 | This study | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarker, S.; Isberg, S.R.; Moran, J.L.; Araujo, R.D.; Elliott, N.; Melville, L.; Beddoe, T.; Helbig, K.J. Crocodilepox Virus Evolutionary Genomics Supports Observed Poxvirus Infection Dynamics on Saltwater Crocodile (Crocodylus porosus). Viruses 2019, 11, 1116. https://doi.org/10.3390/v11121116
Sarker S, Isberg SR, Moran JL, Araujo RD, Elliott N, Melville L, Beddoe T, Helbig KJ. Crocodilepox Virus Evolutionary Genomics Supports Observed Poxvirus Infection Dynamics on Saltwater Crocodile (Crocodylus porosus). Viruses. 2019; 11(12):1116. https://doi.org/10.3390/v11121116
Chicago/Turabian StyleSarker, Subir, Sally R. Isberg, Jasmin L. Moran, Rachel De Araujo, Nikki Elliott, Lorna Melville, Travis Beddoe, and Karla J. Helbig. 2019. "Crocodilepox Virus Evolutionary Genomics Supports Observed Poxvirus Infection Dynamics on Saltwater Crocodile (Crocodylus porosus)" Viruses 11, no. 12: 1116. https://doi.org/10.3390/v11121116
APA StyleSarker, S., Isberg, S. R., Moran, J. L., Araujo, R. D., Elliott, N., Melville, L., Beddoe, T., & Helbig, K. J. (2019). Crocodilepox Virus Evolutionary Genomics Supports Observed Poxvirus Infection Dynamics on Saltwater Crocodile (Crocodylus porosus). Viruses, 11(12), 1116. https://doi.org/10.3390/v11121116