Promises and Pitfalls of In Vivo Evolution to Improve Phage Therapy


Abstract
1. Introduction
1.1. A Precedent for the Need to Choose Phages Wisely
1.2. Phage Properties Subject to Selection in Patients
2. Methods: Models
2.1. The Standard Model and Anomalies from Phage Therapy Results
2.2. A Dynamics Model to Accommodate Spatial Structure
3. Results
3.1. Growth on a Single Bacterium
3.2. Phage Decay Rates
3.3. Matrix Degrading Activities: Depolymerases
3.4. Phage Evolution to Overcome Bacterial Resistance
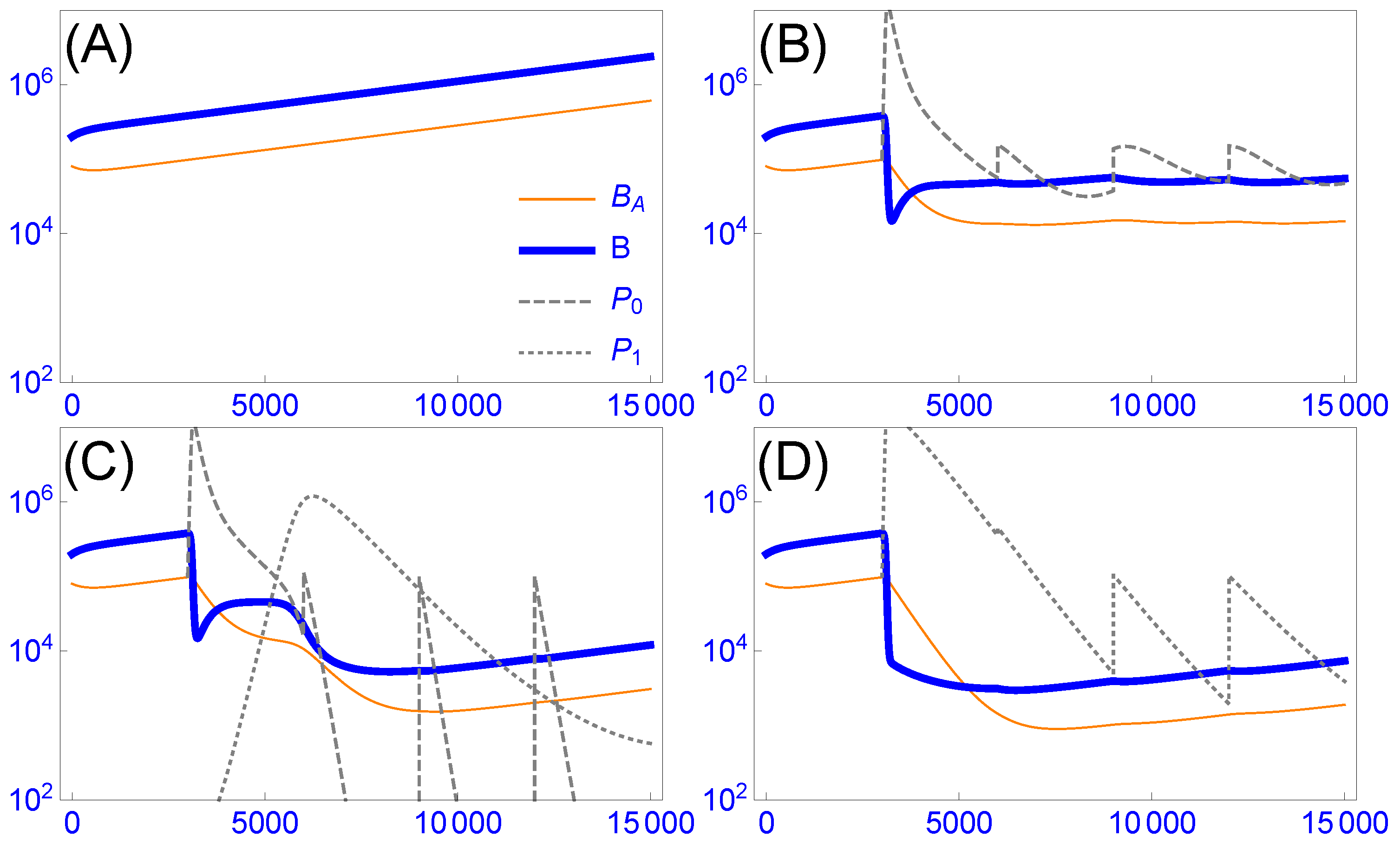
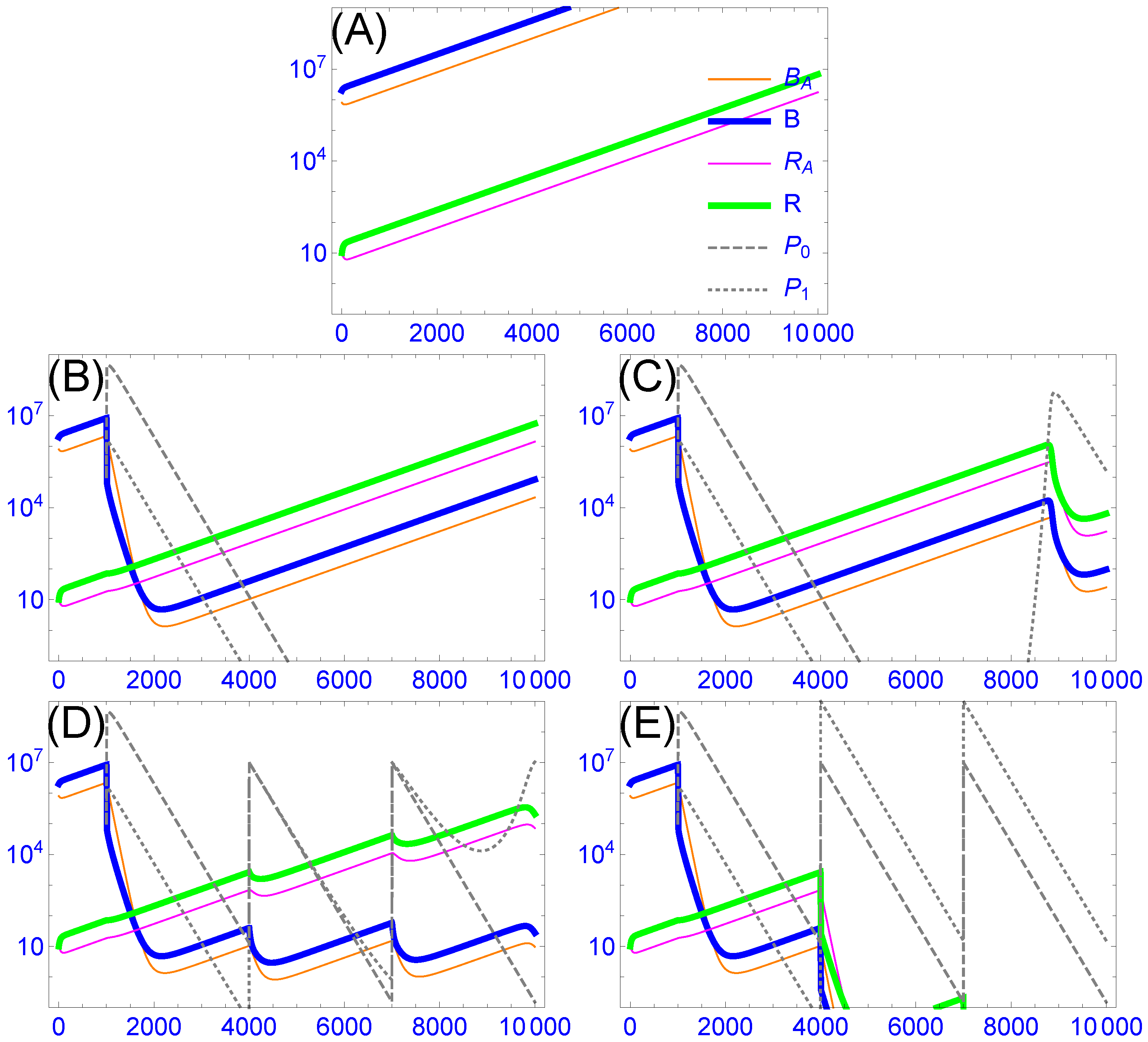
3.4.1. Intrinsic Phage Evolution to Overcome Bacterial Resistance Is Not Assured
3.4.2. Cocktails Can Be Designed to Block Stepwise Bacterial Escape, but They Can Experience Lags and Phage Loss
3.4.3. Resistance-Proof Phages Can Avoid Evolutionary Arms Races, but In Vivo Dynamics Do Not Ensure Their Ascendancy in Cocktails
4. Discussion
Depressing Bacterial Densities versus Improving Infection Outcomes
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A. Model Details
Appendix A.1. One Bacterial Strain with Two Bacterial States and Two Phages

{kind=link}
{kind=link}
| Notation | Description | Units |
| Variables (Functions of Time) | Description | Units |
| density of bacteria in aggregates (protected) | /mL | |
| B | density of susceptible, planktonic bacteria | /mL |
| density of strain 0 phage | /mL | |
| density of strain 1 phage | /mL | |
| Parameters | Description | Units |
| adsorption rate of phage strain i to planktonic bacteria | mL/min | |
| death rate of phage strain i | /min | |
| loss rate of bacteria from death or conversion to aggregates () | /min | |
| burst size of phage strain i | individuals | |
| c | conversion rate of aggregates to planktonic bacteria | |
| loss rate of aggregates to become planktonic bacteria | /min | |
| aggregate formation rate by planktonic bacteria | /min |
Appendix A.2. Two Bacterial States with Two Bacterial States and Two Phages
| Notation | Description | Units |
|---|---|---|
| Variables (Functions of Time) | Description | Units |
| density of resistant bacteria in aggregates | /mL | |
| R | density of resistant, planktonic bacteria | /mL |
References
- Brüssow, H. Phage therapy for the treatment of human intestinal bacterial infections: Soon to be a reality? Expert Rev. Gastroenterol. Hepatol. 2017, 11, 785–788. [Google Scholar] [CrossRef]
- Abedon, S.T. Use of phage therapy to treat long-standing, persistent, or chronic bacterial infections. Adv. Drug Deliv. Rev. 2018. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, C. Phage therapy’s latest makeover. Nat. Biotechnol. 2019, 37, 581–586. [Google Scholar] [CrossRef] [PubMed]
- Kortright, K.E.; Chan, B.K.; Koff, J.L.; Turner, P.E. Phage therapy: A renewed approach to combat antibiotic-resistant bacteria. Cell Host Microbe 2019, 25, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Schooley, R.T.; Biswas, B.; Gill, J.J.; Hernandez-Morales, A.; Lancaster, J.; Lessor, L.; Barr, J.J.; Reed, S.L.; Rohwer, F.; Benler, S.; et al. Development and use of personalized bacteriophage-based therapeutic cocktails to treat a patient with a disseminated resistant Acinetobacter baumannii infection. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Schooley, R.T.; Biswas, B.; Gill, J.J.; Hernandez-Morales, A.; Lancaster, J.; Lessor, L.; Barr, J.J.; Reed, S.L.; Rohwer, F.; Benler, S.; et al. Erratum for Schooley et al., “Development and use of personalized bacteriophage-based therapeutic cocktails to treat a patient with a disseminated resistant Acinetobacter baumannii infection”. Antimicrob. Agents Chemother. 2018, 62, e02221-18. [Google Scholar] [CrossRef]
- Dedrick, R.M.; Guerrero-Bustamante, C.A.; Garlena, R.A.; Russell, D.A.; Ford, K.; Harris, K.; Gilmour, K.C.; Soothill, J.; Jacobs-Sera, D.; Schooley, R.T.; et al. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat. Med. 2019, 25, 730–733. [Google Scholar] [CrossRef]
- Chan, B.K.; Turner, P.E.; Kim, S.; Mojibian, H.R.; Elefteriades, J.A.; Narayan, D. Phage treatment of an aortic graft infected with Pseudomonas aeruginosa. Evol. Med. Public Health 2018, 2018, 60–66. [Google Scholar] [CrossRef]
- Rhoads, D.D.; Wolcott, R.D.; Kuskowski, M.A.; Wolcott, B.M.; Ward, L.S.; Sulakvelidze, A. Bacteriophage therapy of venous leg ulcers in humans: Results of a phase I safety trial. J. Wound Care 2009, 18, 237–243. [Google Scholar] [CrossRef]
- Sarker, S.A.; Sultana, S.; Reuteler, G.; Moine, D.; Descombes, P.; Charton, F.; Bourdin, G.; McCallin, S.; Ngom-Bru, C.; Neville, T.; et al. Oral phage therapy of acute bacterial diarrhea with two coliphage preparations: A randomized trial in children from Bangladesh. EBioMedicine 2016, 4, 124–137. [Google Scholar] [CrossRef]
- Jault, P.; Leclerc, T.; Jennes, S.; Pirnay, J.P.; Que, Y.A.; Resch, G.; Rousseau, A.F.; Ravat, F.; Carsin, H.; Le Floch, R.; et al. Efficacy and tolerability of a cocktail of bacteriophages to treat burn wounds infected by Pseudomonas aeruginosa (PhagoBurn): A randomised, controlled, double-blind phase 1/2 trial. Lancet Infect. Dis. 2019, 19, 35–45. [Google Scholar] [CrossRef]
- Smith, H.W.; Huggins, M.B. Successful treatment of experimental Escherichia coli Infect. Mice Using Phage: Its Gen. Super. Antibiot. J. Gen. Microbiol. 1982, 128, 307–318. [Google Scholar] [PubMed]
- Henry, M.; Lavigne, R.; Debarbieux, L. Predicting in vivo efficacy of therapeutic bacteriophages used to treat pulmonary infections. Antimicrob. Agents Chemother. 2013, 57, 5961–5968. [Google Scholar] [CrossRef] [PubMed]
- Arnold, F.H. Innovation by evolution: Bringing new chemistry to life (Nobel Lecture). Angew. Chem. (Int. Ed. Engl.) 2019. [Google Scholar] [CrossRef]
- D’Herelle, F. Immunity in Natural Infectious Disease; Authorized English ed.; Smith, G.H., Ed.; Williams & Wilkins Co.: Baltimore, MD, USA, 1924. [Google Scholar]
- McEwen, S.; Collignon, P. Antimicrobial resistance: A One Health perspective. Microbiol Spectr. 2018, 6, ARBA-0009-2017. [Google Scholar] [CrossRef]
- Bull, J.J.; Levin, B.R.; DeRouin, T.; Walker, N.; Bloch, C.A. Dynamics of success and failure in phage and antibiotic therapy in experimental infections. BMC Microbiol. 2002, 2, 35. [Google Scholar] [CrossRef]
- Bull, J.J.; Otto, G.; Molineux, I.J. In vivo growth rates are poorly correlated with phage therapy success in a mouse infection model. Antimicrob. Agents Chemother. 2012, 56, 949–954. [Google Scholar] [CrossRef]
- Merril, C.R.; Biswas, B.; Carlton, R.; Jensen, N.C.; Creed, G.J.; Zullo, S.; Adhya, S. Long-circulating bacteriophage as antibacterial agents. Proc. Natl. Acad. Sci. USA 1996, 93, 3188–3192. [Google Scholar] [CrossRef]
- Sutherland, I.W.; Hughes, K.A.; Skillman, L.C.; Tait, K. The interaction of phage and biofilms. FEMS Microbiol. Lett. 2004, 232, 1–6. [Google Scholar] [CrossRef]
- Lu, T.K.; Collins, J.J. Dispersing biofilms with engineered enzymatic bacteriophage. Proc. Natl. Acad. Sci. USA 2007, 104, 11197–11202. [Google Scholar] [CrossRef]
- Chan, B.K.; Abedon, S.T. Phage therapy pharmacology: Phage cocktails. In Advances in Applied Microbiology; Laskin, A.I., Sariaslani, S., Gadd, G.M., Eds.; Academic Press: San Diego, CA, USA, 2012; Volume 78, pp. 1–23. [Google Scholar] [CrossRef]
- Gladstone, E.G.; Molineux, I.J.; Bull, J.J. Evolutionary principles and synthetic biology: Avoiding a molecular tragedy of the commons with an engineered phage. J. Biol. Eng. 2012, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.H. Bacteriophages; Interscience Publishers: New York, NY, USA, 1959. [Google Scholar]
- Campbell, A. Conditions for the existence of bacteriophage. Evolution 1961, 15, 143–165. [Google Scholar] [CrossRef]
- Levin, B.R.; Stewart, F.M.; Chao, L. Resource—Limited growth, competition, and predation: A model and experimental studies with bacteria and bacteriophage. Am. Nat. 1977, 977, 3–24. [Google Scholar] [CrossRef]
- Roach, D.R.; Leung, C.Y.; Henry, M.; Morello, E.; Singh, D.; Di Santo, J.P.; Weitz, J.S.; Debarbieux, L. Synergy between the host immune system and bacteriophage is essential for successful phage therapy against an acute respiratory pathogen. Cell Host Microbe 2017, 22, 38–47.e4. [Google Scholar] [CrossRef] [PubMed]
- Abedon, S.T. Bacteriophage-mediated biocontrol of wound infections, and ecological exploitation of biofilms by phages. In Recent Clinical Techniques, Results, and Research in Wounds; Springer: Cham, Switzerland, 2018; pp. 1–38. [Google Scholar]
- Darch, S.E.; Kragh, K.N.; Abbott, E.A.; Bjarnsholt, T.; Bull, J.J.; Whiteley, M. Phage inhibit pathogen dissemination by targeting bacterial migrants in a chronic infection model. mBio 2017, 8. [Google Scholar] [CrossRef]
- Bull, J.J. Optimality models of phage life history and parallels in disease evolution. J. Theor. Biol. 2006, 241, 928–938. [Google Scholar] [CrossRef]
- Heineman, R.H.; Springman, R.; Bull, J.J. Optimal foraging by bacteriophages through host avoidance. Am. Nat. 2008, 171, E149–E157. [Google Scholar] [CrossRef]
- Westwater, C.; Kasman, L.M.; Schofield, D.A.; Werner, P.A.; Dolan, J.W.; Schmidt, M.G.; Norris, J.S. Use of genetically engineered phage to deliver antimicrobial agents to bacteria: An alternative therapy for treatment of bacterial infections. Antimicrob. Agents Chemother. 2003, 47, 1301–1307. [Google Scholar] [CrossRef]
- Bull, J.J.; Regoes, R.R. Pharmacodynamics of non-replicating viruses, bacteriocins and lysins. Proc. Biol. Sci. R. Soc. 2006, 273, 2703–2712. [Google Scholar] [CrossRef]
- Hodyra-Stefaniak, K.; Lahutta, K.; Majewska, J.; Kaźmierczak, Z.; Lecion, D.; Harhala, M.; Kęska, W.; Owczarek, B.; Jończyk-Matysiak, E.; Kłopot, A.; et al. Bacteriophages engineered to display foreign peptides may become short-circulating phages. Microb. Biotechnol. 2019, 12, 730–741. [Google Scholar] [CrossRef]
- Sutherland, I.W.; Wilkinson, J.F. Depolymerases for bacterial exopolysaccharides obtained from phage-infected bacteria. J. Gen. Microbiol. 1965, 39, 373–383. [Google Scholar] [CrossRef]
- Sutherland, I.W. Polysaccharide lyases. FEMS Microbiol. Rev. 1995, 16, 323–347. [Google Scholar] [CrossRef] [PubMed]
- Hughes, K.A.; Sutherland, I.W.; Clark, J.; Jones, M.V. Bacteriophage and associated polysaccharide depolymerases–novel tools for study of bacterial biofilms. J. Appl. Microbiol. 1998, 85, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Hanlon, G.W.; Denyer, S.P.; Olliff, C.J.; Ibrahim, L.J. Reduction in exopolysaccharide viscosity as an aid to bacteriophage penetration through Pseudomonas aeruginosa biofilms. Appl. Environ. Microbiol. 2001, 67, 2746–2753. [Google Scholar] [CrossRef] [PubMed]
- Azeredo, J.; Sutherland, I.W. The use of phages for the removal of infectious biofilms. Curr. Pharm. Biotechnol. 2008, 9, 261–266. [Google Scholar] [CrossRef]
- Bull, J.J.; Vimr, E.R.; Molineux, I.J. A tale of tails: Sialidase is key to success in a model of phage therapy against K1-capsulated Escherichia coli. Virology 2010, 398, 79–86. [Google Scholar] [CrossRef]
- Mushtaq, N.; Redpath, M.B.; Luzio, J.P.; Taylor, P.W. Prevention and cure of systemic Escherichia coli K1 infection by modification of the bacterial phenotype. Antimicrob. Agents Chemother. 2004, 48, 1503–1508. [Google Scholar] [CrossRef]
- Mushtaq, N.; Redpath, M.B.; Luzio, J.P.; Taylor, P.W. Treatment of experimental Escherichia coli infection with recombinant bacteriophage-derived capsule depolymerase. J. Antimicrob. Chemother. 2005, 56, 160–165. [Google Scholar] [CrossRef]
- Lin, T.L.; Hsieh, P.F.; Huang, Y.T.; Lee, W.C.; Tsai, Y.T.; Su, P.A.; Pan, Y.J.; Hsu, C.R.; Wu, M.C.; Wang, J.T. Isolation of a bacteriophage and its depolymerase specific for K1 capsule of Klebsiella pneumoniae: Implication in typing and treatment. J. Infect. Dis. 2014, 210, 1734–1744. [Google Scholar] [CrossRef]
- Lin, H.; Paff, M.L.; Molineux, I.J.; Bull, J.J. Therapeutic application of phage capsule depolymerases against K1, K5, and K30 capsulated E. coli in mice. Front. Microbiol. 2017, 8, 2257. [Google Scholar] [CrossRef]
- Lin, H.; Paff, M.L.; Molineux, I.J.; Bull, J.J. Antibiotic therapy using phage depolymerases: Robustness across a range of conditions. Viruses 2018, 10, 622. [Google Scholar] [CrossRef] [PubMed]
- Cornelissen, A.; Ceyssens, P.J.; T’Syen, J.; Van Praet, H.; Noben, J.P.; Shaburova, O.V.; Krylov, V.N.; Volckaert, G.; Lavigne, R. The T7-related Pseudomonas putida phage ϕ15 displays virion-associated biofilm degradation properties. PLoS ONE 2011, 6, e18597. [Google Scholar] [CrossRef] [PubMed]
- Donlan, R.M.; Costerton, J.W. Biofilms: Survival mechanisms of clinically relevant microorganisms. Clin. Microbiol. Rev. 2002, 15, 167–193. [Google Scholar] [CrossRef] [PubMed]
- Tseng, B.S.; Zhang, W.; Harrison, J.J.; Quach, T.P.; Song, J.L.; Penterman, J.; Singh, P.K.; Chopp, D.L.; Packman, A.I.; Parsek, M.R. The extracellular matrix protects Pseudomonas aeruginosa biofilms by limiting the penetration of tobramycin. Environ. Microbiol. 2013, 15, 2865–2878. [Google Scholar] [CrossRef] [PubMed]
- Pei, R.; Lamas-Samanamud, G.R. Inhibition of biofilm formation by T7 bacteriophages producing quorum-quenching enzymes. Appl. Environ. Microbiol. 2014, 80, 5340–5348. [Google Scholar] [CrossRef] [PubMed]
- Bárdy, P.; Pantůček, R.; Benešíkk, M.; Doškař, J. Genetically modified bacteriophages in applied microbiology. J. Appl. Microbiol. 2016, 121, 618–633. [Google Scholar] [CrossRef] [PubMed]
- Bessler, W.; Fehmel, F.; Freund-Mölbert, E.; Knüfermann, H.; Stirm, S. Escherichia coli capsule bacteriophages. IV. Free capsule depolymerase 29. J. Virol. 1975, 15, 976–984. [Google Scholar]
- Kassa, T.; Chhibber, S. Thermal treatment of the bacteriophage lysate of Klebsiella pneumoniae B5055 as a step for the purification of capsular depolymerase enzyme. J. Virol. Methods 2012, 179, 135–141. [Google Scholar] [CrossRef]
- Schmerer, M.; Molineux, I.J.; Bull, J.J. Synergy as a rationale for phage therapy using phage cocktails. PeerJ 2014, 2, e590. [Google Scholar] [CrossRef]
- Fleming, D.; Chahin, L.; Rumbaugh, K. Glycoside hydrolases degrade polymicrobial bacterial biofilms in wounds. Antimicrob. Agents Chemother. 2017, 61, e01998-16. [Google Scholar] [CrossRef]
- Hashemolhosseini, S.; Holmes, Z.; Mutschler, B.; Henning, U. Alterations of receptor specificities of coliphages of the T2 family. J. Mol. Biol. 1994, 240, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Hashemolhosseini, S.; Montag, D.; Krämer, L.; Henning, U. Determinants of receptor specificity of coliphages of the T4 family. A chaperone alters the host range. J. Mol. Biol. 1994, 241, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Dinsmore, P.K.; Klaenhammer, T.R. Bacteriophage resistance in Lactococcus. Mol. Biotechnol. 1995, 4, 297–314. [Google Scholar] [CrossRef] [PubMed]
- Durmaz, E.; Klaenhammer, T.R. Abortive phage resistance mechanism AbiZ speeds the lysis clock to cause premature lysis of phage-infected Lactococcus lactis. J. Bacteriol. 2007, 189, 1417–1425. [Google Scholar] [CrossRef]
- Labrie, S.J.; Samson, J.E.; Moineau, S. Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 2010, 8, 317–327. [Google Scholar] [CrossRef]
- Doron, S.; Melamed, S.; Ofir, G.; Leavitt, A.; Lopatina, A.; Keren, M.; Amitai, G.; Sorek, R. Systematic discovery of antiphage defense systems in the microbial pangenome. Science 2018, 359. [Google Scholar] [CrossRef]
- Oechslin, F. Resistance development to bacteriophages occurring during bacteriophage therapy. Viruses 2018, 10, 351. [Google Scholar] [CrossRef]
- Rostøl, J.T.; Marraffini, L. (Ph)ighting phages: How bacteria resist their parasites. Cell Host Microbe 2019, 25, 184–194. [Google Scholar] [CrossRef]
- Levin, B.R. Frequency-dependent selection in bacterial populations. Philos. Trans. R. Soc. London. Ser. B Biol. Sci. 1988, 319, 459–472. [Google Scholar] [CrossRef]
- Bohannan, B.J.M.; Lenski, R.E. Linking genetic change to community evolution: Insights from studies of bacteria and bacteriophage. Ecol. Lett. 2000, 3, 362–377. [Google Scholar] [CrossRef]
- Mizoguchi, K.; Morita, M.; Fischer, C.R.; Yoichi, M.; Tanji, Y.; Unno, H. Coevolution of bacteriophage PP01 and Escherichia coli O157:H7 in continuous culture. Appl. Environ. Microbiol. 2003, 69, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Gómez, P.; Buckling, A. Bacteria-phage antagonistic coevolution in soil. Science 2011, 332, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Muñoz, S.L.; Koskella, B. Bacteria-phage interactions in natural environments. Adv. Appl. Microbiol. 2014, 89, 135–183. [Google Scholar] [CrossRef]
- Fortuna, M.A.; Barbour, M.A.; Zaman, L.; Hall, A.R.; Buckling, A.; Bascompte, J. Coevolutionary dynamics shape the structure of bacteria-phage infection networks. Evol. Int. J. Org. Evol. 2019, 73, 1001–1011. [Google Scholar] [CrossRef] [PubMed]
- Ershova, A.S.; Rusinov, I.S.; Spirin, S.A.; Karyagina, A.S.; Alexeevski, A.V. Role of restriction-modification systems in prokaryotic evolution and ecology. Biochem. Biokhimiia 2015, 80, 1373–1386. [Google Scholar] [CrossRef] [PubMed]
- Iordanescu, S.; Surdeanu, M. Two restriction and modification systems in Staphylococcus aureus NCTC8325. J. Gen. Microbiol. 1976, 96, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Korona, R.; Levin, B.R. Phage-mediated selection and the evolution and maintenance of restriction-modification. Evol. Int. J. Org. Evol. 1993, 47, 556–575. [Google Scholar] [CrossRef] [PubMed]
- Korona, R.; Korona, B.; Levin, B.R. Sensitivity of naturally occurring coliphages to type I and type II restriction and modification. J. Gener. Microbiol. 1993, 139, 1283–1290. [Google Scholar] [CrossRef]
- Jiang, W.; Maniv, I.; Arain, F.; Wang, Y.; Levin, B.R.; Marraffini, L.A. Dealing with the evolutionary downside of CRISPR immunity: Bacteria and beneficial plasmids. PLoS Genet. 2013, 9, e1003844. [Google Scholar] [CrossRef]
- Paez-Espino, D.; Sharon, I.; Morovic, W.; Stahl, B.; Thomas, B.C.; Barrangou, R.; Banfield, J.F. CRISPR immunity drives rapid phage genome evolution in Streptococcus thermophilus. mBio 2015, 6, e00262-15. [Google Scholar] [CrossRef]
- Weissman, J.L.; Holmes, R.; Barrangou, R.; Moineau, S.; Fagan, W.F.; Levin, B.; Johnson, P.L.F. Immune loss as a driver of coexistence during host-phage coevolution. ISME J. 2018, 12, 585–597. [Google Scholar] [CrossRef] [PubMed]
- Gurney, J.; Pleška, M.; Levin, B.R. Why put up with immunity when there is resistance: An excursion into the population and evolutionary dynamics of restriction-modification and CRISPR-Cas. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2019, 374, 20180096. [Google Scholar] [CrossRef] [PubMed]
- Palmer, K.L.; Gilmore, M.S. Multidrug-resistant enterococci lack CRISPR-cas. mBio 2010, 1, e00227-10. [Google Scholar] [CrossRef] [PubMed]
- Kos, V.N.; Déraspe, M.; McLaughlin, R.E.; Whiteaker, J.D.; Roy, P.H.; Alm, R.A.; Corbeil, J.; Gardner, H. The resistome of Pseudomonas aeruginosa in relationship to phenotypic susceptibility. Antimicrob. Agents Chemother. 2015, 59, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Harcombe, W.R.; Bull, J.J. Impact of phages on two-species bacterial communities. Appl. Environ. Microbiol. 2005, 71, 5254–5259. [Google Scholar] [CrossRef]
- Hernandez, C.A.; Koskella, B. Phage resistance evolution in vitro is not reflective of in vivo outcome in a plant-bacteria-phage system. Evol. Int. J. Org. Evol. 2019. [Google Scholar] [CrossRef]
- LaVergne, S.; Hamilton, T.; Biswas, B.; Kumaraswamy, M.; Schooley, R.T.; Wooten, D. Phage therapy for a multidrug-resistant Acinetobacter baumannii craniectomy site infection. Open Forum Infect. Dis. 2018, 5, ofy064. [Google Scholar] [CrossRef]
- Yoichi, M.; Abe, M.; Miyanaga, K.; Unno, H.; Tanji, Y. Alteration of tail fiber protein gp38 enables T2 phage to infect Escherichia coli O157:H7. J. Biotechnol. 2005, 115, 101–107. [Google Scholar] [CrossRef]
- Mahichi, F.; Synnott, A.J.; Yamamichi, K.; Osada, T.; Tanji, Y. Site-specific recombination of T2 phage using IP008 long tail fiber genes provides a targeted method for expanding host range while retaining lytic activity. FEMS Microbiol. Lett. 2009, 295, 211–217. [Google Scholar] [CrossRef]
- Pouillot, F.; Blois, H.; Iris, F. Genetically engineered virulent phage banks in the detection and control of emergent pathogenic bacteria. Biosecur. Bioterrorism Biodefense Strateg. Pract. Sci. 2010, 8, 155–169. [Google Scholar] [CrossRef]
- Yehl, K.; Lemire, S.; Yang, A.C.; Ando, H.; Mimee, M.; Torres, M.D.T.; de la Fuente-Nunez, C.; Lu, T.K. Engineering phage host-range and suppressing bacterial resistance through phage tail fiber mutagenesis. Cell 2019, 179, 459–469.e9. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.K.; Sistrom, M.; Wertz, J.E.; Kortright, K.E.; Narayan, D.; Turner, P.E. Phage selection restores antibiotic sensitivity in MDR Pseudomonas aeruginosa. Sci. Rep. 2016, 6, 26717. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, W.N.; Concepción-Acevedo, J.; Park, T.; Andleeb, S.; Bull, J.J.; Levin, B.R. Synergy and order effects of antibiotics and phages in killing Pseudomonas aeruginosa biofilms. PLoS ONE 2017, 12, e0168615. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, J.; Shigehisa, R.; Nasukawa, T.; Mizukami, K.; Takemura-Uchiyama, I.; Ujihara, T.; Murakami, H.; Imanishi, I.; Nishifuji, K.; Sakaguchi, M.; et al. Piperacillin and ceftazidime produce the strongest synergistic phage-antibiotic effect in Pseudomonas aeruginosa. Arch. Virol. 2018, 163, 1941–1948. [Google Scholar] [CrossRef] [PubMed]
- Segall, A.M.; Roach, D.R.; Strathdee, S.A. Stronger together? Perspectives on phage-antibiotic synergy in clinical applications of phage therapy. Curr. Opin. Microbiol. 2019, 51, 46–50. [Google Scholar] [CrossRef]
- Abedon, S.T. Phage-antibiotic combination treatments: Antagonistic impacts of antibiotics on the pharmacodynamics of phage therapy? Antibiotics 2019, 8, 182. [Google Scholar] [CrossRef]
- Matsuda, T.; Freeman, T.A.; Hilbert, D.W.; Duff, M.; Fuortes, M.; Stapleton, P.P.; Daly, J.M. Lysis-deficient bacteriophage therapy decreases endotoxin and inflammatory mediator release and improves survival in a murine peritonitis model. Surgery 2005, 137, 639–646. [Google Scholar] [CrossRef]


| Characteristic | How Beneficial |
|---|---|
| Broad host range | can be applied rapidly, with minimal testing of pathogen sensitivity |
| Good in vivo growth and persistence | single dosing sufficient for treatment |
| Bacterial resistance difficult | single phage type sufficient for treatment |
| Synergistic with antibiotics * | can be used in combination with standard treatment |
| Disrupts bacterial extracellular protections | makes vulnerable bacterial clusters (e.g., biofilms and aggregates) that are otherwise recalcitrant to treatment |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bull, J.J.; Levin, B.R.; Molineux, I.J. Promises and Pitfalls of In Vivo Evolution to Improve Phage Therapy. Viruses 2019, 11, 1083. https://doi.org/10.3390/v11121083
Bull JJ, Levin BR, Molineux IJ. Promises and Pitfalls of In Vivo Evolution to Improve Phage Therapy. Viruses. 2019; 11(12):1083. https://doi.org/10.3390/v11121083
Chicago/Turabian StyleBull, James J., Bruce R. Levin, and Ian J. Molineux. 2019. "Promises and Pitfalls of In Vivo Evolution to Improve Phage Therapy" Viruses 11, no. 12: 1083. https://doi.org/10.3390/v11121083
APA StyleBull, J. J., Levin, B. R., & Molineux, I. J. (2019). Promises and Pitfalls of In Vivo Evolution to Improve Phage Therapy. Viruses, 11(12), 1083. https://doi.org/10.3390/v11121083