Npro of Classical Swine Fever Virus Suppresses Type III Interferon Production by Inhibiting IRF1 Expression and Its Nuclear Translocation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Virus, Cells and Recombinant Plasmids
2.2. Virus Rescue and Identification
2.3. Single Step Growth Curve
2.4. Virus Titration and Indirect Immunofluorescence
2.5. Virus Infection, Treatments with IFN-λ3 and Chemicals, and Transfection
2.6. RNA Isolation and qRT-PCR
2.7. Dual Luciferase Reporter Assay
2.8. Western Blotting
2.9. Confocal Microscopic Analysis of IRF1 Nuclear Translocation Affected by CSFV
2.10. Statistical Analysis
3. Results
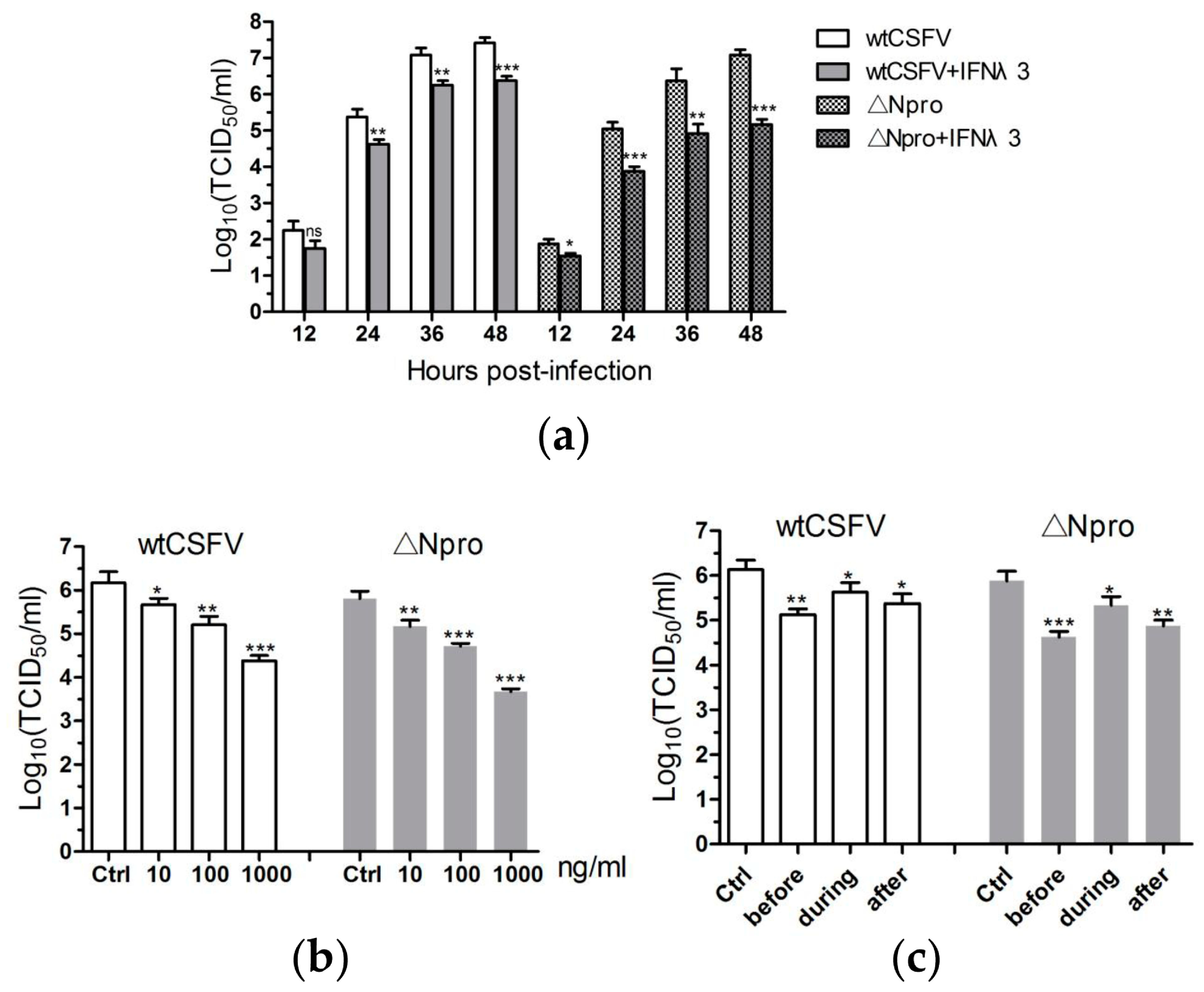
3.1. Identification and Growth Kinetics of the ∆Npro Mutant
3.2. Antiviral Activities of IFN-λ3 against CSFV
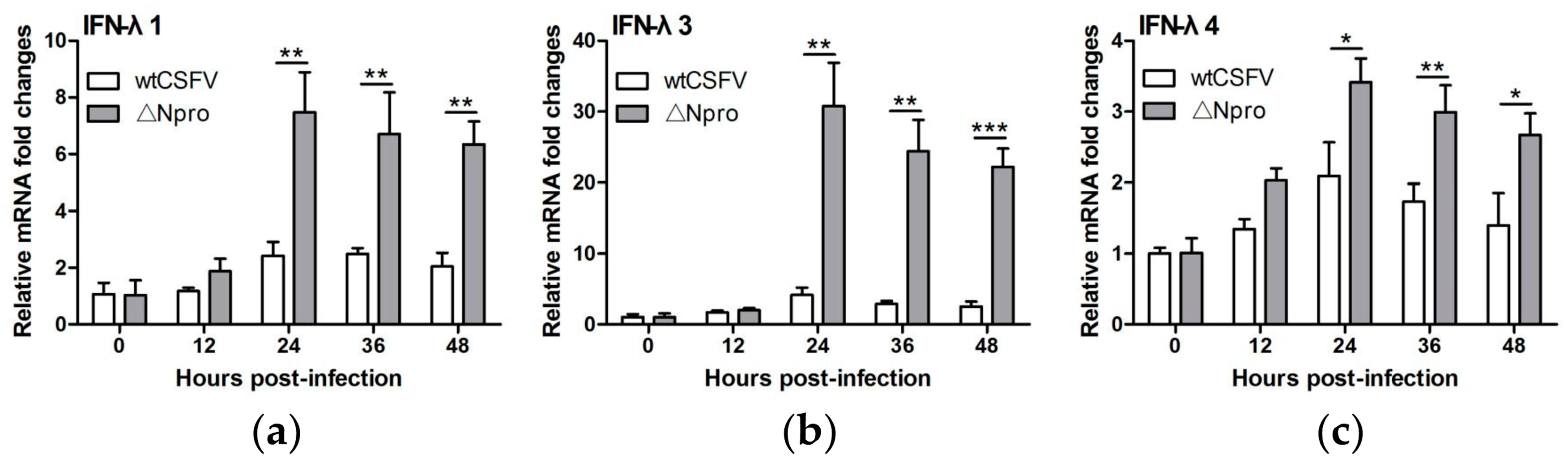
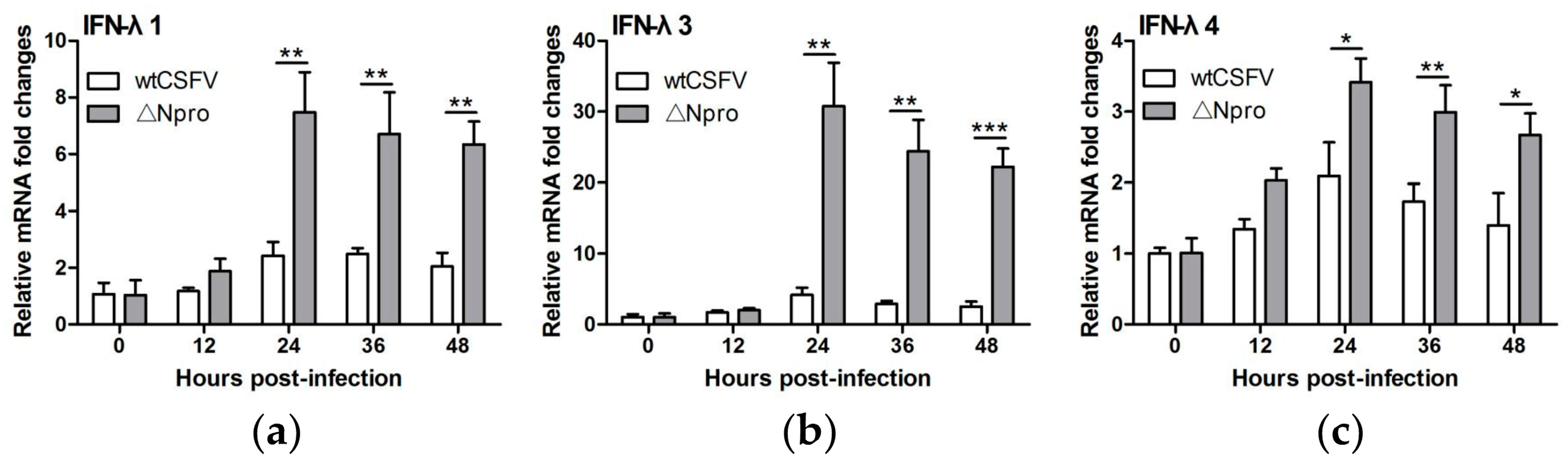
3.3. Npro Deletion Mutant Induced Much Higher Levels of Type III IFNs Transcription than Its Parental CSFV Strain
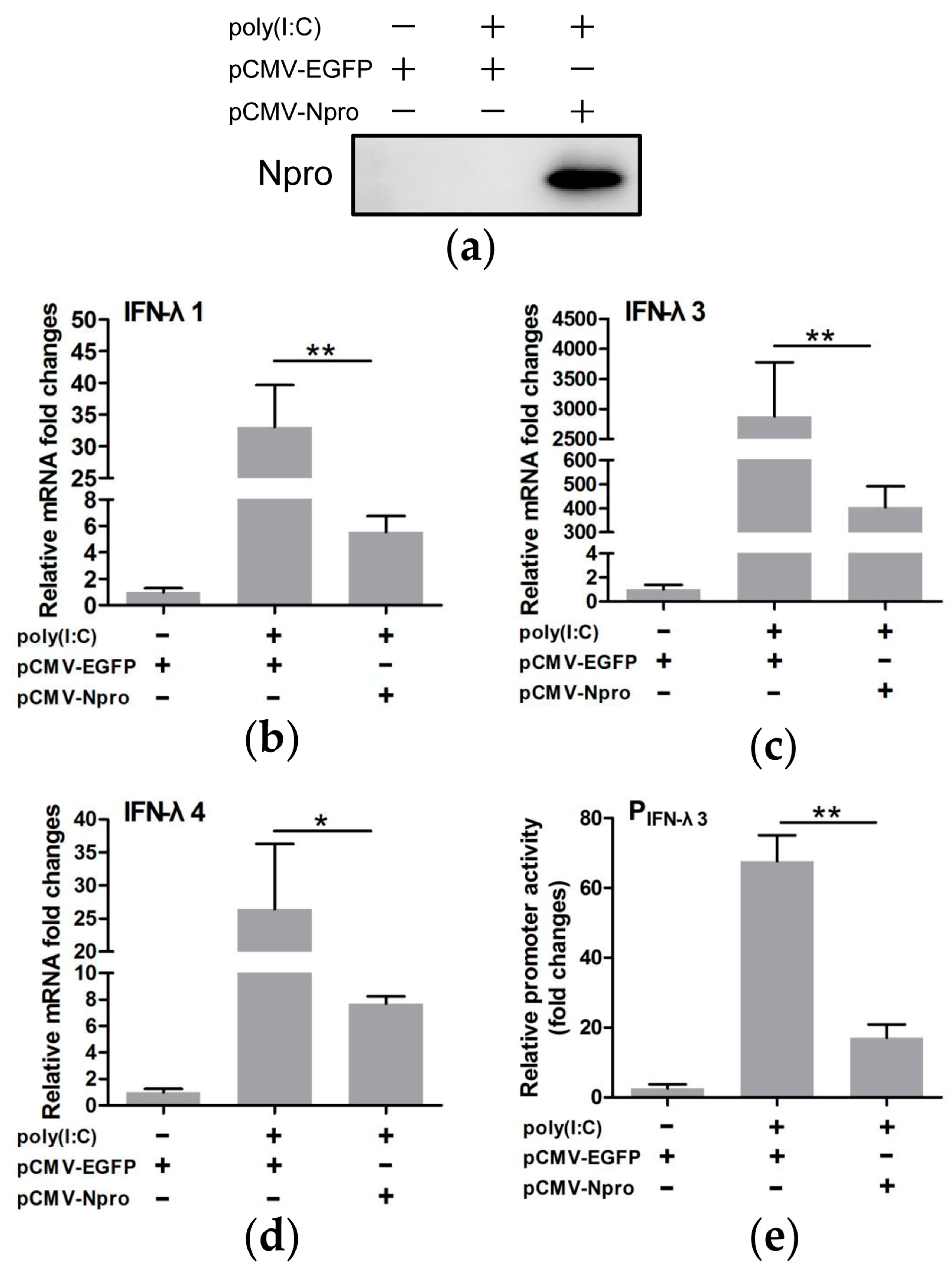
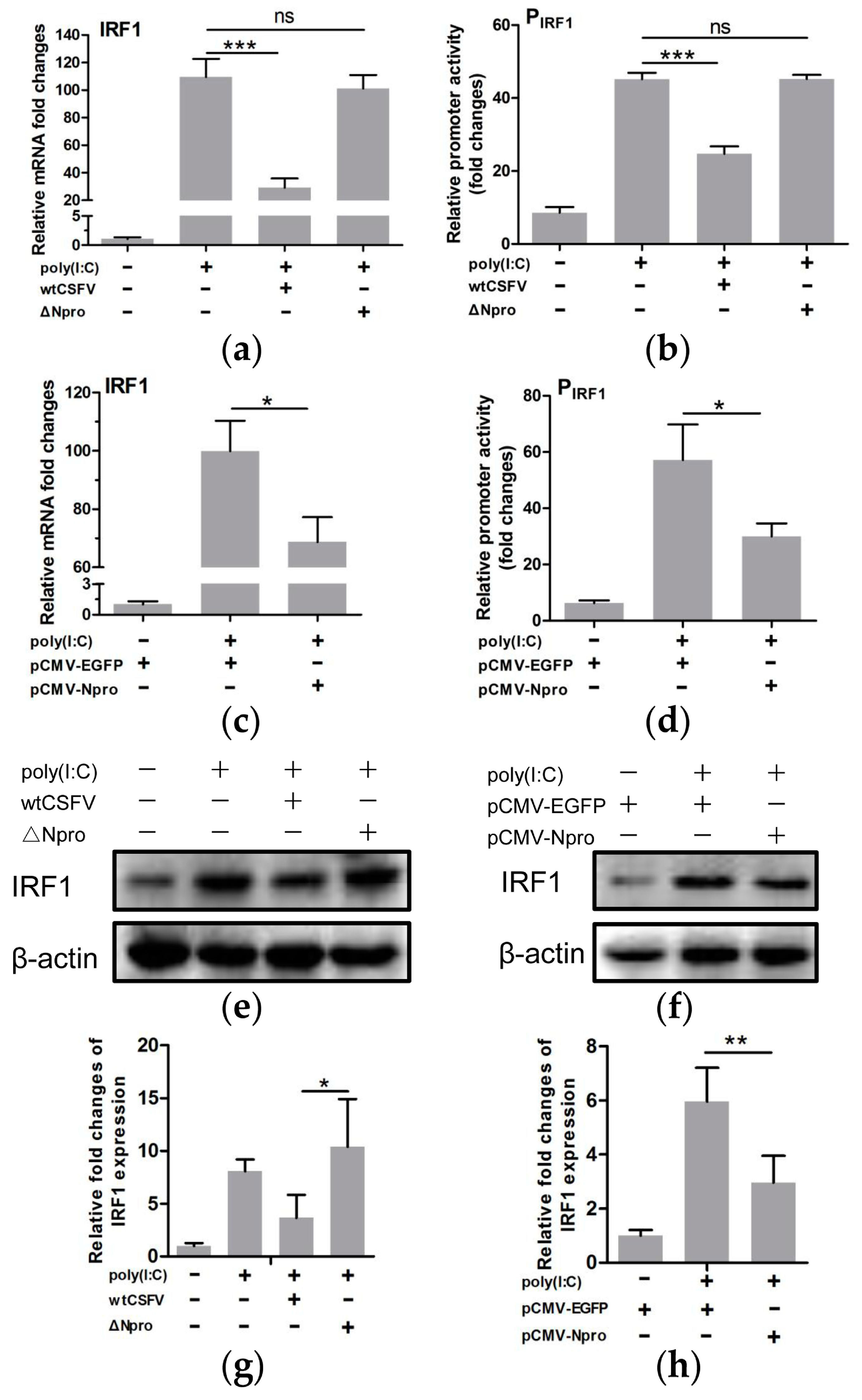
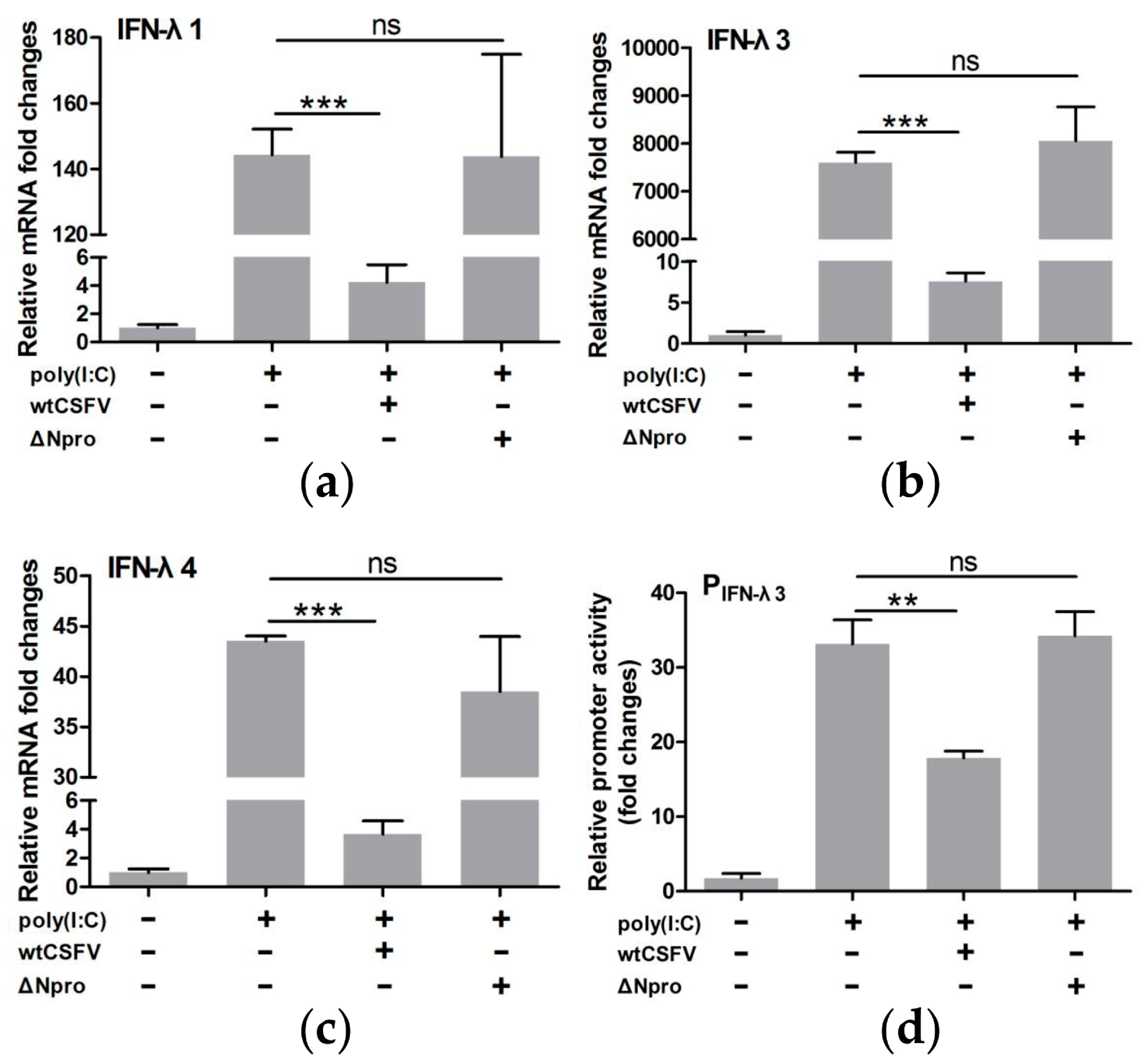
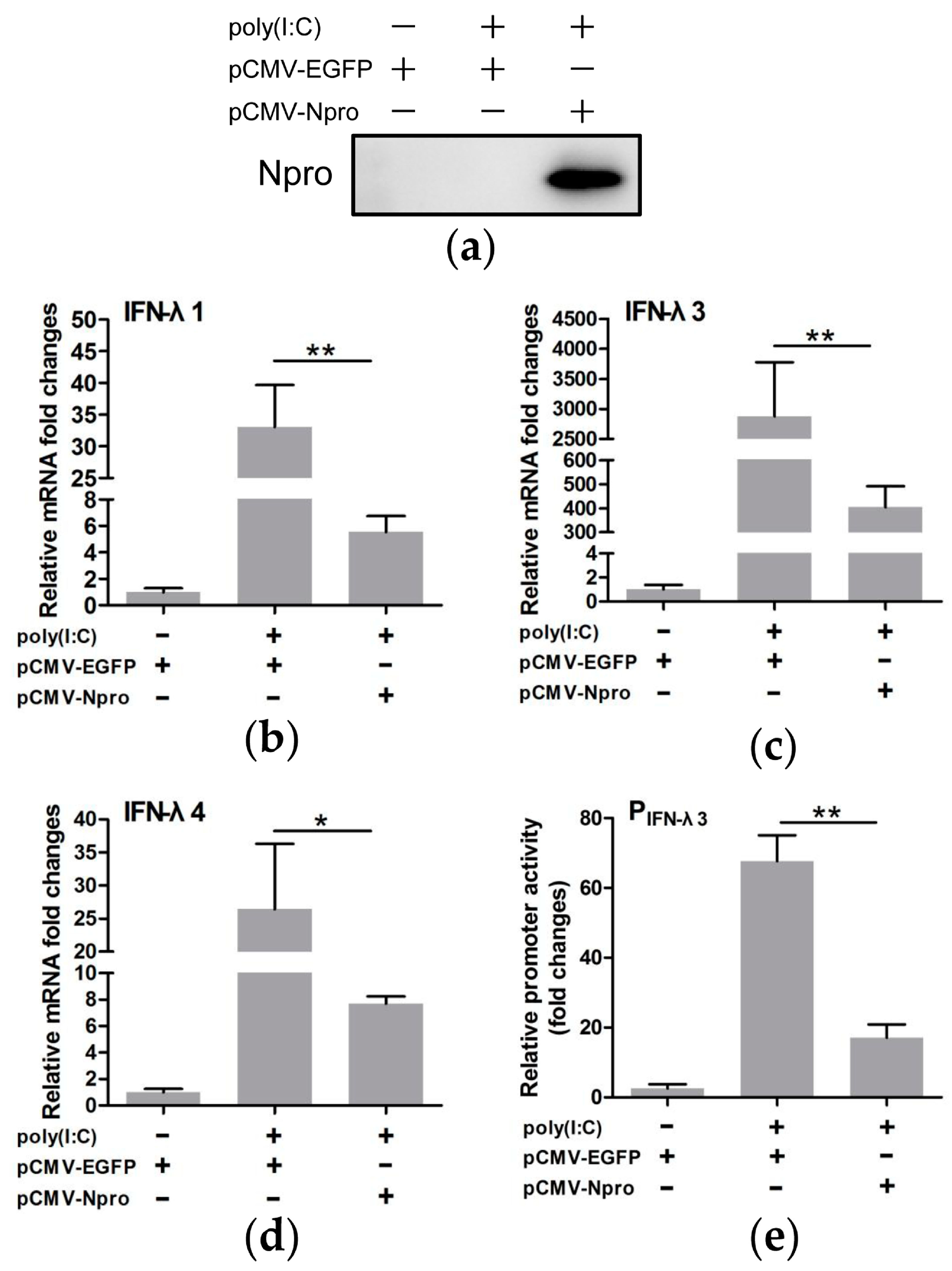
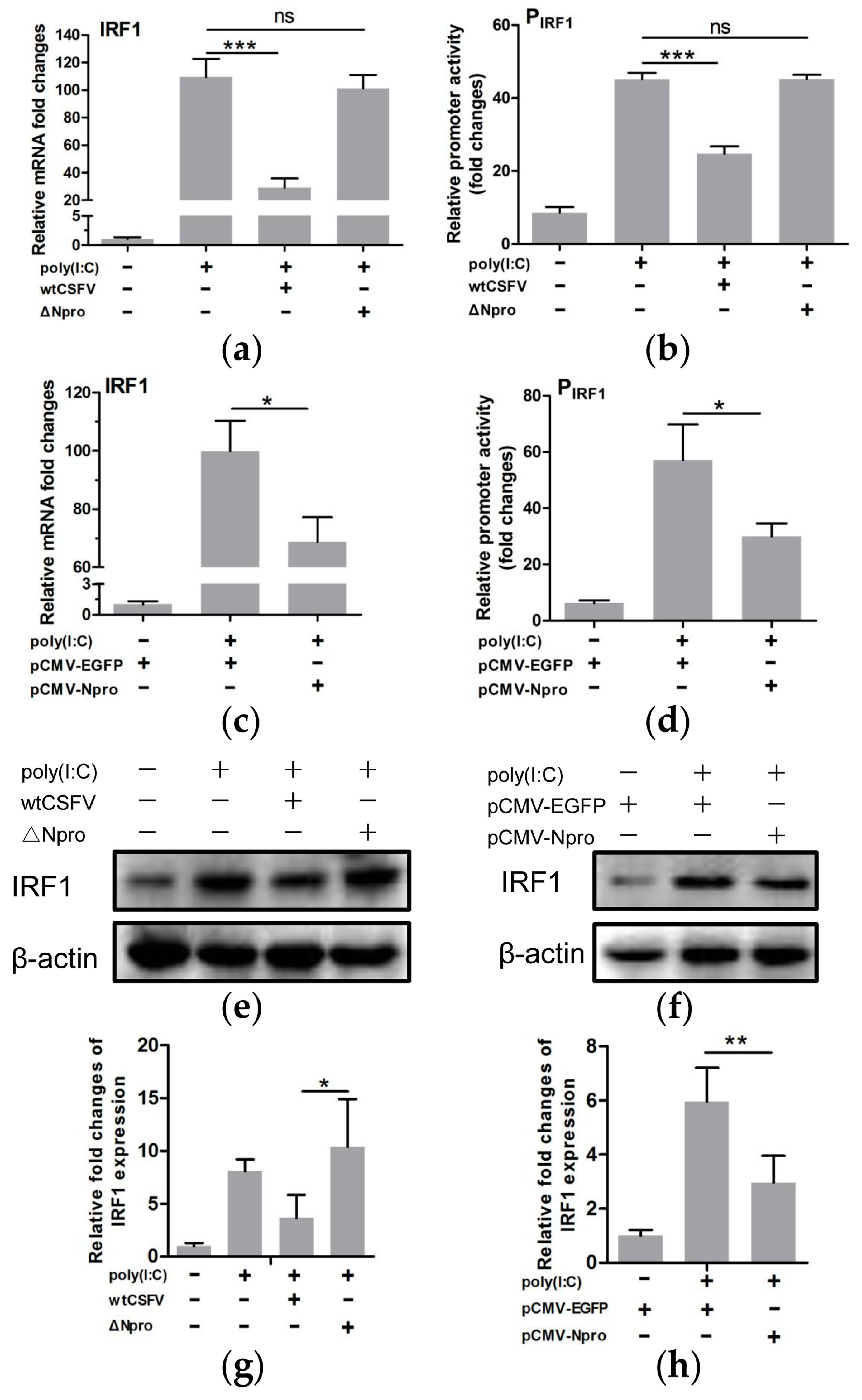
3.4. CSFV Inhibited poly(I:C)-Induced Production of Type III IFNs by Its Npro
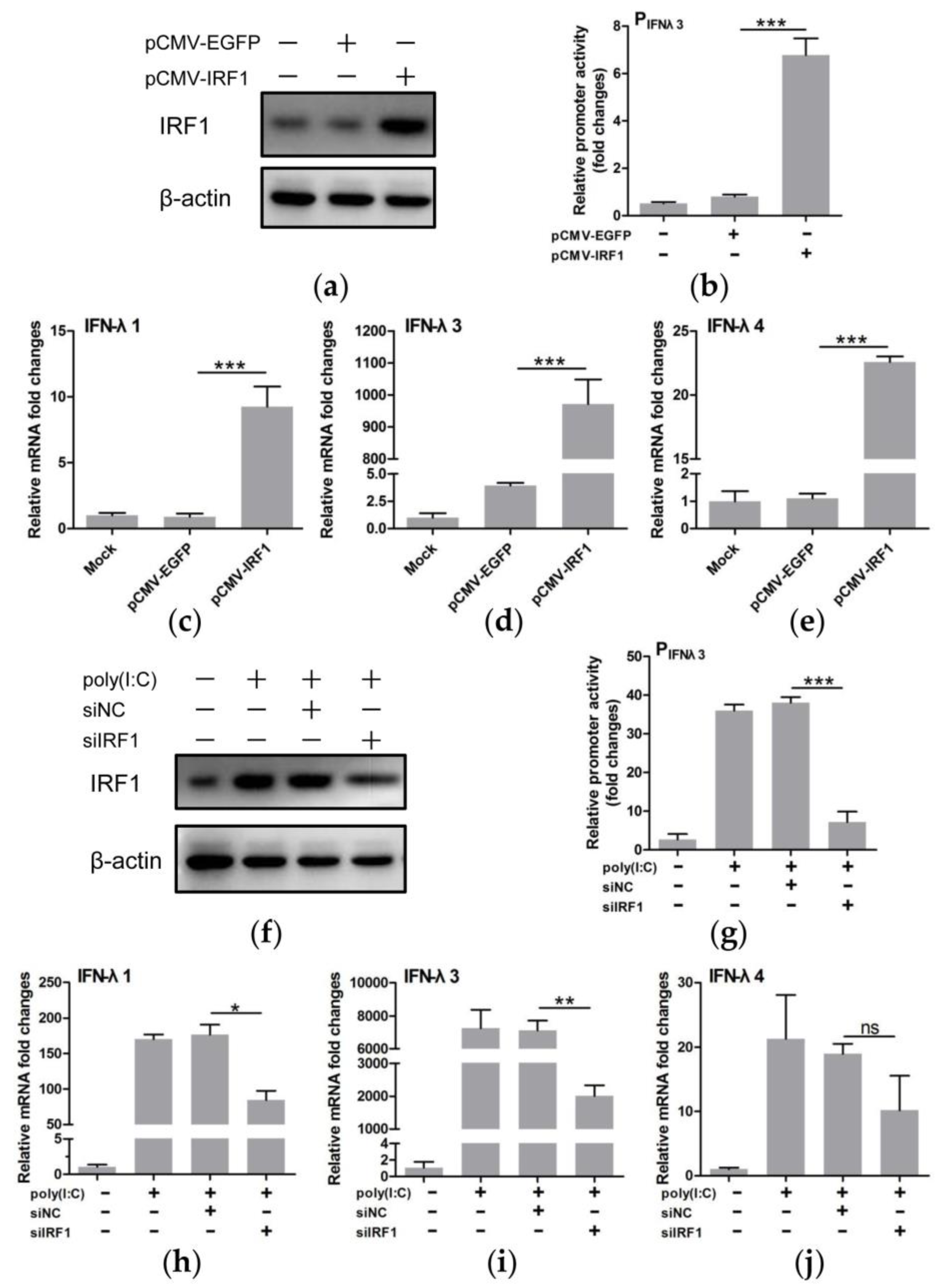
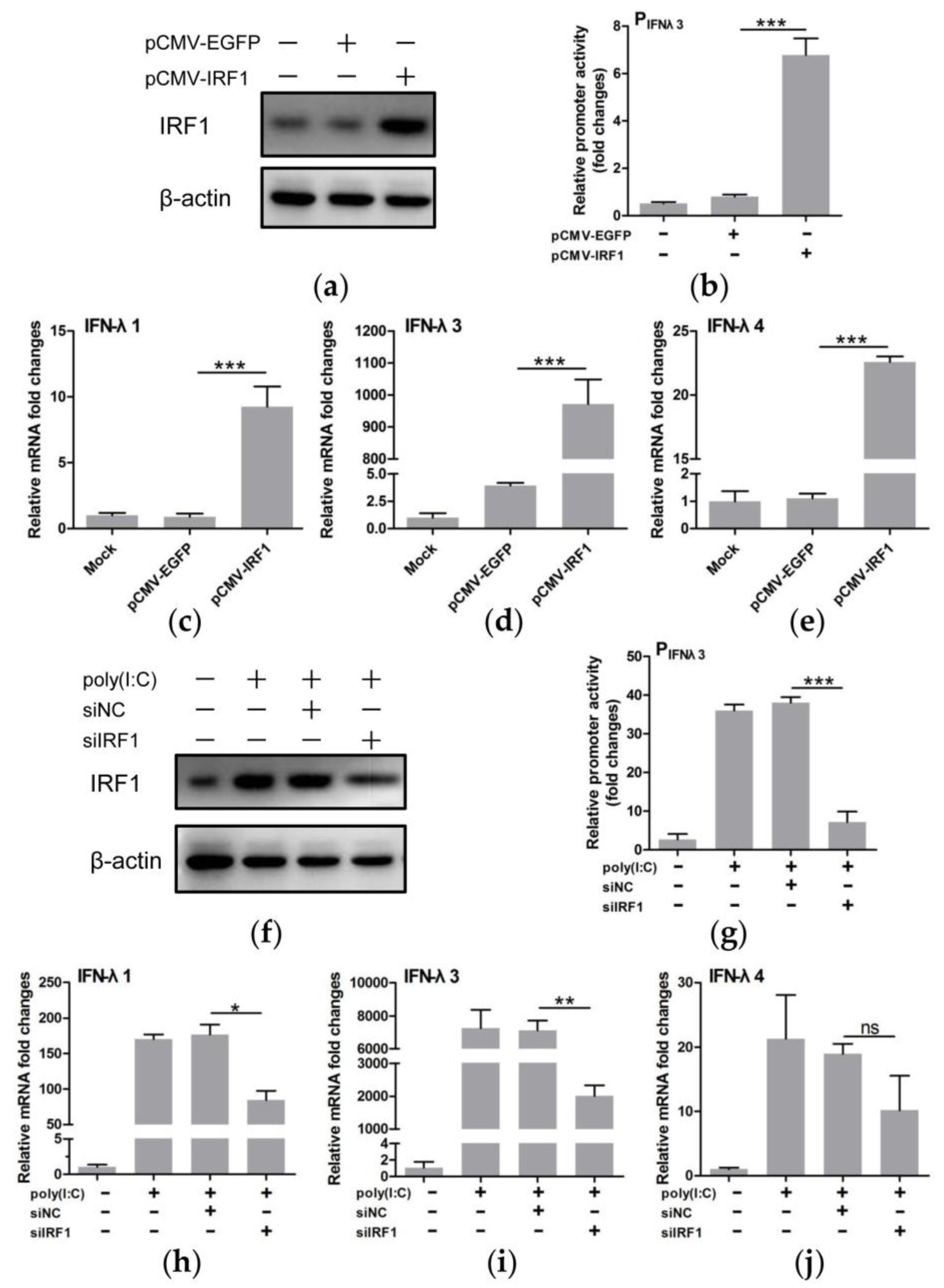
3.5. IRF1 Was a Positive Regulator of Type III IFNs
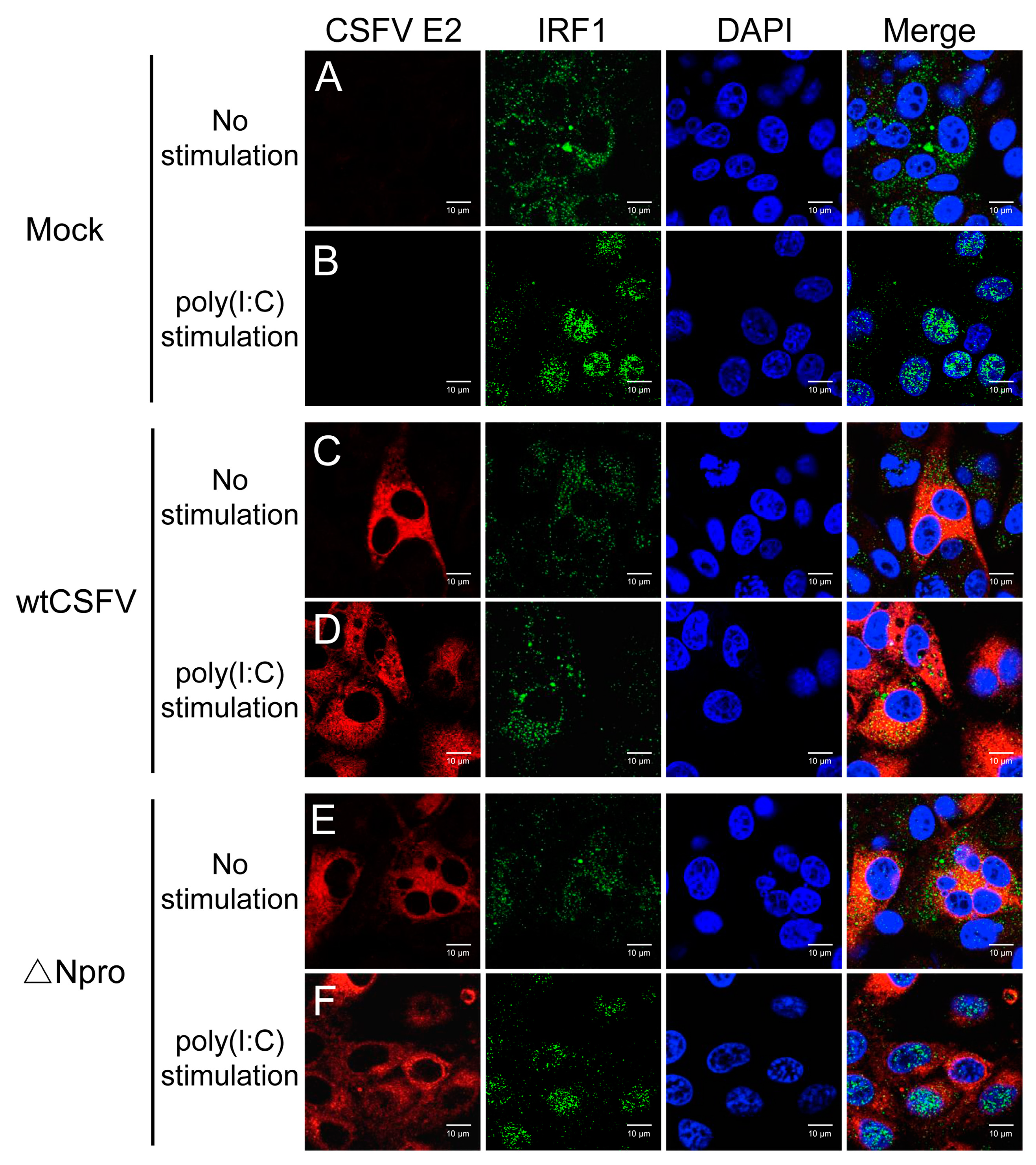
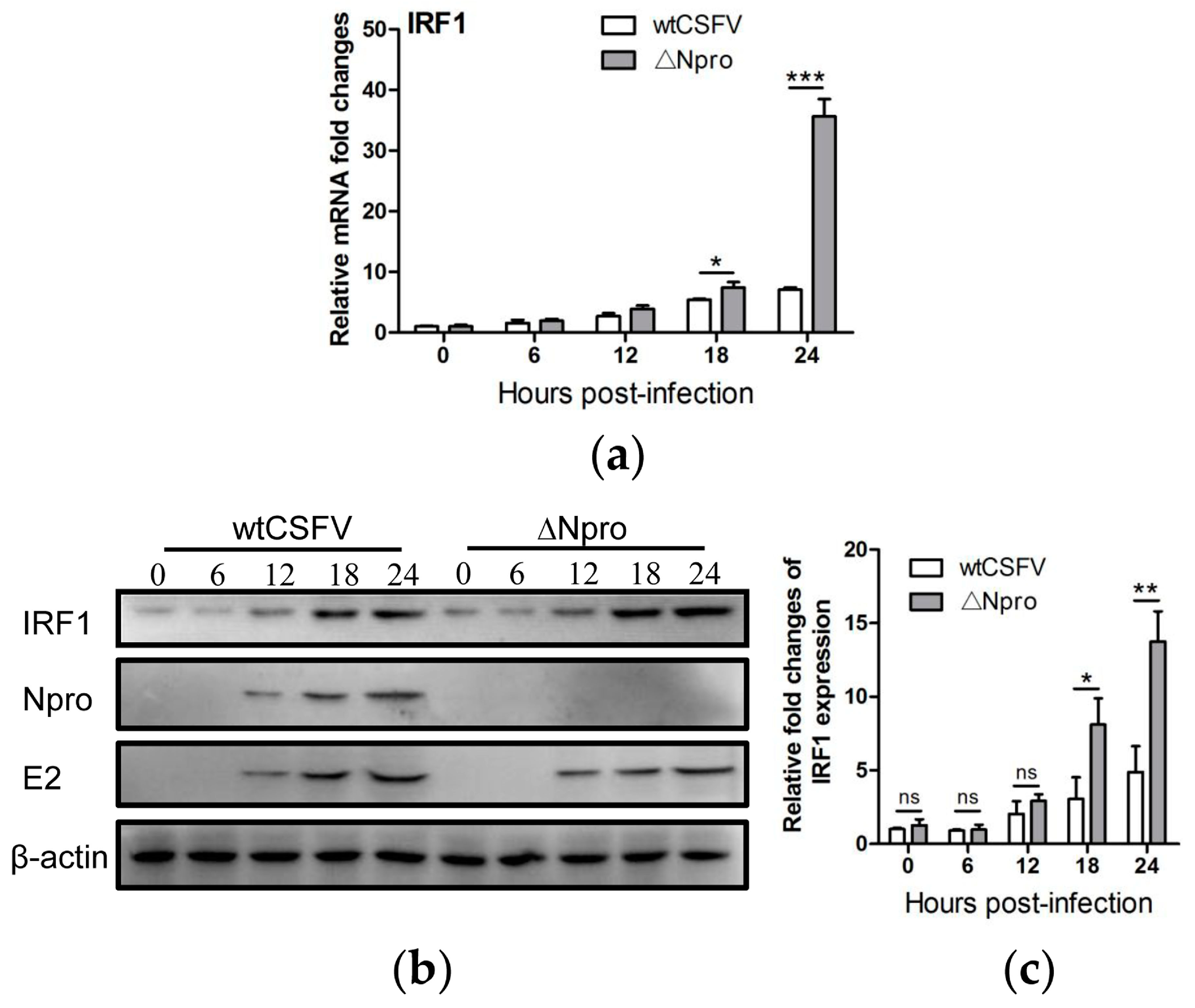
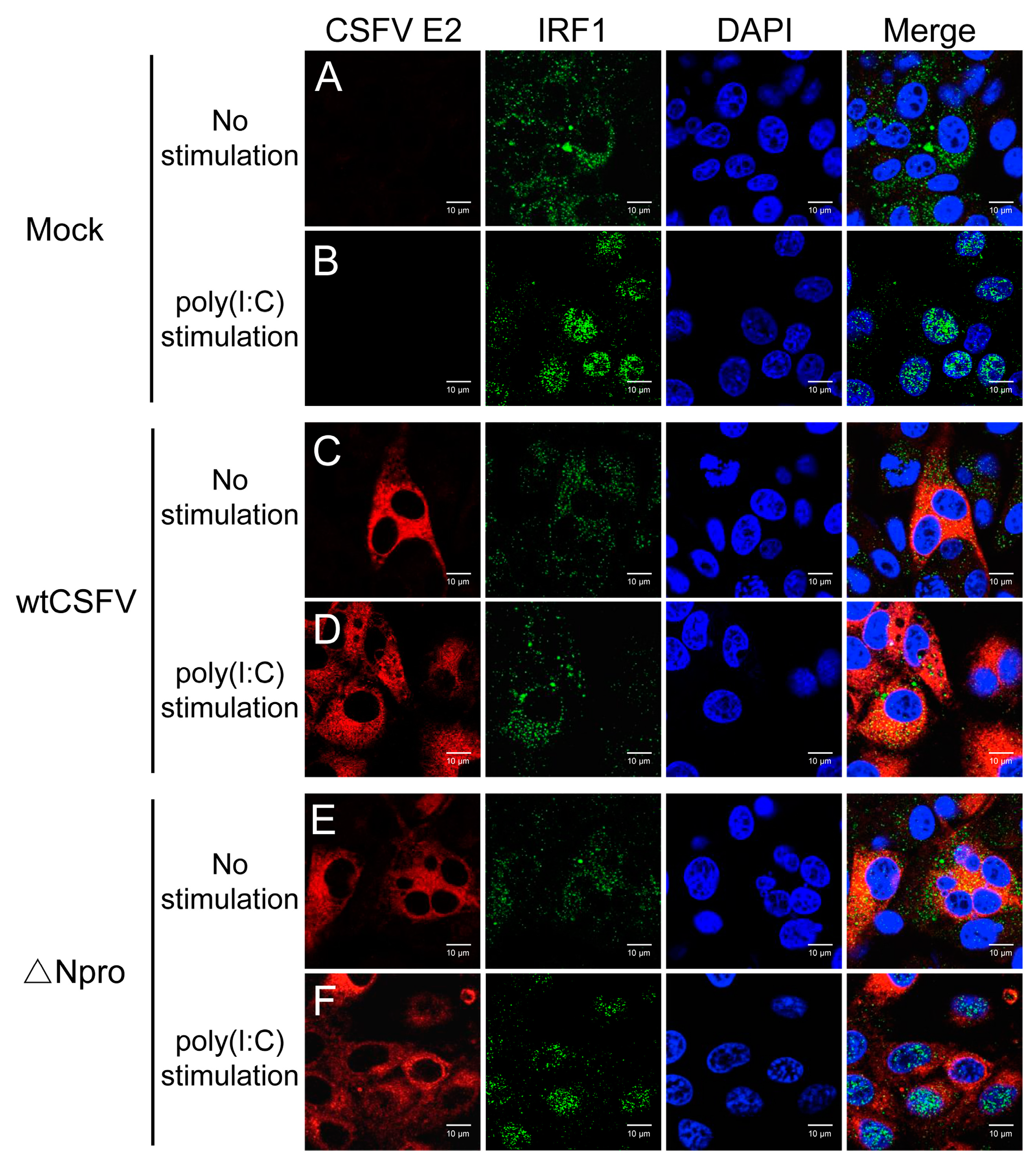
3.6. IRF1 Expression Profiles Were Different between wtCSFV and ΔNpro Infected Cells
3.7. Down-Regulation of IRF1 Expression by CSFV Npro
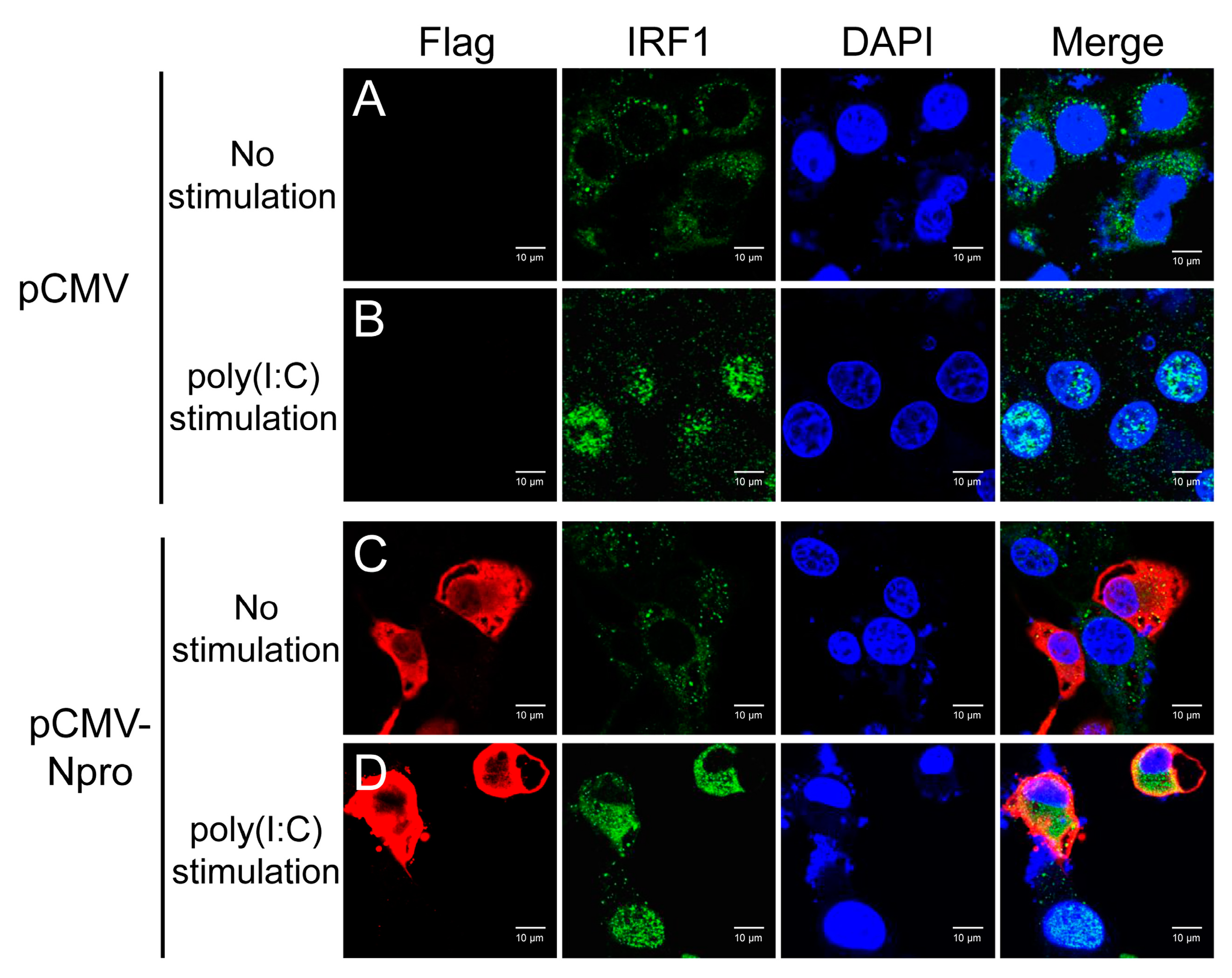
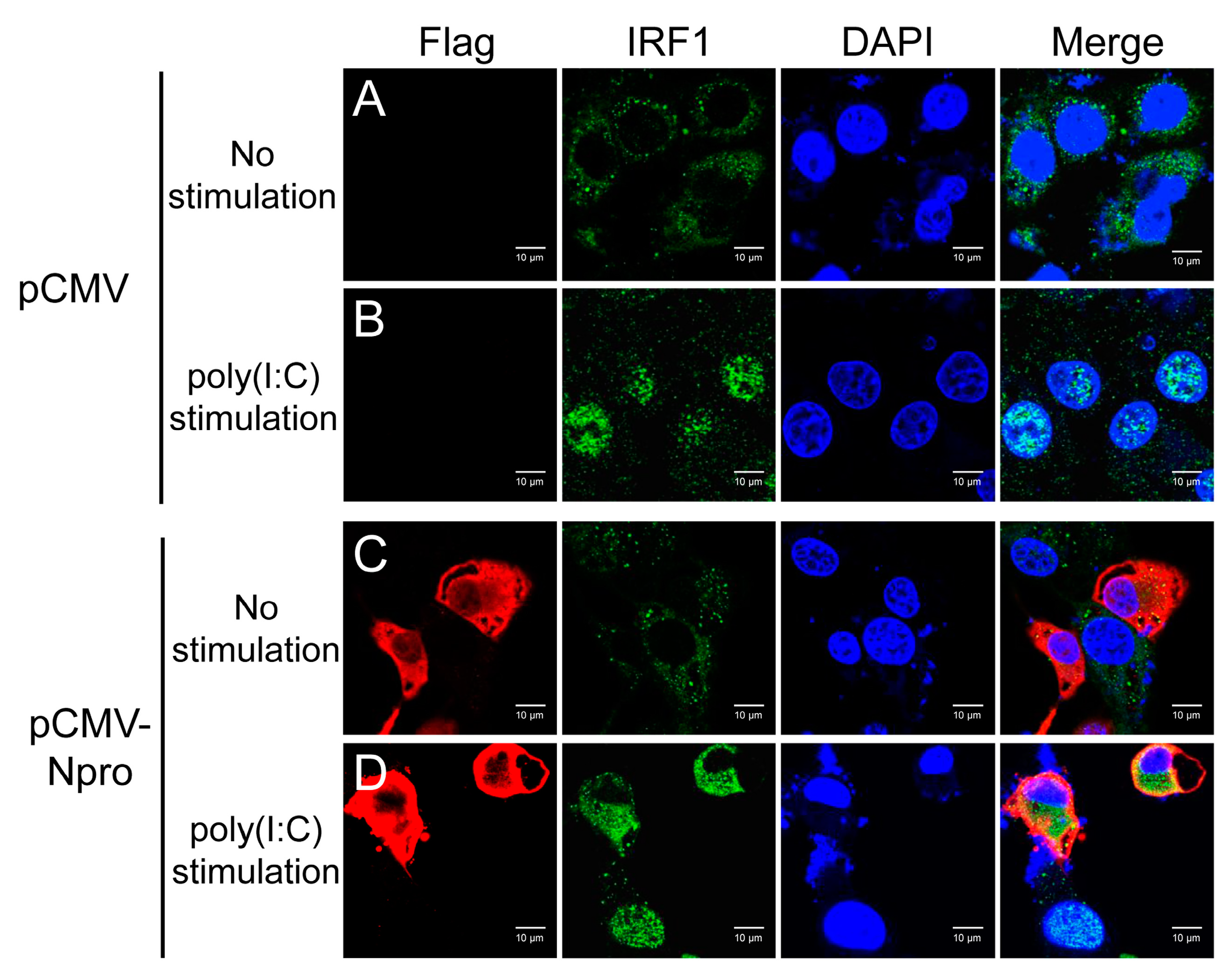
3.8. Inhibition of IRF1 Nuclear Translocation by CSFV Npro
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Iwasaki, A.; Medzhitov, R. Regulation of adaptive immunity by the innate immune system. Science 2010, 327, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, A.; Yanai, H. Interferon signalling network in innate defence. Cell Microbiol. 2006, 8, 907–922. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Liu, J.; Cao, X. Regulation of type I interferon signaling in immunity and inflammation: A comprehensive review. J. Autoimmun. 2017, 83, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hermant, P.; Michiels, T. Interferon-lambda in the context of viral infections: Production, response and therapeutic implications. J. Innate Immun. 2014, 6, 563–574. [Google Scholar] [CrossRef]
- Kotenko, S.V.; Gallagher, G.; Baurin, V.V.; Lewis-Antes, A.; Shen, M.L.; Shah, N.K.; Langer, J.A.; Sheikh, F.; Dickensheets, H.; Donnelly, R.P. IFN-lambda s mediate antiviral protection through a distinct class II cytokine receptor complex. Nat. Immunol. 2003, 4, 69–77. [Google Scholar] [CrossRef]
- Prokunina-Olsson, L.; Muchmore, B.; Tang, W.; Pfeiffer, R.M.; Park, H.; Dickensheets, H.; Hergott, D.; Porter-Gill, P.; Mumy, A.; Kohaar, I.; et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat. Genet. 2013, 45, 164–171. [Google Scholar] [CrossRef]
- Sheppard, P.; Kindsvogel, W.; Xu, W.; Henderson, K.; Schlutsmeyer, S.; Whitmore, T.E.; Kuestner, R.; Garrigues, U.; Birks, C.; Roraback, J.; et al. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat. Immunol. 2003, 4, 63–68. [Google Scholar] [CrossRef]
- Sang, Y.; Rowland, R.R.; Blecha, F. Molecular characterization and antiviral analyses of porcine type III interferons. J. Interferon Cytokine Res. 2010, 30, 801–807. [Google Scholar] [CrossRef]
- Zhang, Q.; Ke, H.; Blikslager, A.; Fujita, T.; Yoo, D. Type III interferon restriction by porcine epidemic diarrhea virus and the role of viral protein nsp1 in IRF1 signaling. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Mordstein, M.; Neugebauer, E.; Ditt, V.; Jessen, B.; Rieger, T.; Falcone, V.; Sorgeloos, F.; Ehl, S.; Mayer, D.; Kochs, G.; et al. Lambda interferon renders epithelial cells of the respiratory and gastrointestinal tracts resistant to viral infections. J. Virol. 2010, 84, 5670–5677. [Google Scholar] [CrossRef]
- Onoguchi, K.; Yoneyama, M.; Takemura, A.; Akira, S.; Taniguchi, T.; Namiki, H.; Fujita, T. Viral infections activate types I and III interferon genes through a common mechanism. J. Biol. Chem. 2007, 282, 7576–7581. [Google Scholar] [CrossRef] [PubMed]
- Thomson, S.J.; Goh, F.G.; Banks, H.; Krausgruber, T.; Kotenko, S.V.; Foxwell, B.M.; Udalova, I.A. The role of transposable elements in the regulation of IFN-lambda1 gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 11564–11569. [Google Scholar] [CrossRef] [PubMed]
- Sommereyns, C.; Paul, S.; Staeheli, P.; Michiels, T. IFN-lambda (IFN-lambda) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog. 2008, 4, e1000017. [Google Scholar] [CrossRef] [PubMed]
- Odendall, C.; Dixit, E.; Stavru, F.; Bierne, H.; Franz, K.M.; Durbin, A.F.; Boulant, S.; Gehrke, L.; Cossart, P.; Kagan, J.C. Diverse intracellular pathogens activate type III interferon expression from peroxisomes. Nat. Immunol. 2014, 15, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Dixit, E.; Boulant, S.; Zhang, Y.J.; Lee, A.S.Y.; Odendall, C.; Shum, B.; Hacohen, N.; Chen, Z.J.; Whelan, S.P.; Fransen, M.; et al. Peroxisomes are signaling platforms for antiviral innate immunity. Cell 2010, 141, 668–681. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Mahlakoiv, T.; Hernandez, P.; Gronke, K.; Diefenbach, A.; Staeheli, P. Leukocyte-derived IFN-alpha/beta and epithelial IFN-lambda constitute a compartmentalized mucosal defense system that restricts enteric virus infections. PLoS Pathog. 2015, 11, e1004782. [Google Scholar] [CrossRef]
- Ji, W.; Guo, Z.; Ding, N.Z.; He, C.Q. Studying classical swine fever virus: Making the best of a bad virus. Virus Res. 2015, 197, 35–47. [Google Scholar] [CrossRef]
- Blome, S.; Staubach, C.; Henke, J.; Carlson, J.; Beer, M. Classical swine fever-an updated review. Viruses 2017, 9, 86. [Google Scholar] [CrossRef]
- Lamp, B.; Riedel, C.; Roman-Sosa, G.; Heimann, M.; Jacobi, S.; Becher, P.; Thiel, H.J.; Rumenapf, T. Biosynthesis of classical swine fever virus nonstructural proteins. J. Virol. 2011, 85, 3607–3620. [Google Scholar] [CrossRef]
- Becher, P.; Avalos Ramirez, R.; Orlich, M.; Cedillo Rosales, S.; König, M.; Schweizer, M.; Stalder, H.; Schirrmeier, H.; Thiel, H.-J. Genetic and antigenic characterization of novel pestivirus genotypes: Implications for classification. Virology 2003, 311, 96–104. [Google Scholar] [CrossRef]
- Rumenapf, T.; Stark, R.; Heimann, M.; Thiel, H.J. N-terminal protease of pestiviruses: Identification of putative catalytic residues by site-directed mutagenesis. J. Virol. 1998, 72, 2544–2547. [Google Scholar] [PubMed]
- Stark, R.; Meyers, G.; Rumenapf, T.; Thiel, H.J. Processing of pestivirus polyprotein - cleavage site between autoprotease and nucleocapsid protein of classical swine fever virus. J. Virol. 1993, 67, 7088–7095. [Google Scholar] [PubMed]
- Wiskerchen, M.; Belzer, S.K.; Collett, M.S. Pestivirus gene-expression - the 1st protein product of the bovine viral diarrhea virus large open reading frame, P20, possesses proteolytic activity. J. Virol. 1991, 65, 4508–4514. [Google Scholar]
- Bensaude, E.; Turner, J.L.; Wakeley, P.R.; Sweetman, D.A.; Pardieu, C.; Drew, T.W.; Wileman, T.; Powell, P.P. Classical swine fever virus induces proinflammatory cytokines and tissue factor expression and inhibits apoptosis and interferon synthesis during the establishment of long-term infection of porcine vascular endothelial cells. J. Gen. Virol. 2004, 85, 1029–1037. [Google Scholar] [CrossRef]
- La Rocca, S.A.; Herbert, R.J.; Crooke, H.; Drew, T.W.; Wileman, T.E.; Powell, P.P. Loss of interferon regulatory factor 3 in cells infected with classical swine fever virus involves the N-terminal protease, Npro. J. Virol. 2005, 79, 7239–7247. [Google Scholar] [CrossRef]
- Ruggli, N.; Bird, B.H.; Liu, L.; Bauhofer, O.; Tratschin, J.D.; Hofmann, M.A. N-pro of classical swine fever virus is an antagonist of double-stranded RNA-mediated apoptosis and IFN-alpha/beta induction. Virology 2005, 340, 265–276. [Google Scholar] [CrossRef]
- Ruggli, N.; Tratschin, J.D.; Schweizer, M.; McCullough, K.C.; Hofmann, M.A.; Summerfield, A. Classical swine fever virus interferes with cellular antiviral defense: Evidence for a novel function of N-pro. J. Virol. 2003, 77, 7645–7654. [Google Scholar] [CrossRef]
- Bauhofer, O.; Summerfield, A.; Sakoda, Y.; Tratschin, J.D.; Hofmann, M.A.; Ruggli, N. Classical swine fever virus Npro interacts with interferon regulatory factor 3 and induces its proteasomal degradation. J. Virol. 2007, 81, 3087–3096. [Google Scholar] [CrossRef]
- Seago, J.; Hilton, L.; Reid, E.; Doceul, V.; Jeyatheesan, J.; Moganeradj, K.; McCauley, J.; Charleston, B.; Goodbourn, S. The Npro product of classical swine fever virus and bovine viral diarrhea virus uses a conserved mechanism to target interferon regulatory factor-3. J. Gen. Virol. 2007, 88, 3002–3006. [Google Scholar] [CrossRef]
- Reid, E.; Juleff, N.; Windsor, M.; Gubbins, S.; Roberts, L.; Morgan, S.; Meyers, G.; Perez-Martin, E.; Tchilian, E.; Charleston, B.; et al. Type I and III IFNs produced by plasmacytoid dendritic cells in response to a member of the flaviviridae suppress cellular immune responses. J. Immunol. 2016, 196, 4214–4226. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Bai, Q.; Chi, X.; Goraya, M.U.; Wang, L.; Wang, S.; Chen, B.; Chen, J.L. Infection with classical swine fever virus induces expression of type III interferons and activates innate immune signaling. Front. Microbiol. 2017, 8, 2558. [Google Scholar] [CrossRef] [PubMed]
- Cao, T.; Zhang, S.; Li, X.; Xu, Y.; Wang, Z.; Chen, C.; Paudyal, N.; Li, X.; Sun, J.; Fang, W. Classical swine fever virus C-strain with eight mutation sites shows enhanced cell adaptation and protects pigs from lethal challenge. Arch. Virol. 2019, 164, 1619–1628. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Tong, C.; Li, D.; Wan, J.; Yuan, X.; Li, X.; Peng, J.; Fang, W. Antigenic analysis of classical swine fever virus E2 glycoprotein using pig antibodies identifies residues contributing to antigenic variation of the vaccine C-strain and group 2 strains circulating in China. Virol. J. 2010, 7, 378. [Google Scholar] [CrossRef]
- Shan, Y.; Liu, Z.Q.; Li, G.W.; Chen, C.; Luo, H.; Liu, Y.J.; Zhuo, X.H.; Shi, X.F.; Fang, W.H.; Li, X.L. Nucleocapsid protein from porcine epidemic diarrhea virus isolates can antagonize interferon-lambda production by blocking the nuclear factor-kappaB nuclear translocation. J. Zhejiang Univ. Sci. B 2018, 19, 570–580. [Google Scholar] [CrossRef]
- Gu, Y.; Qi, B.; Zhou, Y.; Jiang, X.; Zhang, X.; Li, X.; Fang, W. Porcine circovirus type 2 activates CaMMKbeta to initiate autophagy in PK-15 cells by increasing cytosolic calcium. Viruses 2016, 8, 135. [Google Scholar] [CrossRef]
- Durantel, D.; Carrouee-Durantel, S.; Branza-Nichita, N.; Dwek, R.A.; Zitzmann, N. Effects of interferon, ribavirin, and iminosugar derivatives on cells persistently infected with noncytopathic bovine viral diarrhea virus. Antimicrob. Agents Chemother. 2004, 48, 497–504. [Google Scholar] [CrossRef]
- Zhou, Z.; Wang, N.; Woodson, S.E.; Dong, Q.; Wang, J.; Liang, Y.; Rijnbrand, R.; Wei, L.; Nichols, J.E.; Guo, J.T.; et al. Antiviral activities of ISG20 in positive-strand RNA virus infections. Virology 2011, 409, 175–188. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Liu, B.M.; Wang, N.; Lee, Y.M.; Liu, C.M.; Li, K. TRIM56 is a virus- and interferon-inducible E3 ubiquitin ligase that restricts pestivirus infection. J. Virol. 2011, 85, 3733–3745. [Google Scholar] [CrossRef]
- Diebold, S.S.; Kaisho, T.; Hemmi, H.; Akira, S.; Sousa, C.R.E. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 2004, 303, 1529–1531. [Google Scholar] [CrossRef]
- Sen, G.C.; Sarkar, S.N. Transcriptional signaling by double-stranded RNA: Role of TLR3. Cytokine Growth Factor Rev. 2005, 16, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Lau, J.F.; Parisien, J.P.; Horvath, C.M. Interferon regulatory factor subcellular localization is determined by a bipartite nuclear localization signal in the DNA-binding domain and interaction with cytoplasmic retention factors. Proc. Natl. Acad. Sci. USA 2000, 97, 7278–7283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szymanski, M.R.; Fiebach, A.R.; Tratschin, J.D.; Gut, M.; Ramanujam, V.M.; Gottipati, K.; Patel, P.; Ye, M.; Ruggli, N.; Choi, K.H. Zinc binding in pestivirus N(pro) is required for interferon regulatory factor 3 interaction and degradation. J. Mol. Biol. 2009, 391, 438–449. [Google Scholar] [CrossRef] [PubMed]
- Gottipati, K.; Ruggli, N.; Gerber, M.; Tratschin, J.D.; Benning, M.; Bellamy, H.; Choi, K.H. The structure of classical swine fever virus N(pro): A novel cysteine Autoprotease and zinc-binding protein involved in subversion of type I interferon induction. PLoS Pathog. 2013, 9, e1003704. [Google Scholar] [CrossRef]
- Pott, J.; Mahlakoiv, T.; Mordstein, M.; Duerr, C.U.; Michiels, T.; Stockinger, S.; Staeheli, P.; Hornef, M.W. IFN-lambda determines the intestinal epithelial antiviral host defense. Proc. Natl. Acad. Sci. USA 2011, 108, 7944–7949. [Google Scholar] [CrossRef]
- Li, L.; Fu, F.; Xue, M.; Chen, W.; Liu, J.; Shi, H.; Chen, J.; Bu, Z.; Feng, L.; Liu, P. IFN-lambda preferably inhibits PEDV infection of porcine intestinal epithelial cells compared with IFN-alpha. Antiviral Res. 2017, 140, 76–82. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, C.H.; Ryu, J.H.; Kim, M.J.; Park, C.Y.; Lee, J.M.; Holtzman, M.J.; Yoon, J.H. Reactive oxygen species induce antiviral innate immune response through IFN-lambda regulation in human nasal epithelial cells. Am. J. Resp. Cell. Mol. 2013, 49, 855–865. [Google Scholar] [CrossRef]
- Tratschin, J.D.; Moser, C.; Ruggli, N.; Hofmann, M.A. Classical swine fever virus leader proteinase Npro is not required for viral replication in cell culture. J. Virol. 1998, 72, 7681–7684. [Google Scholar]
- Wang, D.; Fang, L.; Liu, L.; Zhong, H.; Chen, Q.; Luo, R.; Liu, X.; Zhang, Z.; Chen, H.; Xiao, S. Foot-and-mouth disease virus (FMDV) leader proteinase negatively regulates the porcine interferon-lambda1 pathway. Mol. Immunol. 2011, 49, 407–412. [Google Scholar] [CrossRef]
- Bandi, P.; Pagliaccetti, N.E.; Robek, M.D. Inhibition of type III interferon activity by orthopoxvirus immunomodulatory proteins. J. Interf. Cytok. Res. 2010, 30, 123–133. [Google Scholar] [CrossRef]
- Huang, J.Y.; Smirnov, S.V.; Lewis-Antes, A.; Balan, M.; Li, W.; Tang, S.; Silke, G.V.; Putz, M.M.; Smith, G.L.; Kotenko, S.V. Inhibition of type I and type III interferons by a secreted glycoprotein from Yaba-like disease virus. Proc. Natl. Acad. Sci. USA 2007, 104, 9822–9827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brosnahan, A.J.; Brown, D.R. Porcine IPEC-J2 intestinal epithelial cells in microbiological investigations. Vet. Microbiol. 2012, 156, 229–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Chang, H.; Yang, X.; Zhao, Y.; Chen, L.; Wang, X.; Liu, H.; Wang, C.; Zhao, J. Antiviral activity of porcine interferon regulatory factor 1 against swine viruses in cell culture. Viruses 2015, 7, 5908–5918. [Google Scholar] [CrossRef] [PubMed]
- Reis, L.F.L.; Ruffner, H.; Stark, G.; Aguet, M.; Weissmann, C. Mice devoid of interferon regulatory factor-1 (Irf-1) show normal expression of type-I interferon genes. Embo J. 1994, 13, 4798–4806. [Google Scholar] [CrossRef]
- Sato, S.; Li, K.; Kameyama, T.; Hayashi, T.; Ishida, Y.; Murakami, S.; Watanabe, T.; Iijima, S.; Sakurai, Y.; Watashi, K.; et al. The RNA sensor RIG-I dually functions as an innate sensor and direct antiviral factor for hepatitis B virus. Immunity 2015, 42, 123–132. [Google Scholar] [CrossRef]
- Park, H.; Serti, E.; Eke, O.; Muchmore, B.; Prokunina-Olsson, L.; Capone, S.; Folgori, A.; Rehermann, B. IL-29 is the dominant type III interferon produced by hepatocytes during acute hepatitis C virus infection. Hepatology 2012, 56, 2060–2070. [Google Scholar] [CrossRef]
- Li, X.Q.; Li, X.N.; Liang, J.J.; Cai, X.B.; Tao, Q.; Li, Y.X.; Qin, Q.; Xu, S.P.; Luo, T.R. IRF1 up-regulates isg15 gene expression in dsRNA stimulation or CSFV infection by targeting nucleotides -487 to -325 in the 5’ flanking region. Mol. Immunol. 2018, 94, 153–165. [Google Scholar] [CrossRef]
- Iwanaszko, M.; Kimmel, M. NF-kappaB and IRF pathways: Cross-regulation on target genes promoter level. BMC Genom. 2015, 16, 307. [Google Scholar] [CrossRef]
- Chen, L.J.; Dong, X.Y.; Zhao, M.Q.; Shen, H.Y.; Wang, J.Y.; Pei, J.J.; Liu, W.J.; Luo, Y.W.; Ju, C.M.; Chen, J.D. Classical swine fever virus failed to activate nuclear factor-kappa b signaling pathway both in vitro and in vivo. Virol. J. 2012, 9, 293. [Google Scholar] [CrossRef]
- Doceul, V.; Charleston, B.; Crooke, H.; Reid, E.; Powell, P.P.; Seago, J. The Npro product of classical swine fever virus interacts with IkappaBalpha, the NF-kappaB inhibitor. J. Gen. Virol. 2008, 89, 1881–1889. [Google Scholar] [CrossRef]
- Coccia, E.M.; Severa, M.; Giacomini, E.; Monneron, D.; Remoli, M.E.; Julkunen, I.; Cella, M.; Lande, R.; Uze, G. Viral infection and Toll-like receptor agonists induce a differential expression of type I and lambda interferons in human plasmacytoid and monocyte-derived dendritic cells. Eur. J. Immunol. 2004, 34, 796–805. [Google Scholar] [CrossRef] [PubMed]
- Ank, N.; Iversen, M.B.; Bartholdy, C.; Staeheli, P.; Hartmann, R.; Jensen, U.B.; Dagnaes-Hansen, F.; Thomsen, A.R.; Chen, Z.; Haugen, H.; et al. An important role for type III interferon (IFN-lambda/IL-28) in TLR-induced antiviral activity. J. Immunol. 2008, 180, 2474–2485. [Google Scholar] [CrossRef] [PubMed]
- Makela, S.M.; Osterlund, P.; Julkunen, I. TLR ligands induce synergistic interferon-beta and interferon-lambda1 gene expression in human monocyte-derived dendritic cells. Mol. Immunol. 2011, 48, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Guo, K.; Zheng, M.; Ning, P.; Li, H.; Kang, K.; Lin, Z.; Zhang, C.; Liang, W.; Zhang, Y. A comparison of the impact of Shimen and C strains of classical swine fever virus on Toll-like receptor expression. J. Gen. Virol. 2015, 96, 1732–1745. [Google Scholar] [CrossRef] [PubMed]
- Zaheer, R.S.; Proud, D. Human rhinovirus-induced epithelial production of CXCL10 is dependent upon IFN regulatory factor-1. Am. J. Resp. Cell. Mol. 2010, 43, 413–421. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, T.; Li, X.; Xu, Y.; Zhang, S.; Wang, Z.; Shan, Y.; Sun, J.; Fang, W.; Li, X. Npro of Classical Swine Fever Virus Suppresses Type III Interferon Production by Inhibiting IRF1 Expression and Its Nuclear Translocation. Viruses 2019, 11, 998. https://doi.org/10.3390/v11110998
Cao T, Li X, Xu Y, Zhang S, Wang Z, Shan Y, Sun J, Fang W, Li X. Npro of Classical Swine Fever Virus Suppresses Type III Interferon Production by Inhibiting IRF1 Expression and Its Nuclear Translocation. Viruses. 2019; 11(11):998. https://doi.org/10.3390/v11110998
Chicago/Turabian StyleCao, Tong, Xiaoye Li, Yonghao Xu, Shengnan Zhang, Zuohuan Wang, Ying Shan, Jianhe Sun, Weihuan Fang, and Xiaoliang Li. 2019. "Npro of Classical Swine Fever Virus Suppresses Type III Interferon Production by Inhibiting IRF1 Expression and Its Nuclear Translocation" Viruses 11, no. 11: 998. https://doi.org/10.3390/v11110998
APA StyleCao, T., Li, X., Xu, Y., Zhang, S., Wang, Z., Shan, Y., Sun, J., Fang, W., & Li, X. (2019). Npro of Classical Swine Fever Virus Suppresses Type III Interferon Production by Inhibiting IRF1 Expression and Its Nuclear Translocation. Viruses, 11(11), 998. https://doi.org/10.3390/v11110998