Emergence of Southern Rice Black-Streaked Dwarf Virus in the Centuries-Old Chinese Yuanyang Agrosystem of Rice Landraces

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Material and Methods
2.1. Study Area
2.2. Plant Sampling
2.3. Genotyping-by-Sequencing of Rice Landraces
2.4. Analysis of Rice Genotyping-by-Sequencing Data
2.5. RNA Extraction and Detection of SRBSDV by Real-Time Quantitative PCR
2.6. Reverse-Transcription PCR, Partial SRBSDV Segment 8 Sequencing and Sequence Analysis
2.7. Phylogenetic Analysis
2.8. Statistical Analyses
3. Results and Discussion
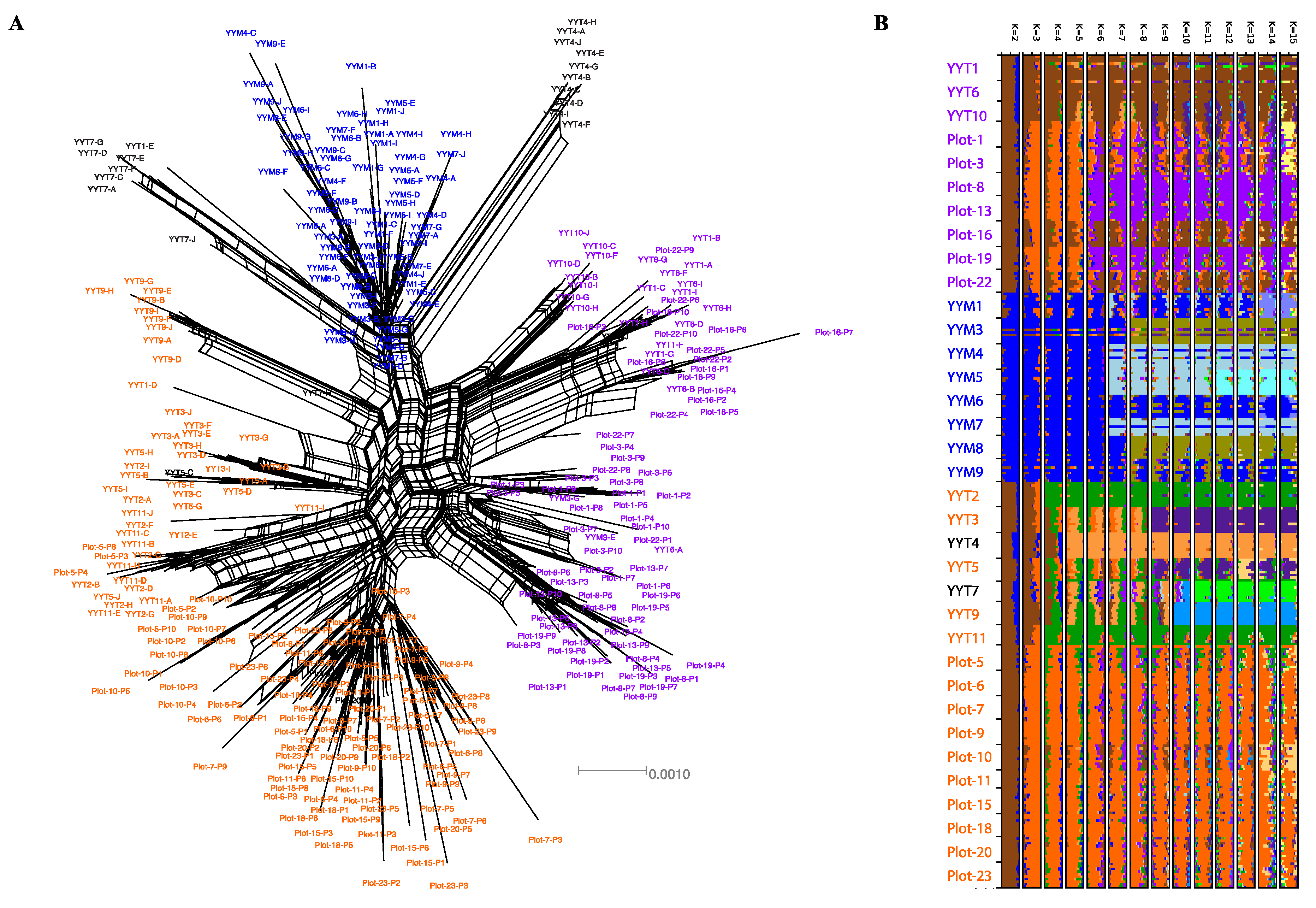
3.1. Highly Genetically Diverse TIL, HY and MH Varieties Are Cultivated in the HHRTS
3.2. SRBSDV Prevalence and Distribution
3.3. SRBSDV Genetic Diversity
3.4. The Emergence of SRBSDV and Long-Term HHRTS Sustainability
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Lv, M.F.; Xie, L.; Wang, H.F.; Wang, H.D.; Chen, J.P.; Zhang, H.M. Biology of Southern rice black-streaked dwarf virus: A novel fijivirus emerging in East Asia. Plant Pathol. 2017, 66, 515–521. [Google Scholar] [CrossRef]
- Zhou, G.; Xu, D.; Xu, D.; Zhang, M. Southern rice black-streaked dwarf virus: A white-backed planthopper-transmitted fijivirus threatening rice production in Asia. Front. Microbiol. 2013, 4, 270. [Google Scholar] [CrossRef] [PubMed]
- Hoang, A.T.; Zhang, H.M.; Yang, J.; Chen, J.P.; Hebrard, E.; Zhou, G.H.; Vien, V.N.; Cheng, J.A. Identification, Characterization, and Distribution of Southern rice black-streaked dwarf virus in Vietnam. Plant Dis. 2011, 95, 1063–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsukura, K.; Towata, T.; Sakai, J.; Onuki, M.; Okuda, M.; Matsumura, M. Dynamics of Southern rice black-streaked dwarf virus in Rice and Implication for Virus Acquisition. Phytopathology 2013, 103, 509–512. [Google Scholar] [CrossRef]
- Zhang, H.M.; Yang, J.; Chen, J.P.; Adams, M.J. A black-streaked dwarf disease on rice in China is caused by a novel fijivirus. Arch. Virol. 2008, 153, 1893–1898. [Google Scholar] [CrossRef]
- Zhou, Y.C.; Noussourou, M.; Kon, T.; Rojas, M.R.; Jiang, H.; Chen, L.F.; Gamby, K.; Foster, R.; Gilbertson, R.L. Evidence of local evolution of tomato-infecting begomovirus species in West Africa: Characterization of tomato leaf curl Mali virus and tomato yellow leaf crumple virus from Mali. Arch.Virol. 2008, 153, 693–706. [Google Scholar] [CrossRef]
- Wang, Q.A.; Yang, J.A.; Zhou, G.H.; Zhang, H.M.; Chen, J.P.; Adams, M.J. The Complete Genome Sequence of Two Isolates of Southern rice black-streaked dwarf virus, a New Member of the Genus Fijivirus. J. Phytopathol. 2010, 158, 733–737. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, X.; Wang, D.; Weng, J.; Di, H.; Zhang, L.; Dong, L.; Zhang, H.; Zu, H.; Li, X.; et al. Differences in Molecular Characteristics of Segment 8 in Rice black-streaked dwarf virus and Southern rice black-streaked dwarf virus. Plant Dis. 2018, 102, 1115–1123. [Google Scholar] [CrossRef]
- Bernardo, P.; Charles-Dominique, T.; Barakat, M.; Ortet, P.; Fernandez, E.; Filloux, D.; Hartnady, P.; Rebelo, T.A.; Cousins, S.R.; Mesleard, F.; et al. Geometagenomics illuminates the impact of agriculture on the distribution and prevalence of plant viruses at the ecosystem scale. ISME J. 2018, 12, 173–184. [Google Scholar] [CrossRef]
- Jiao, Y.M.; Li, X.Z.; Liang, L.H.; Takeuchi, K.; Okuro, T.; Zhang, D.D.; Sun, L.F. Indigenous ecological knowledge and natural resource management in the cultural landscape of China’s Hani Terraces. Ecol. Res. 2012, 27, 247–263. [Google Scholar] [CrossRef]
- Liao, J.; Huang, H.; Meusnier, I.; Adreit, H.; Ducasse, A.; Bonnot, F.; Pan, L.; He, X.; Kroj, T.; Fournier, E.; et al. Pathogen effectors and plant immunity determine specialization of the blast fungus to rice subspecies. eLife 2016, 5. [Google Scholar] [CrossRef]
- Yang, L.; Liu, M.C.; Lun, F.; Yuan, Z.; Zhang, Y.X.; Min, Q.W. An Analysis on Crops Choice and Its Driving Factors in Agricultural Heritage Systems-A Case of Honghe Hani Rice Terraces System. Sustainability 2017, 9. [Google Scholar] [CrossRef]
- Dedeurwaerdere, T.; Hannachi, M. Socio-economic drivers of coexistence of landraces and modern crop varieties in agro-biodiversity rich Yunnan rice fields. Ecol. Econ. 2019, 159, 177–188. [Google Scholar] [CrossRef]
- Ben Saad, R.; Fabre, D.; Mieulet, D.; Meynard, D.; Dingkuhn, M.; Al-Doss, A.; Guiderdoni, E.; Hassairi, A. Expression of the Aeluropus littoralis AlSAP gene in rice confers broad tolerance to abiotic stresses through maintenance of photosynthesis. Plant Cell Environ. 2012, 35, 626–643. [Google Scholar] [CrossRef] [PubMed]
- Risterucci, A.M.; Hippolyte, I.; Perrier, X.; Xia, L.; Caig, V.; Evers, M.; Huttner, E.; Kilian, A.; Glaszmann, J.C. Development and assessment of Diversity Arrays Technology for high-throughput DNA analyses in Musa. Tag. Theor. Appl. Genetics. Theor. Und Angew. Genet. 2009, 119, 1093–1103. [Google Scholar] [CrossRef] [PubMed]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef]
- Palanga, E.; Filloux, D.; Martin, D.P.; Fernandes, E.; Gargani, D.; Ferdinand, R.; Zabre, J.; Bouda, Z.; Neya, J.B.; Sawadogo, M.; et al. Metagenomic-based screening and molecular characterization of cowpea-infecting viruses in Burkina Faso. PLoS ONE 2016, 11, e0165188. [Google Scholar] [CrossRef]
- Wang, Z.C.; Yu, L.; Jin, L.H.; Wang, W.L.; Zhao, Q.; Ran, L.L.; Li, X.Y.; Chen, Z.; Guo, R.; Wei, Y.T.; et al. Evaluation of Rice Resistance to Southern Rice Black-Streaked Dwarf Virus and Rice Ragged Stunt Virus through Combined Field Tests, Quantitative Real-Time PCR, and Proteome Analysis. Viruses 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: a virus classification tool based on pairwise sequence alignment and identity calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Delsuc, F.; Dufayard, J.F.; Gascuel, O. Estimating maximum likelihood phylogenies with PhyML. Methods Mol. Biol. 2009, 537, 113–137. [Google Scholar] [CrossRef]
- Frichot, E.; Mathieu, F.; Trouillon, T.; Bouchard, G.; Francois, O. Fast and efficient estimation of individual ancestry coefficients. Genetics 2014, 196, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Carpi, G.; Holmes, E.C.; Kitchen, A. The evolutionary dynamics of bluetongue virus. J. Mol. Evol. 2010, 70, 583–592. [Google Scholar] [CrossRef]
- Hon, C.C.; Lam, T.Y.; Drummond, A.; Rambaut, A.; Lee, Y.F.; Yip, C.W.; Zeng, F.; Lam, P.Y.; Ng, P.T.; Leung, F.C. Phylogenetic analysis reveals a correlation between the expansion of very virulent infectious bursal disease virus and reassortment of its genome segment B. J. Virol. 2006, 80, 8503–8509. [Google Scholar] [CrossRef]
- Lu, Z.; Liu, H.; Fu, S.; Lu, X.; Dong, Q.; Zhang, S.; Tong, S.; Li, M.; Li, W.; Tang, Q.; et al. Liao ning virus in China. Virol. J. 2011, 8, 282. [Google Scholar] [CrossRef]
- Matthijnssens, J.; Heylen, E.; Zeller, M.; Rahman, M.; Lemey, P.; Van Ranst, M. Phylodynamic analyses of rotavirus genotypes G9 and G12 underscore their potential for swift global spread. Mol. Biol. Evol. 2010, 27, 2431–2436. [Google Scholar] [CrossRef]
- Stenger, D.C.; Sisterson, M.S.; French, R. Population genetics of Homalodisca vitripennis reovirus validates timing and limited introduction to California of its invasive insect host, the glassy-winged sharpshooter. Virology 2010, 407, 53–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, L.; Rang, C.U.; Wong, L.E. Distribution of spontaneous mutants and inferences about the replication mode of the RNA bacteriophage phi6. J. Virol. 2002, 76, 3276–3281. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Sampling Field | Variety Group | Year | Rice Variety Name | Village | Number of Samples | SRBSDV Positive Samples | SRBSDV Negative Samples |
|---|---|---|---|---|---|---|---|
| YYT1 | HY | 2016 | Chepugu | Malizhai | 9 | 5 | 4 |
| YYT6 | HY | 2016 | Chepugu | Malizhai | 9 | 4 | 5 |
| YYT10 | HY | 2016 | Jinpinggu | Malizhai | 10 | 4 | 6 |
| Plot_1 | HY | 2018 | Hongyang | Gingko | 10 | 1 | 9 |
| Plot_3 | HY | 2018 | Hongyang | Gingko | 10 | 3 | 7 |
| Plot_8 | HY | 2018 | Hongyang | Huangcaolin | 9 | 2 | 7 |
| Plot_13 | HY | 2018 | Hongyang | Xiaoshuijing | 10 | 0 | 10 |
| Plot_16 | HY | 2018 | Hongyang | Tuguozhai | 10 | 4 | 6 |
| Plot_19 | HY | 2018 | Hongyang | Shuibulong | 9 | 0 | 9 |
| Plot_22 | HY | 2018 | Hongyang | Dayutang | 10 | 6 | 4 |
| YYM1 | MH | 2016 | Mingliangyou 527 | Malizhai | 10 | 1 | 9 |
| YYM3 | MH | 2016 | Mingliangyou 528 | Malizhai | 8 | 1 | 7 |
| YYM4 | MH | 2016 | Hefeng 177 | Malizhai | 10 | 2 | 8 |
| YYM5 | MH | 2016 | Zhongyou 177 | Malizhai | 10 | 0 | 10 |
| YYM6 | MH | 2016 | Liangyou 2186 | Malizhai | 10 | 0 | 10 |
| YYM7 | MH | 2016 | Guofeng 1 | Malizhai | 10 | 0 | 10 |
| YYM8 | MH | 2016 | Liangyou 725 | Malizhai | 10 | 0 | 10 |
| YYM9 | MH | 2016 | Liangyou 2161 | Malizhai | 10 | 0 | 10 |
| YYT2 | TIL | 2016 | Lubaigu | Malizhai | 10 | 1 | 9 |
| YYT3 | TIL | 2016 | Zaogu | Malizhai | 10 | 0 | 10 |
| YYT5 | TIL | 2016 | Epugu | Malizhai | 10 | 1 | 9 |
| YYT9 | TIL | 2016 | Nuogu | Malizhai | 10 | 4 | 6 |
| YYT11 | TIL | 2016 | Luhonggu | Malizhai | 10 | 0 | 10 |
| Plot_5 | TIL | 2018 | Acuce | Gingko | 10 | 2 | 8 |
| Plot_6 | TIL | 2018 | Acuce | Gingko | 10 | 3 | 7 |
| Plot_7 | TIL | 2018 | Acuce | Huangcaolin | 9 | 1 | 8 |
| Plot_9 | TIL | 2018 | Acuce | Huangcaolin | 10 | 5 | 5 |
| Plot_10 | TIL | 2018 | Acuce | Xiaoshuijing | 10 | 0 | 10 |
| Plot_11 | TIL | 2018 | Acuce | Xiaoshuijing | 7 | 3 | 4 |
| Plot_15 | TIL | 2018 | Acuce | Tuguozhai | 10 | 0 | 10 |
| Plot_18 | TIL | 2018 | Acuce | Shuibulong | 9 | 0 | 9 |
| Plot_20 | TIL | 2018 | Acuce | Shuibulong | 10 | 2 | 8 |
| Plot_23 | TIL | 2018 | Acuce | Dayutang | 10 | 4 | 6 |
| YYT4 | Na (outlier) | 2016 | Nuogu | Malizhai | 10 | 0 | 10 |
| YYT7 | Na (outlier) | 2016 | Jianshuigu | Malizhai | 10 | 0 | 10 |
| YYT8 | japonica | 2016 | Honglueduolu | Malizhai | 10 | 0 | 10 |
| Plot_12 | japonica | 2018 | Unknown | Xiaoshuijing | 10 | 1 | 9 |
| Year | 2016 | 2018 | |||||
|---|---|---|---|---|---|---|---|
| Village | Malizhai (12.4%) | Xiaoshuijing (10.8%) | Tuguozhai (20%) | Shuibulong (7.1%) | Huangcaolin (28.6%) | Gingko (22.5%) | Dayutang (50%) |
| TIL (17.9%) | 6/50 (12%) | 3/17 (17.6%) | 0/10 | 2/19 (10.5%) | 6/19 (31.6%) | 5/20 (25%) | 4/10 (40%) |
| HY (30.2%) | 13/28 (46.4%) | 0/10 | 4/10 (40%) | 0/9 | 2/9 (22.2%) | 4/20 (20%) | 6/10 (60%) |
| MH (5.1%) | 4/78 (5.1%) | ||||||
| Na (outlier) | 0/20 | ||||||
| Japonica (5.3%) | 0/10 | 1/10 (11.1%) | |||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alonso, P.; Gladieux, P.; Moubset, O.; Shih, P.-J.; Mournet, P.; Frouin, J.; Blondin, L.; Ferdinand, R.; Fernandez, E.; Julian, C.; et al. Emergence of Southern Rice Black-Streaked Dwarf Virus in the Centuries-Old Chinese Yuanyang Agrosystem of Rice Landraces. Viruses 2019, 11, 985. https://doi.org/10.3390/v11110985
Alonso P, Gladieux P, Moubset O, Shih P-J, Mournet P, Frouin J, Blondin L, Ferdinand R, Fernandez E, Julian C, et al. Emergence of Southern Rice Black-Streaked Dwarf Virus in the Centuries-Old Chinese Yuanyang Agrosystem of Rice Landraces. Viruses. 2019; 11(11):985. https://doi.org/10.3390/v11110985
Chicago/Turabian StyleAlonso, Pascal, Pierre Gladieux, Oumaima Moubset, Pei-Jung Shih, Pierre Mournet, Julien Frouin, Laurence Blondin, Romain Ferdinand, Emmanuel Fernandez, Charlotte Julian, and et al. 2019. "Emergence of Southern Rice Black-Streaked Dwarf Virus in the Centuries-Old Chinese Yuanyang Agrosystem of Rice Landraces" Viruses 11, no. 11: 985. https://doi.org/10.3390/v11110985
APA StyleAlonso, P., Gladieux, P., Moubset, O., Shih, P.-J., Mournet, P., Frouin, J., Blondin, L., Ferdinand, R., Fernandez, E., Julian, C., Filloux, D., Adreit, H., Fournier, E., Ducasse, A., Grosbois, V., Morel, J.-B., Huang, H., Jin, B., He, X., ... Roumagnac, P. (2019). Emergence of Southern Rice Black-Streaked Dwarf Virus in the Centuries-Old Chinese Yuanyang Agrosystem of Rice Landraces. Viruses, 11(11), 985. https://doi.org/10.3390/v11110985