A Toxicity Screening Approach to Identify Bacteriophage-Encoded Anti-Microbial Proteins

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains, Plasmids, Bacteriophages and Culture Conditions
2.2. Mass Spectrometry (MS) Data Analysis of Phage Proteins
2.3. Comparative Protein Analysis of Hypothetical Proteins of Unknown Function (HPUFs) and Genome Annotation
2.4. PCR Amplification of Control and Hypothetical Proteins of Unknown Function Genes for Cloning
2.5. Cloning
2.6. Screening for Toxic Hypothetical Proteins of Unknown Function Genes
2.7. Confirmation of Protein Toxicity
2.8. Structure Prediction of Toxic HPUFs
2.9. Statistics
3. Results
3.1. Elimination of Hypothetical Proteins of Unknown Function (HPUFs) by LC-MS/MS Analysis of Phage Particle Proteomes
3.2. Further Elimination of Hypothetical Proteins of Unknown Function (HPUFs) by BLASTp and HHpred Analyses
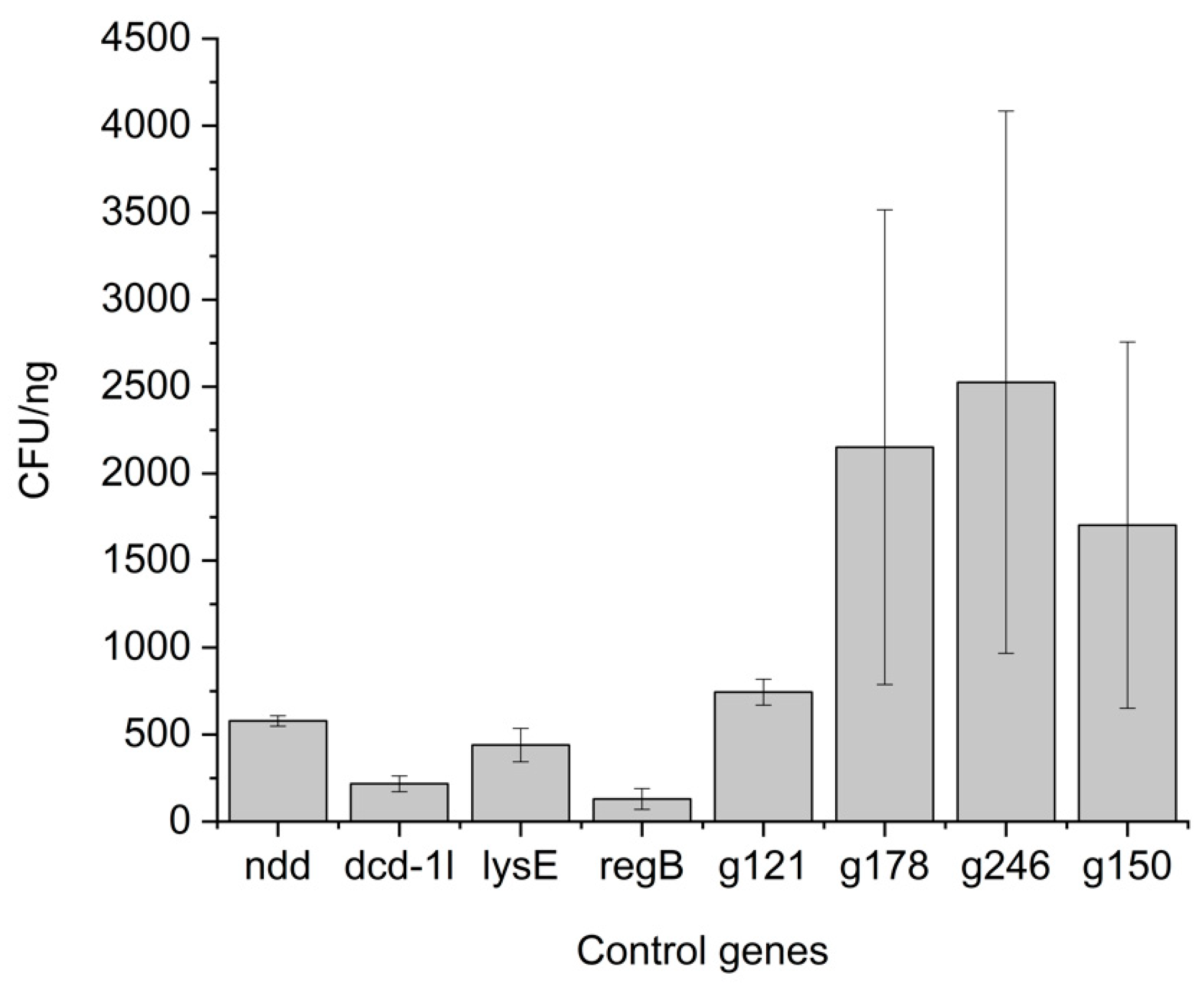
3.3. Ligations of Toxic and Non-Toxic Control Genes Show Differences in Plating Efficiency
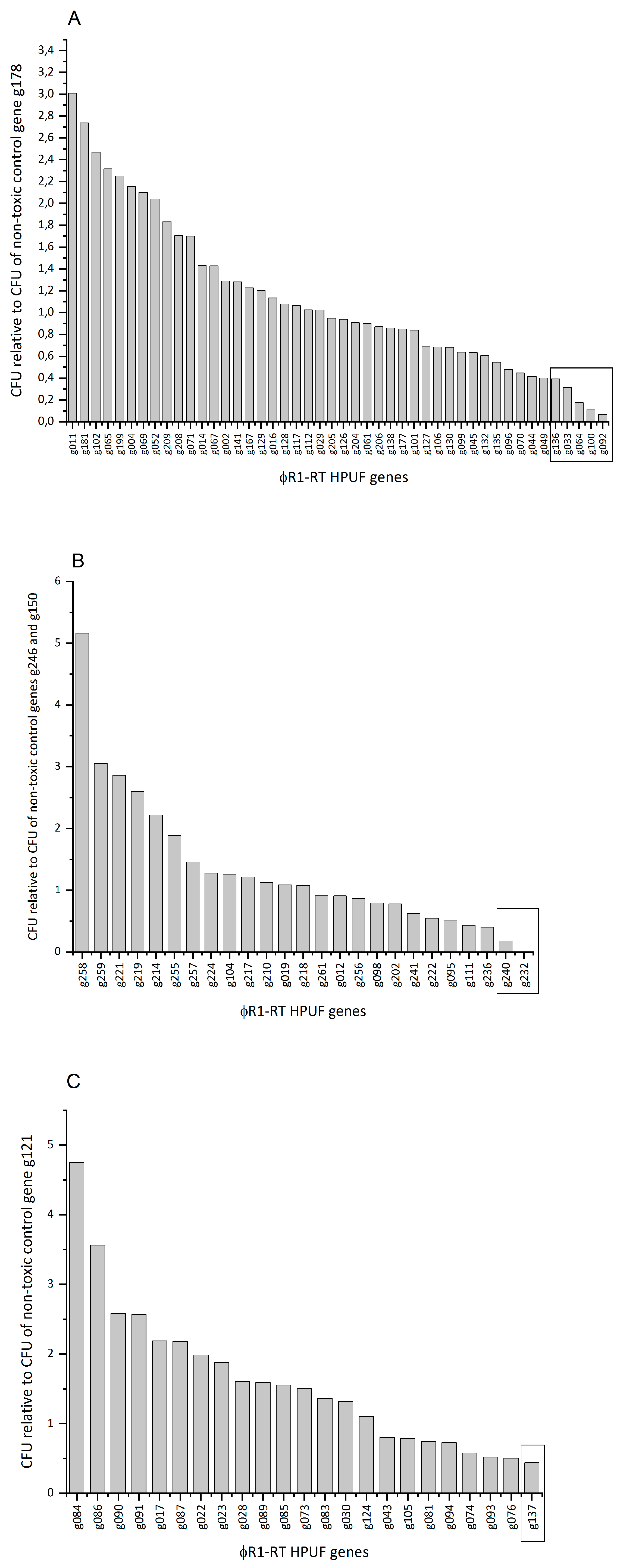
3.4. Eight Potential Toxic Protein Hits Identified from an Initial CFU-Based Screening Approach
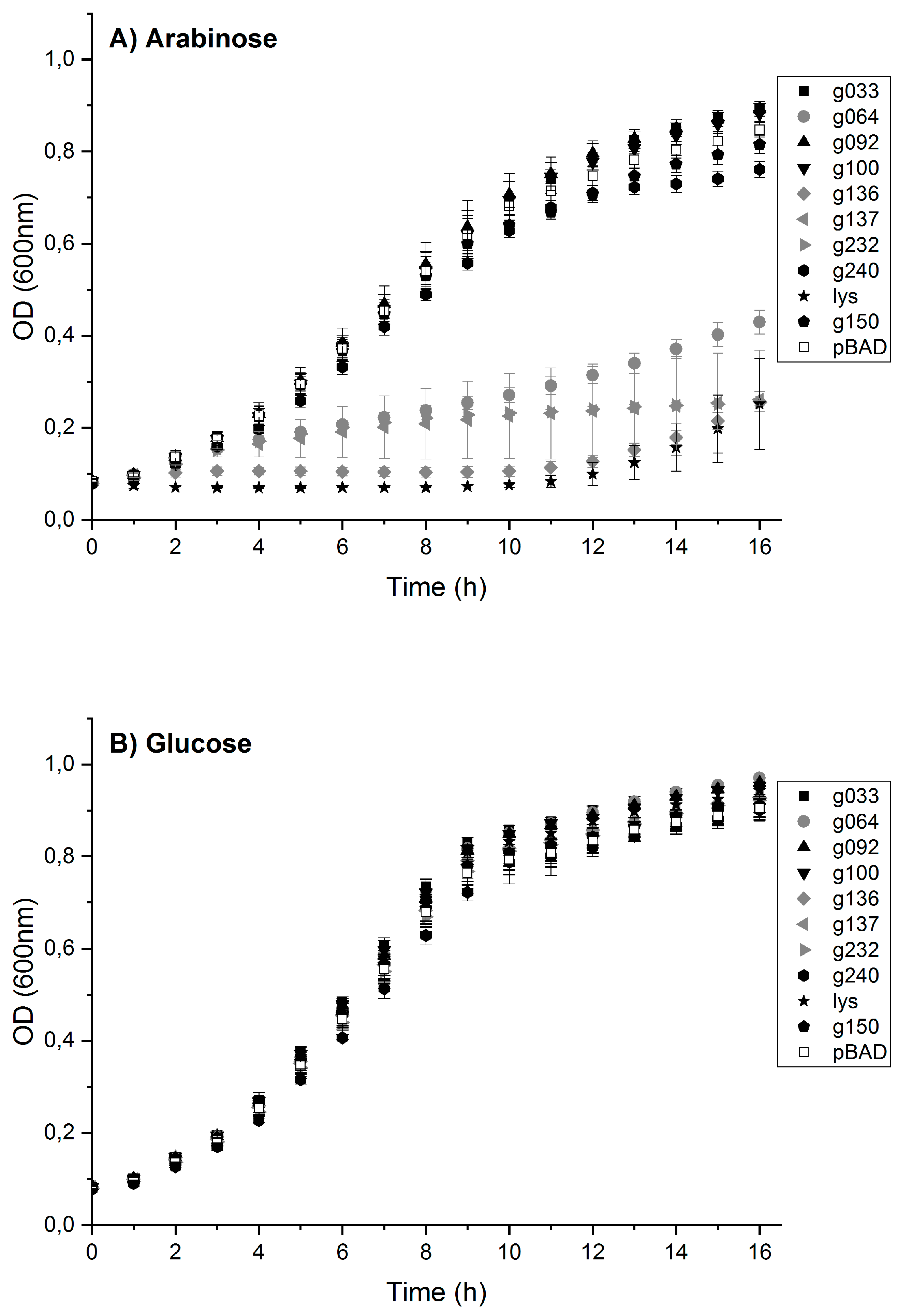
3.5. Toxic Protein Hits Gp064, Gp136, Gp137 and Gp232 Inhibit the Growth of E. coli Cells
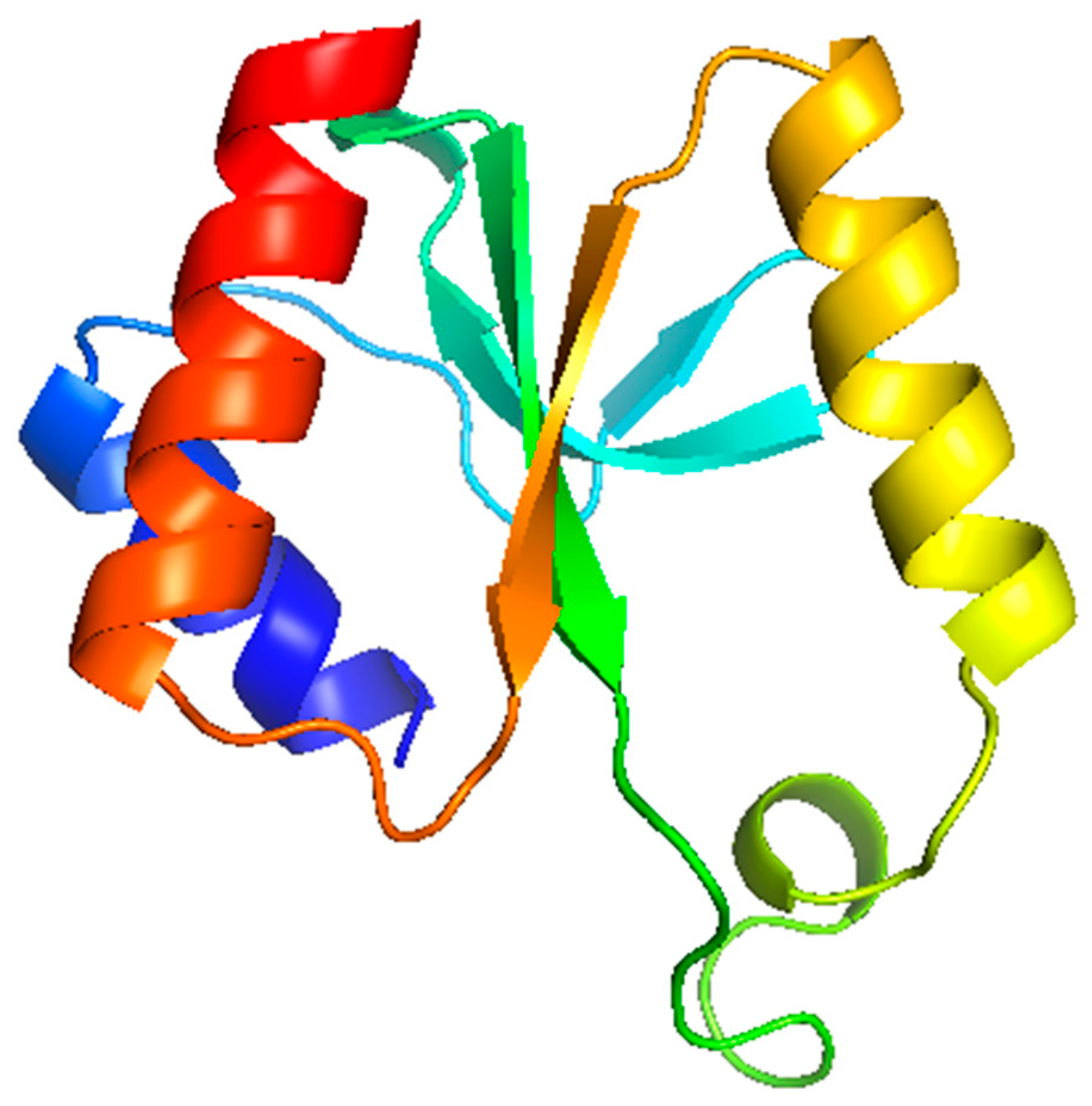
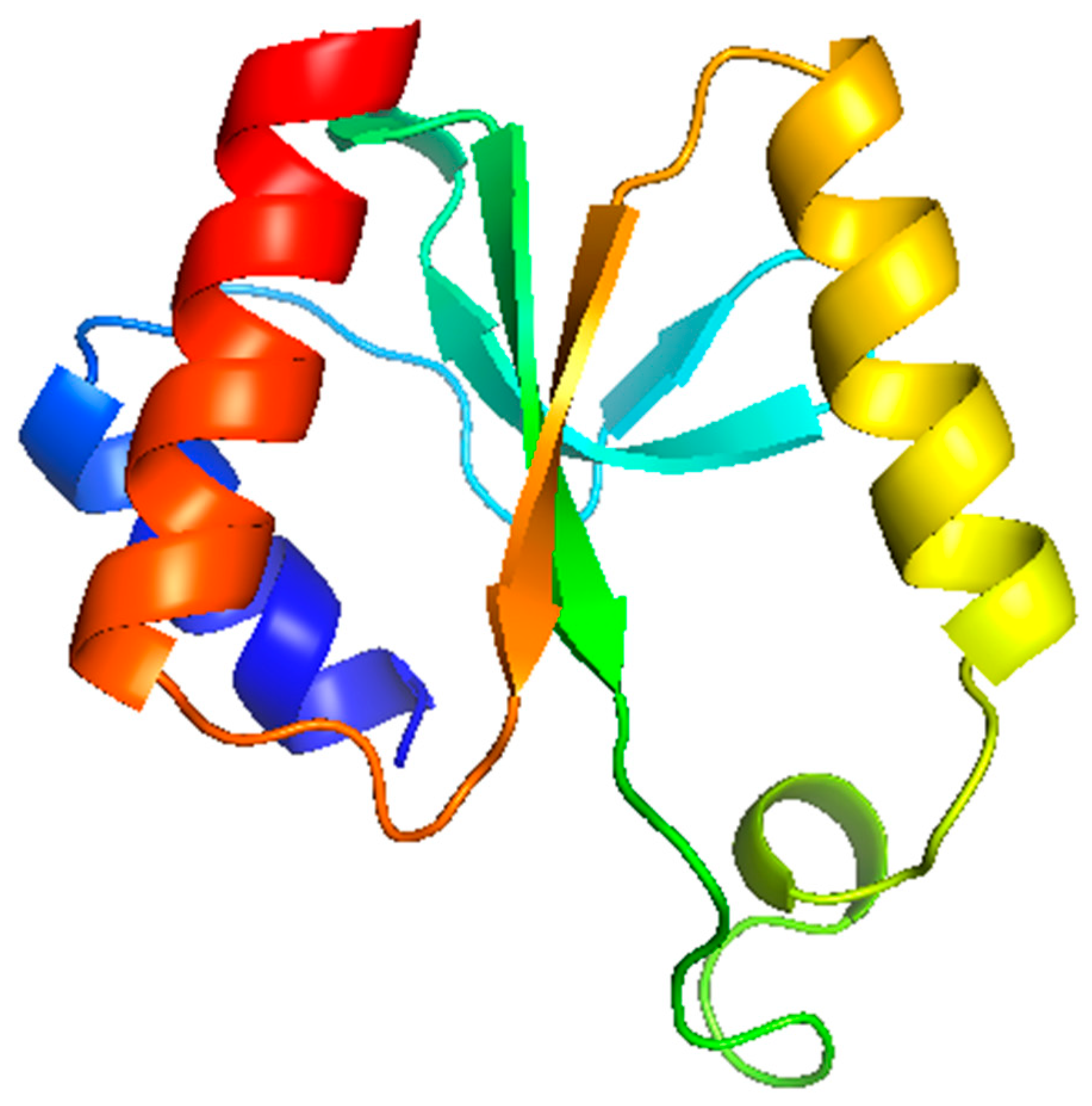
3.6. Modelling Studies
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Prestinaci, F.; Pezzotti, P.; Pantosti, A. Antimicrobial resistance: A global multifaceted phenomenon. Pathog. Glob. Health 2015, 109, 309–318. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the last 25 years. J. Nat. Prod. 2007, 70, 461–477. [Google Scholar] [CrossRef]
- Roach, D.R.; Donovan, D.M. Antimicrobial bacteriophage-derived proteins and therapeutic applications. Bacteriophage 2015, 5, e1062590. [Google Scholar] [CrossRef] [PubMed]
- Abedon, S.T.; Thomas-Abedon, C.; Thomas, A.; Mazure, H. Bacteriophage prehistory: Is or is not Hankin, 1896, a phage reference? Bacteriophage 2011, 1, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Young, R. Bacteriophage lysis: Mechanism and regulation. Microbiol. Rev. 1992, 56, 430–481. [Google Scholar] [PubMed]
- Shahbaaz, M.; Hassan, M.I.; Ahmad, F. Functional annotation of conserved hypothetical proteins from Haemophilus influenzae Rd KW20. PLoS ONE 2013, 8, e84263. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Dehbi, M.; Moeck, G.; Arhin, F.; Bauda, P.; Bergeron, D.; Callejo, M.; Ferretti, V.; Ha, N.; Kwan, T.; et al. Antimicrobial drug discovery through bacteriophage genomics. Nat. Biotechnol. 2004, 22, 185–191. [Google Scholar] [CrossRef]
- Shibayama, Y.; Dabbs, E.R. Phage as a source of antibacterial genes: Multiple inhibitory products encoded by Rhodococcus phage YF1. Bacteriophage 2011, 1, 195–197. [Google Scholar] [CrossRef]
- Van den Bossche, A.; Ceyssens, P.J.; De Smet, J.; Hendrix, H.; Bellon, H.; Leimer, N.; Wagemans, J.; Delattre, A.-S.; Cenens, W.; Aertsen, A.; et al. Systematic identification of hypothetical bacteriophage proteins targeting key protein complexes of Pseudomonas aeruginosa. J. Proteome Res. 2014, 13, 4446–4456. [Google Scholar] [CrossRef]
- Klimke, W.; O’Donovan, C.; White, O.; Brister, J.R.; Clark, K.; Fedorov, B.; Mizrachi, I.; Pruitt, K.D.; Tatusova, T. Solving the problem: Genome annotation standards before the data deluge. Stand. Genom. Sci. 2011, 5, 168–193. [Google Scholar] [CrossRef]
- Leon-Velarde, C.G.; Happonen, L.; Pajunen, M.; Leskinen, K.; Kropinski, A.M.; Mattinen, L.; Rajtor, M.; Zur, J.; Smith, D.; Chen, S.; et al. Yersinia enterocolitica-specific infection by bacteriophages TG1 and ϕR1-RT is dependent on temperature-regulated expression of the phage host receptor OmpF. Appl. Environ. Microbiol. 2016, 82, 5340–5353. [Google Scholar] [CrossRef] [PubMed]
- Fàbrega, A.; Vila, J. Yersinia enterocolitica: Pathogenesis, virulence and antimicrobial resistance. Enferm. Infecc. Microbiol. Clin. 2012, 30, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Lamberg, A.; Nieminen, S.; Qiao, M.; Savilahti, H. Efficient insertion mutagenesis strategy for bacterial genomes involving electroporation of in vitro-assembled DNA transposition complexes of bacteriophage Mu. Appl. Environ. Microbiol. 2002, 68, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Pirhonen, M.; Heino, P.; Helander, I.; Harju, P.; Palva, E.T. Bacteriophage T4 resistant mutants of the plant pathogen Erwinia carotovora. Microb. Pathog. 1988, 4, 359–367. [Google Scholar] [CrossRef]
- Mohanraj, U.; Kinnunen, O.; Kaya, M.E.; Aranko, A.S.; Viskari, H.; Linder, M. SUMO-based expression and purification of dermcidin-derived DCD-1L, a human antimicrobial peptide, in Escherichia coli. BioRxiv 2018, 343418. [Google Scholar] [CrossRef]
- Guzman, L.M.; Belin, D.; Carson, M.J.; Beckwith, J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 1995, 177, 4121–4130. [Google Scholar] [CrossRef]
- Leskinen, K.; Tuomala, H.; Wicklund, A.; Horsma-Heikkinen, J.; Kuusela, P.; Skurnik, M.; Kiljunen, S. Characterization of vB_SauM-fRuSau02, a Twort-like bacteriophage isolated from a therapeutic phage cocktail. Viruses 2017, 9, 258. [Google Scholar] [CrossRef]
- Gish, W.; States, D.J. Identification of protein coding regions by database similarity search. Nat. Genet. 1993, 3, 266–272. [Google Scholar] [CrossRef]
- Soding, J.; Biegert, A.; Lupas, A.N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005, 33, W244–W248. [Google Scholar] [CrossRef]
- Chuang, L.-Y.; Cheng, Y.-H.; Yang, C.-H. Specific primer design for the polymerase chain reaction. Biotechnol. Lett. 2013, 35, 1541–1549. [Google Scholar] [CrossRef]
- Institute for Molecular Medicine Finland Technology Centre. Sequencing Unit. Available online: https://www.fimm.fi/en/services/technology-centre/sequencing/ (accessed on 17 June 2019).
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protocol. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Hatfull, G.F.; Pedulla, M.L.; Jacobs-Sera, D.; Cichon, P.M.; Foley, A.; Ford, M.E.; Gonda, R.M.; Houtz, J.M.; Hryckowian, A.J.; Kelchner, V.A.; et al. Exploring the mycobacteriophage metaproteome: Phage genomics as an educational platform. PLoS Genet. 2006, 2, e92. [Google Scholar] [CrossRef] [PubMed]
- Kwan, T.; Liu, J.; DuBow, M.; Gros, P.; Pelletier, J. The complete genomes and proteomes of 27 Staphylococcus aureus bacteriophages. Proc. Natl. Acad. Sci. USA 2005, 102, 5174–5179. [Google Scholar] [CrossRef] [PubMed]
- Howard-Varona, C.; Roux, S.; Dore, H.; Solonenko, N.E.; Holmfeldt, K.; Markillie, L.M.; Orr, G.; Sullivan, M.B. Regulation of infection efficiency in a globally abundant marine Bacteriodetes virus. ISME J. 2017, 11, 284–295. [Google Scholar] [CrossRef]
- Stevens, R.H.; Zhang, H.; Hsiao, C.; Kachlany, S.; Tinoco, E.M.B.; DePew, J.; Fouts, D.E. Structural proteins of Enterococcus faecalis bacteriophage φEf11. Bacteriophage 2016, 6, e1251381. [Google Scholar] [CrossRef]
- Pickard, D.J.J. Preparation of bacteriophage lysates and pure DNA. In Bacteriophages; Springer: Berlin/Heidelberg, Germany, 2009; pp. 3–9. [Google Scholar]
- Thomas, J.A.; Quintana, A.D.B.; Bosch, M.A.; De Peña, A.C.; Aguilera, E.; Coulibaly, A.; Wu, W.; Osier, M.V.; Hudson, A.O.; Weintraub, S.T. Identification of essential genes in the Salmonella phage SPN3US reveals novel insights into giant phage head structure and assembly. J. Virol. 2016, 90, 10284–10298. [Google Scholar] [CrossRef]
- Yang, H.; Ma, Y.; Wang, Y.; Yang, H.; Shen, W.; Chen, X. Transcription regulation mechanisms of bacteriophages: Recent advances and future prospects. Bioengineered 2014, 5, 300–304. [Google Scholar] [CrossRef]
- Miller, E.S.; Kutter, E.; Mosig, G.; Arisaka, F.; Kunisawa, T.; Rüger, W. Bacteriophage T4 genome. Microbiol. Mol. Biol. Rev. 2003, 67, 86–156. [Google Scholar] [CrossRef]
- Singh, S.; Godavarthi, S.; Kumar, A.; Sen, R. A mycobacteriophage genomics approach to identify novel mycobacteriophage proteins with mycobactericidal properties. Microbiology 2019, 165, 722–736. [Google Scholar] [CrossRef]
- Preston, A. Choosing a cloning vector. In E. coli Plasmid Vectors: Methods and Applications; Casali, N., Preston, A., Eds.; Humana Press: Totowa, NJ, USA, 2003; pp. 19–26. [Google Scholar]
- Kutter, E.; Raya, R.; Carlson, K. Molecular mechanisms of phage infection. In Bacteriophages: Biology and Applications, 1st ed.; Kutter, E., Sulakvelidze, A., Eds.; CRC Press: Boca Raton, FL, USA, 2004; pp. 165–222. [Google Scholar]
- Khakhum, N.; Yordpratum, U.; Boonmee, A.; Tattawasart, U.; Rodrigues, J.L.M.; Sermswan, R.W. Identification of the Burkholderia pseudomallei bacteriophage ST79 lysis gene cassette. J. Appl. Microbiol. 2016, 121, 364–372. [Google Scholar] [CrossRef]
- Shi, Y.; Li, N.; Yan, Y.; Wang, H.; Li, Y.; Lu, C.; Sun, J. Combined antibacterial activity of phage lytic proteins holin and lysin from Streptococcus suis bacteriophage SMP. Curr. Microbiol. 2012, 65, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Kinch, L.N.; Ginalski, K.; Rychlewski, L.; Grishin, N.V. Identification of novel restriction endonuclease-like fold families among hypothetical proteins. Nucleic Acids Res. 2005, 33, 3598–3605. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Description | Source |
|---|---|---|
| E. coli strains | ||
| DH10B | Used for HPUFs screening | NEB, USA |
| DH5α | Used for pBAD30 cloning | NEB, USA |
| Bacteriophages | ||
| ϕR1-RT | Used as a template for amplifying all the ϕR1-RT encoded genes used in the current study | [11] |
| T4 | Used as a template for amplifying regB and ndd control genes | [14] |
| Plasmids | ||
| pETSmt3- DCD-1L | Used as template to amplify the dcd-1l control gene | [15] |
| pU11L4 | This plasmid consists of pUC19 with a KpnI-PstI linker in the MCS and luxAB genes under expression of ompF promoter of YeO:3 at the SapI site (Figure S1. | This study |
| ϕX174 RF1 | Used as template to amplify the lysE control gene | Thermo Fisher Scientific, USA |
| pBAD30 | Plasmid with arabinose-inducible promoter to express ϕR1-RT HPUFs in E. coli for confirmatory assay | [16] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohanraj, U.; Wan, X.; Spruit, C.M.; Skurnik, M.; Pajunen, M.I. A Toxicity Screening Approach to Identify Bacteriophage-Encoded Anti-Microbial Proteins. Viruses 2019, 11, 1057. https://doi.org/10.3390/v11111057
Mohanraj U, Wan X, Spruit CM, Skurnik M, Pajunen MI. A Toxicity Screening Approach to Identify Bacteriophage-Encoded Anti-Microbial Proteins. Viruses. 2019; 11(11):1057. https://doi.org/10.3390/v11111057
Chicago/Turabian StyleMohanraj, Ushanandini, Xing Wan, Cindy M. Spruit, Mikael Skurnik, and Maria I. Pajunen. 2019. "A Toxicity Screening Approach to Identify Bacteriophage-Encoded Anti-Microbial Proteins" Viruses 11, no. 11: 1057. https://doi.org/10.3390/v11111057