Viruses in the Invasive Hornet Vespa velutina

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Asian Hornet and Honey Bee Sampling
2.2. RNA Extraction
2.3. Next Generation Sequencing (NGS)
2.4. General Analysis of Viral Content
2.5. Analysis of Viral Genomes
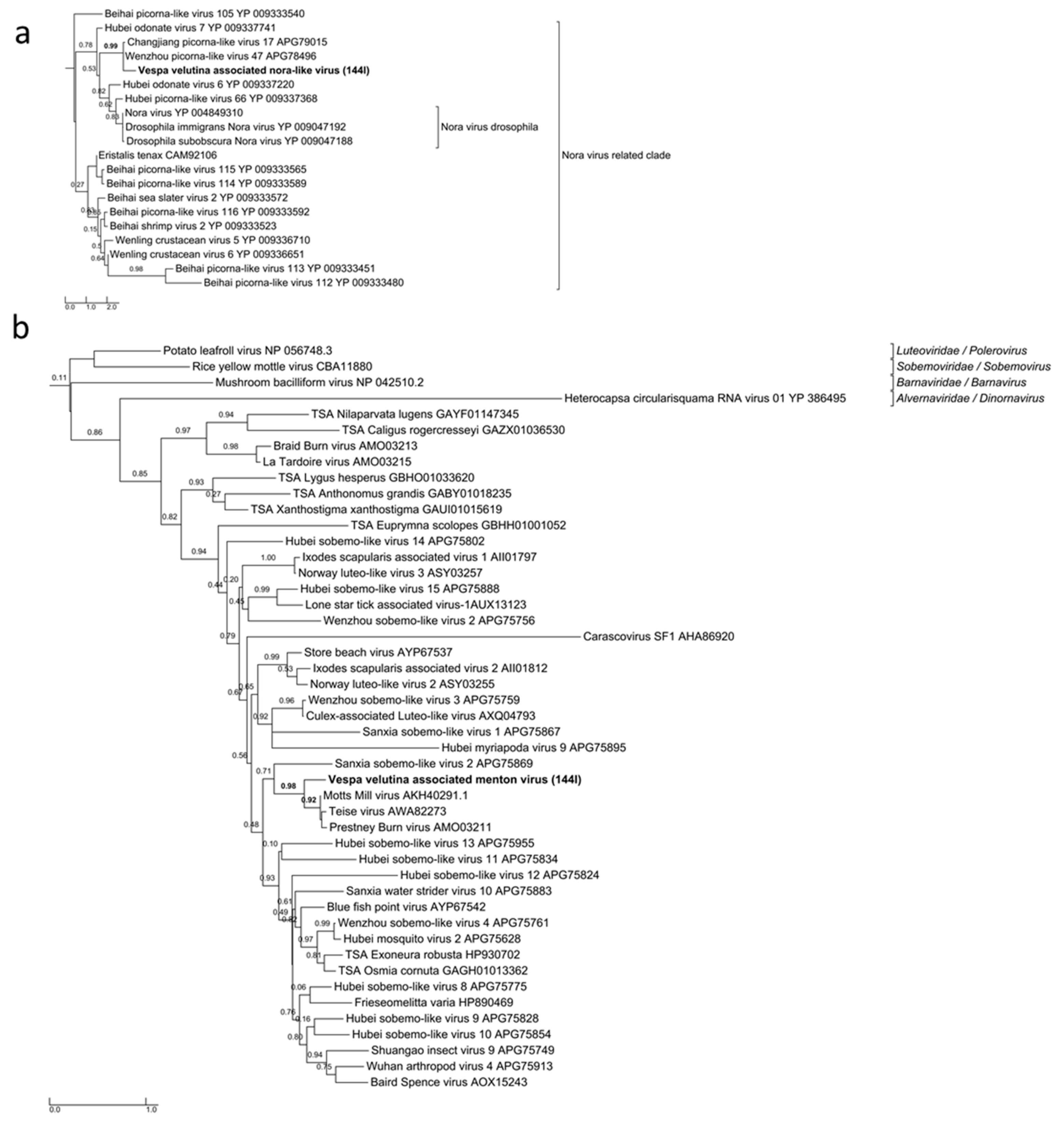
2.6. Phylogenetic Analysis
2.7. RT-PCR
3. Results
3.1. Virus Discovery from RNAseq Experiments
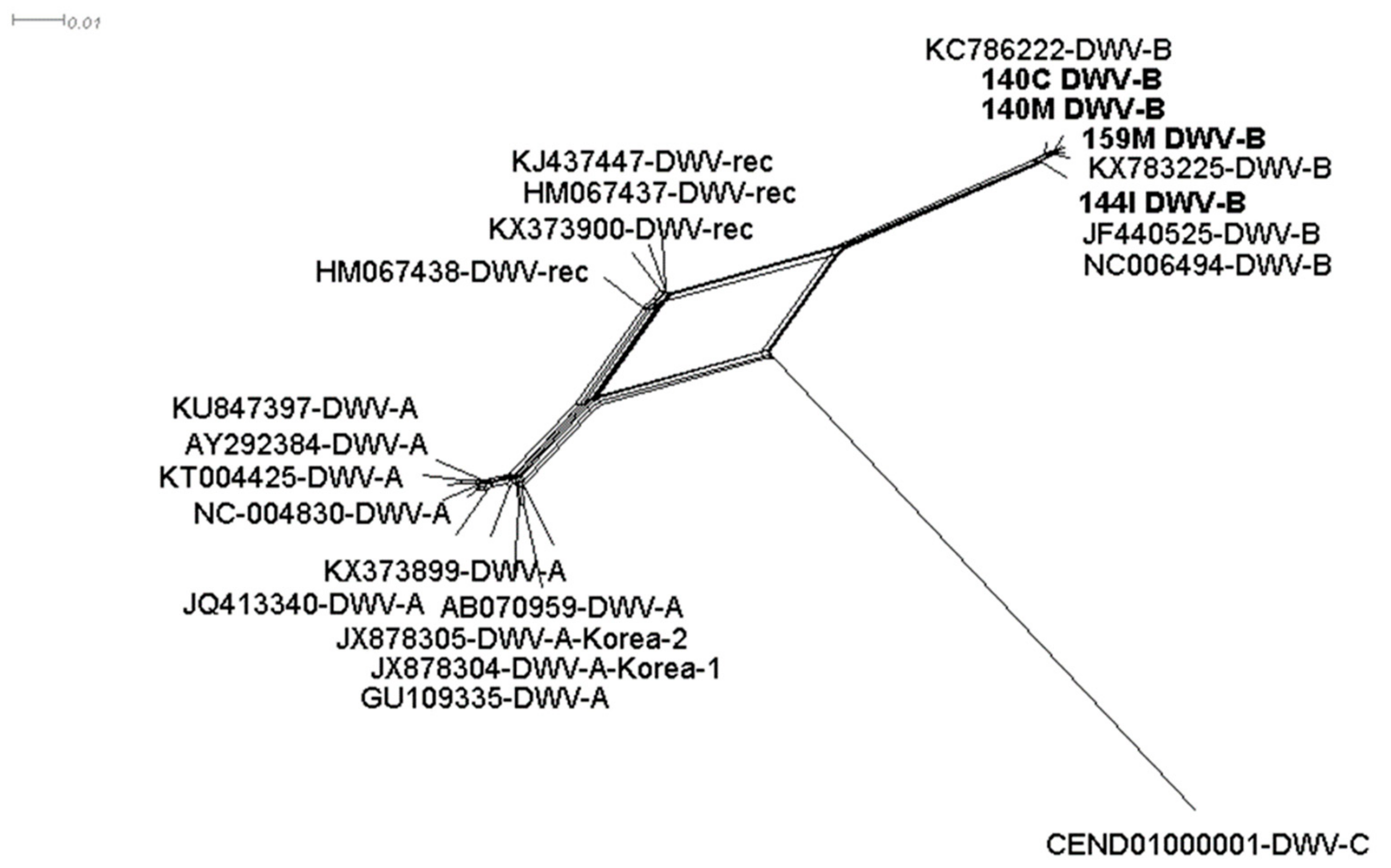
3.2. DWV-B is Highly Prevalent in Symptomatic and Asymptomatic Hornets
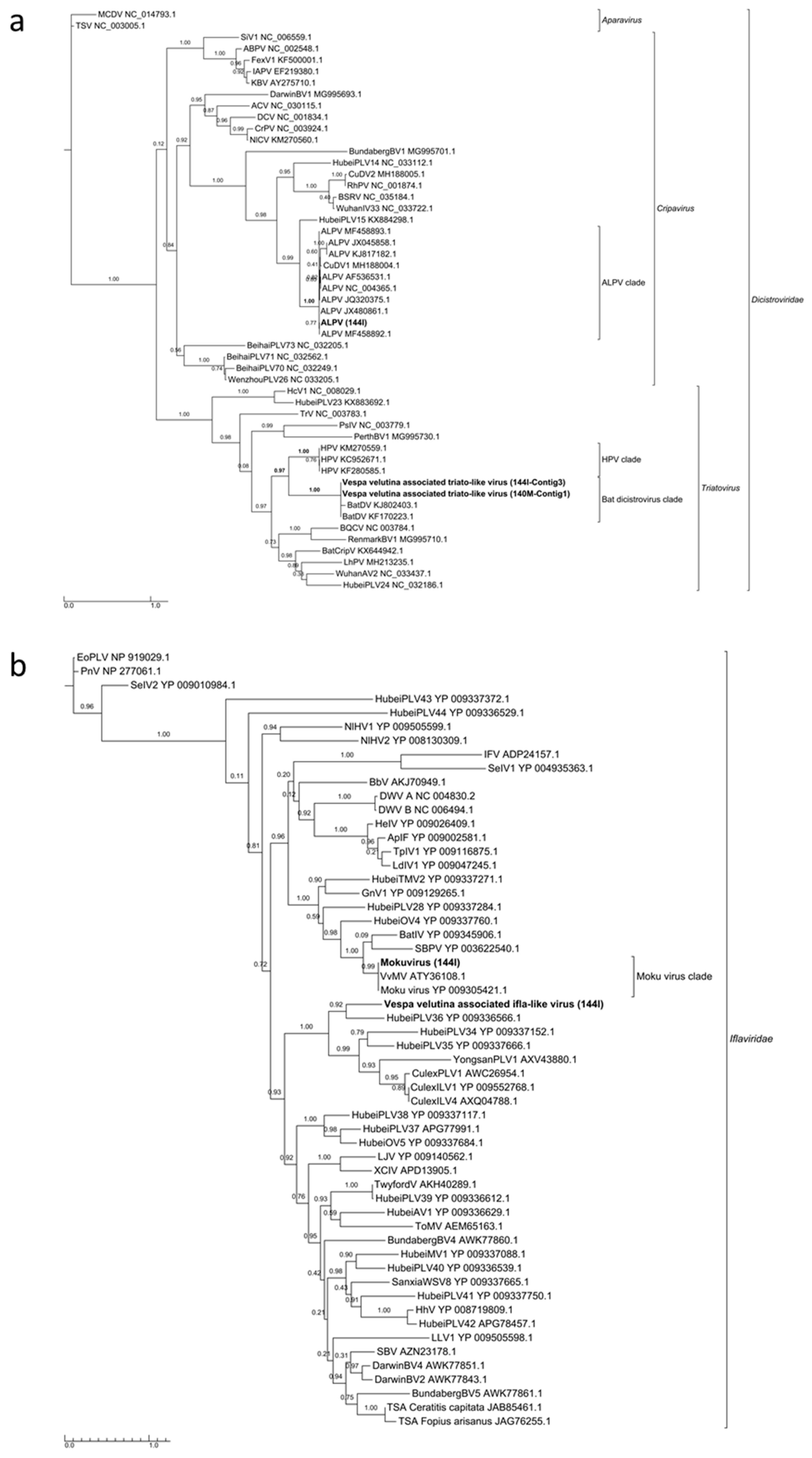
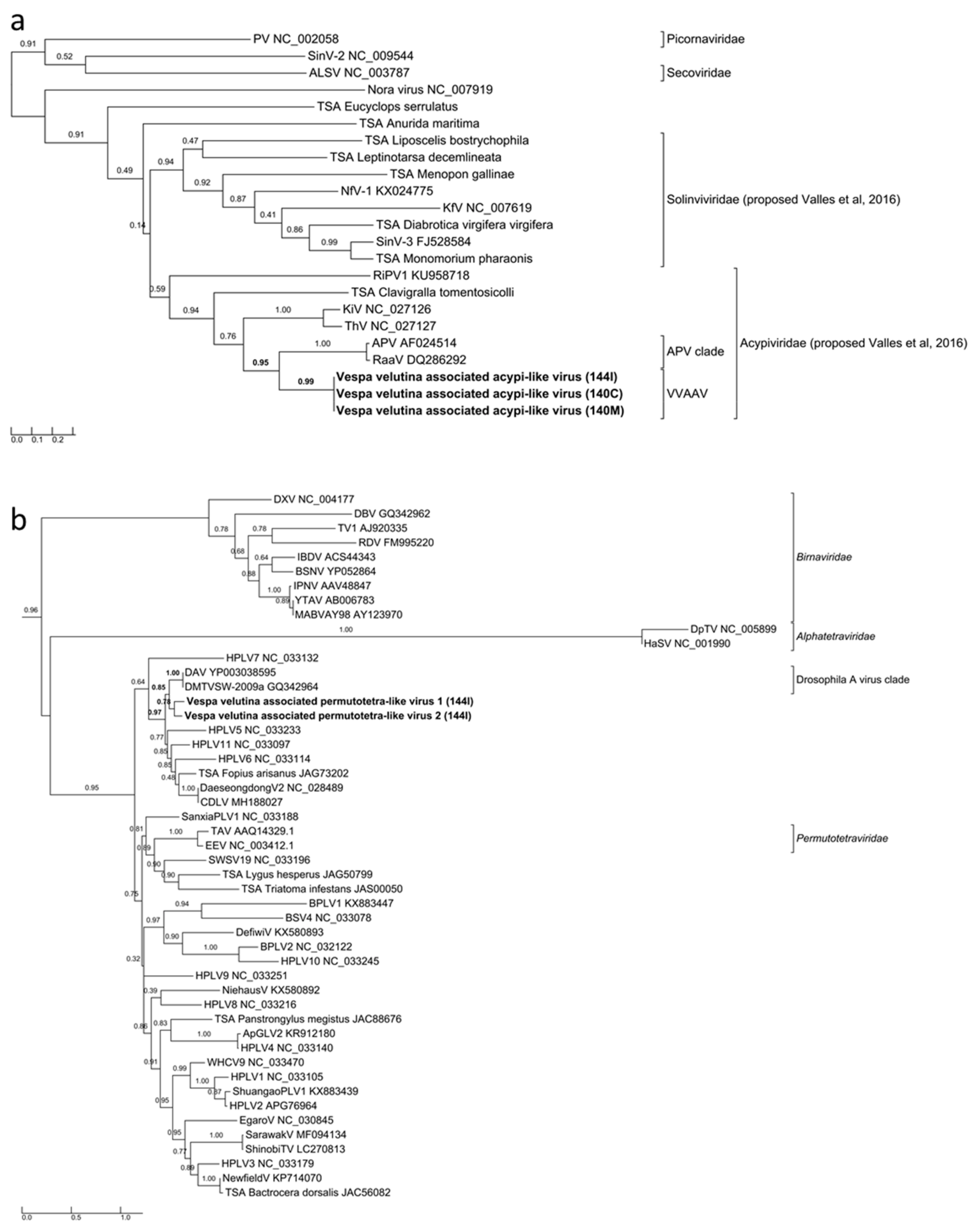
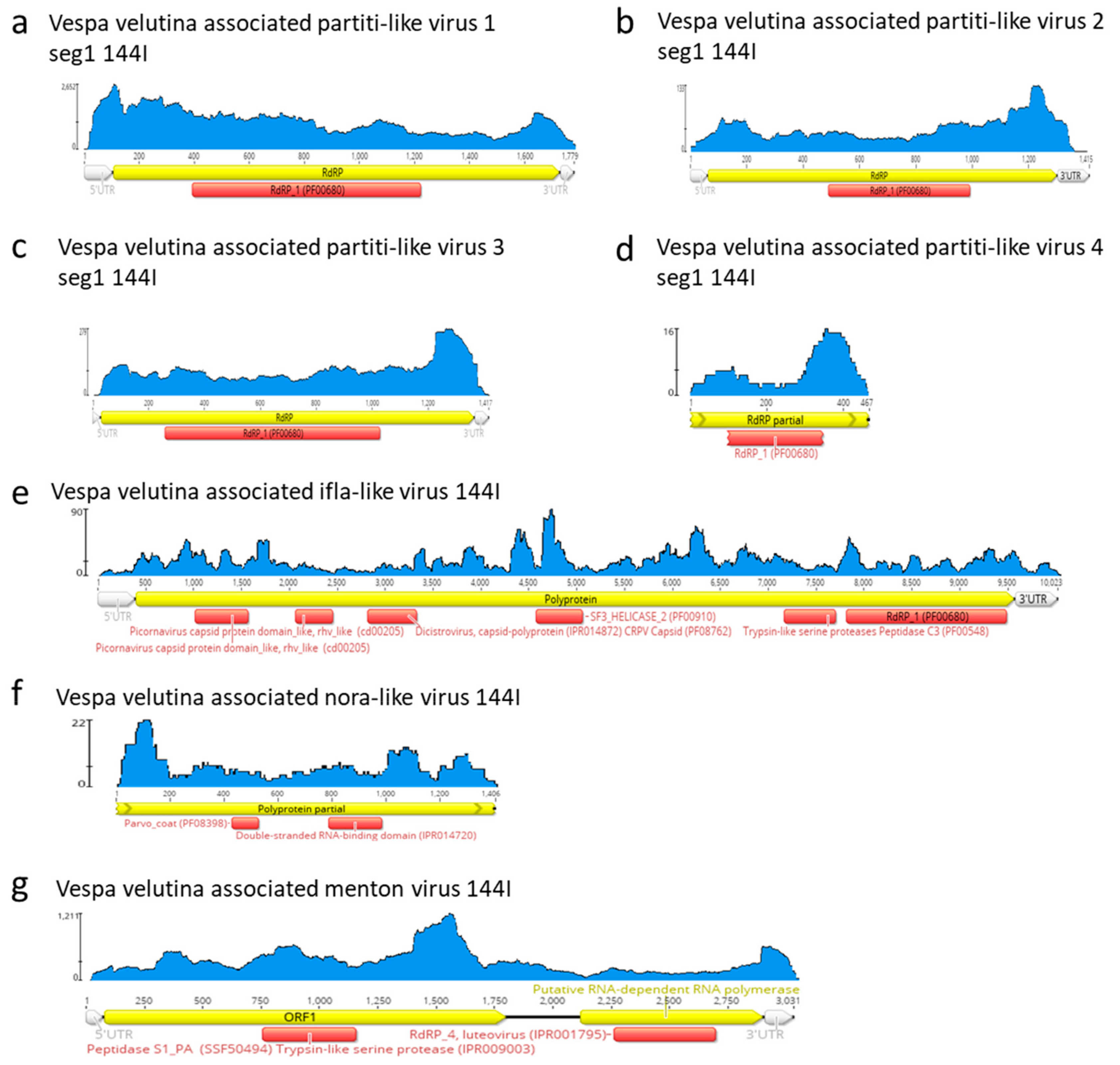
3.3. Six Other Bee-Associated Viruses Detected in Hornets by RNAseq
3.4. Eleven Other Insect Viruses Detected in Hornets from RNAseq
3.5. Viral Polymorphism Study Confirmed Genuine Hornet Infections
3.6. Distribution of Bee-Associated Viruses in Hornets Detected by Multiplex RT-PCR
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Villemant, C.; Barbet-Massin, M.; Perrard, A.; Muller, F.; Gargominy, O.; Jiguet, F.; Rome, Q. Predicting the invasion risk by the alien bee-hawking yellow-legged hornet Vespa velutina nigrithorax across Europe and other continents with niche models. Biol. Conserv. 2011, 144, 2142–2150. [Google Scholar] [CrossRef]
- Haxaire, J.; Bouguet, J.-P.; Jean-Philippe, T. Vespa velutina Lepeletier, 1836, une redoutable nouveauté pour la faune de France (Hym., Vespidae). Bull. Soc. Entomol. Fr. 2006, 111, 194. [Google Scholar]
- Villemant, C.; Haxaire, J.; Streito, J.-C. La découverte du frelon asiatique Vespa velutina, en France. Insectes 2006, 143, 3–7. [Google Scholar]
- Arca, M.; Capdevielle-Dulac, C.; Villemant, C.; Mougel, F.; Arnold, G.; Silvain, J.F. Development of microsatellite markers for the yellow-legged Asian hornet, Vespa velutina, a major threat for European bees. Conserv. Genet. Resour. 2012, 4, 283–286. [Google Scholar] [CrossRef]
- Arca, M.; Mougel, F.; Guillemaud, T.; Dupas, S.; Rome, Q.; Perrard, A.; Muller, F.; Fossoud, A.; Capdevielle-Dulac, C.; Torres-Leguizamon, M.; et al. Reconstructing the invasion and the demographic history of the yellow-legged hornet, Vespa velutina, in Europe. Biol. Invasions 2015, 17, 2357–2371. [Google Scholar] [CrossRef]
- Rome, Q.; Muller, F.J.; Touret-Alby, A.; Darrouzet, E.; Perrard, A.; Villemant, C. Caste differentiation and seasonal changes in Vespa velutina (Hym.: Vespidae) colonies in its introduced range. J. Appl. Entomol. 2015, 139, 771–782. [Google Scholar] [CrossRef]
- Darrouzet, E.; Gevar, J.; Guignard, Q.; Aron, S. Production of early diploid males by European colonies of the invasive hornet Vespa velutina nigrithorax. PLoS ONE 2015, 10, e0136680. [Google Scholar] [CrossRef]
- Rome, Q.; Dambrine, L.; Onate, C.; Muller, F.; Villemant, C.; Garcia Pérez, A.L.; Maia, M.; Carvalho Esteves, P.; Bruneau, E. Spread of the invasive hornet Vespa velutina Lepeletier, 1836, in Europe. Bull. Soc. Entomol. Fr. 2013, 118, 21–22. [Google Scholar]
- Monceau, K.; Bonnard, O.; Moreau, J.; Thiery, D. Spatial distribution of Vespa velutina individuals hunting at domestic honeybee hives: Heterogeneity at a local scale. Insect Sci. 2014, 21, 765–774. [Google Scholar] [CrossRef]
- Bertolino, S.; Lioy, S.; Laurino, D.; Manino, A.; Porporato, M. Spread of the invasive yellow-legged hornet Vespa velutina (Hymenoptera: Vespidae) in Italy. Appl. Entomol. Zool. 2016, 51, 589–597. [Google Scholar] [CrossRef]
- Barbet-Massin, M.; Rome, Q.; Muller, F.; Perrard, A.; Villemant, C.; Jiguet, F. Climate change increases the risk of invasion by the yellow-legged hornet. Biol. Conserv. 2013, 157, 4–10. [Google Scholar] [CrossRef]
- Goldarazena, A.; de Heredia, I.P.; Romon, P.; Iturrondobeitia, J.C.; Gonzalez, M.; Lopez, S. Spread of the yellow-legged hornet Vespa velutina nigrithorax du Buysson (Hymenoptera: Vespidae) across Northern Spain. EPPO Bull. 2015, 45, 133–138. [Google Scholar] [CrossRef]
- Bessa, A.S.; Carvalho, J.; Gomes, A.; Santarem, F. Climate and land-use drivers of invasion: Predicting the expansion of Vespa velutina nigrithorax into the Iberian Peninsula. Insect Conserv. Diver. 2016, 9, 27–37. [Google Scholar] [CrossRef]
- Keeling, M.J.; Franklin, D.N.; Datta, S.; Brown, M.A.; Budge, G.E. Predicting the spread of the Asian hornet (Vespa velutina) following its incursion into Great Britain. Sci. Rep. 2017, 7, 6240. [Google Scholar] [CrossRef]
- Lioy, S.; Manino, A.; Porporato, M.; Laurino, D.; Romano, A.; Capello, M.; Bertolino, S. Establishing surveillance areas for tackling the invasion of Vespa velutina in outbreaks and over the border of its expanding range. Neobiota 2019, 46, 51–69. [Google Scholar] [CrossRef]
- Robinet, C.; Suppo, C.; Darrouzet, E. Rapid spread of the invasive yellow-legged hornet in France: The role of human-mediated dispersal and the effects of control measures. J. Appl. Ecol. 2017, 54, 205–215. [Google Scholar] [CrossRef]
- Fournier, A.; Barbet-Massin, M.; Rome, Q.; Courchamp, F. Predicting species distribution combining multi-scale drivers. Glob. Ecol. Conserv. 2017, 12, 215–226. [Google Scholar] [CrossRef]
- Rome, Q.; Perrard, A.; Muller, F.; Villemant, C. Aliens: The Invasive Species Bulletin: Newsletter of the IUCN/SSC Invasive Species Specialist Group. Available online: http://www.issg.org/pdf/aliens_newsletters/A31.pdf (accessed on 7 November 2019).
- Requier, F.; Rome, Q.; Chiron, G.; Decante, D.; Marion, S.; Menard, M.; Muller, F.; Villemant, C.; Henry, M. Predation of the invasive Asian hornet affects foraging activity and survival probability of honey bees in Western Europe. J. Pest Sci. 2019, 92, 567–578. [Google Scholar] [CrossRef]
- Monceau, K.; Moreau, J.; Poidatz, J.; Bonnard, O.; Thiéry, D. Behavioral syndrome in a native and an invasive hymenoptera species. Insect Sci. 2015, 22, 541–548. [Google Scholar] [CrossRef]
- Turchi, L.; Derijard, B. Options for the biological and physical control of Vespa velutina nigrithorax (Hym.: Vespidae) in Europe: A review. J. Appl. Entomol. 2018, 142, 553–562. [Google Scholar] [CrossRef]
- Monceau, K.; Bonnard, O.; Thiery, D. Chasing the queens of the alien predator of honeybees: A water drop in the invasiveness ocean. Open J. Ecol. 2012, 2, 183–191. [Google Scholar] [CrossRef]
- Monceau, K.; Thiéry, D. Vespa velutina nest distribution at a local scale: An 8-year survey of the invasive honeybee predator. Insect Sci. 2017, 24, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Haxaire, J.; Villemant, C. Impact sur l’entomofaune des “pièges à frelon asiatique”. Insectes 2010, 159, 6. [Google Scholar]
- Rojas-Nossa, S.V.; Novoa, N.; Serrano, A.; Calvino-Cancela, M. Performance of baited traps used as control tools for the invasive hornet Vespa velutina and their impact on non-target insects. Apidologie 2018, 49, 872–885. [Google Scholar] [CrossRef]
- Decante, D.; ITSAP. Lutte Contre le Frelon Asiatique Vespa velutina. Available online: http://itsap.asso.fr/wp-content/uploads/2016/03/cr_evaluation_piegeage_vvelutina_2014.pdf (accessed on 18 April 2015).
- Milanesio, D.; Maurice, S.; Maggiora, R.; Laurino, D.; Porporato, M. Recent upgrades of the harmonic radar for the tracking of the Asian yellow-legged hornet. Ecol. Evol. 2017, 7, 4599–4606. [Google Scholar] [CrossRef]
- Milanesio, D.; Saccani, M.; Maggiora, R.; Laurino, D.; Porporato, M. Design of an harmonic radar for the tracking of the Asian yellow-legged hornet. Ecol. Evol. 2016, 6, 2170–2178. [Google Scholar] [CrossRef]
- Kennedy, P.J.; Ford, S.M.; Poidatz, J.; Thiery, D.; Osborne, J.L. Searching for nests of the invasive Asian hornet (Vespa velutina) using radio-telemetry. Commun. Biol. 2018, 1, 88. [Google Scholar] [CrossRef]
- Blot, J.; ITSAP. Localisation et Destruction des Nids de Frelons Asiatiques. Available online: http://www.itsap.asso.fr/downloads/publications/frelondestuction_light.pdf (accessed on 9 September 2015).
- Beggs, J.R.; Brockerhoff, E.G.; Corley, J.C.; Kenis, M.; Masciocchi, M.; Muller, F.; Rome, Q.; Villemant, C. Ecological effects and management of invasive alien Vespidae. Biocontrol 2011, 56, 505–526. [Google Scholar] [CrossRef]
- Mazzei, M.; Forzan, M.; Cilia, G.; Sagona, S.; Bortolotti, L.; Felicioli, A. First detection of replicative deformed wing virus (DWV) in Vespa velutina nigrithorax. Bull. Insectol. 2018, 71, 211–216. [Google Scholar]
- Mutien, G.; Bernard, T.; Noëmie El, A.; Daniel, C.; Gautier, G.; Marie, H.; Daniel, D.; Georges, D.; Annick, L.; Frédéric, F.; et al. Moku virus in invasive Asian hornets, Belgium, 2016. Emeg. Infect. Dis. J. 2017, 23, 2109. [Google Scholar]
- Yanez, O.; Zheng, H.Q.; Hu, F.L.; Neumann, P.; Dietemann, V. A scientific note on Israeli acute paralysis virus infection of Eastern honeybee Apis cerana and vespine predator Vespa velutina. Apidologie 2012, 43, 587–589. [Google Scholar] [CrossRef]
- Tehel, A.; Brown, M.J.F.; Paxton, R.J. Impact of managed honey bee viruses on wild bees. Curr. Opin. Virol. 2016, 19, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Evison, S.E.; Roberts, K.E.; Laurenson, L.; Pietravalle, S.; Hui, J.; Biesmeijer, J.C.; Smith, J.E.; Budge, G.; Hughes, W.O. Pervasiveness of parasites in pollinators. PLoS ONE 2012, 7, e30641. [Google Scholar] [CrossRef] [PubMed]
- Levitt, A.L.; Singh, R.; Cox-Foster, D.L.; Rajotte, E.; Hoover, K.; Ostiguy, N.; Holmes, E.C. Cross-species transmission of honey bee viruses in associated arthropods. Virus Res. 2013, 176, 232–240. [Google Scholar] [CrossRef]
- Singh, R.; Levitt, A.L.; Rajotte, E.G.; Holmes, E.C.; Ostiguy, N.; Vanengelsdorp, D.; Lipkin, W.I.; Depamphilis, C.W.; Toth, A.L.; Cox-Foster, D.L. RNA viruses in hymenopteran pollinators: Evidence of inter-taxa virus transmission via pollen and potential impact on non-apis hymenopteran species. PLoS ONE 2010, 5, e14357. [Google Scholar] [CrossRef]
- Manley, R.; Boots, M.; Wilfert, L. Emerging viral disease risk to pollinating insects: Ecological, evolutionary and anthropogenic factors. J. Appl. Ecol. 2015, 52, 331–340. [Google Scholar] [CrossRef]
- Tentcheva, D.; Gauthier, L.; Zappulla, N.; Dainat, B.; Cousserans, F.; Colin, M.E.; Bergoin, M. Prevalence and seasonal variations of six bee viruses in Apis mellifera L. and Varroa destructor mite populations in France. Appl. Environ. Microbiol. 2004, 70, 7185–7191. [Google Scholar] [CrossRef]
- Traynor, K.S.; Rennich, K.; Forsgren, E.; Rose, R.; Pettis, J.; Kunkel, G.; Madella, S.; Evans, J.; Lopez, D.; van Engelsdorp, D. Multiyear survey targeting disease incidence in US honey bees. Apidologie 2016, 47, 325–347. [Google Scholar] [CrossRef]
- Dainat, B.; Evans, J.D.; Chen, Y.P.; Gauthier, L.; Neumann, P. Dead or alive: Deformed wing virus and Varroa destructor reduce the life span of winter honeybees. Appl. Environ. Microbiol. 2012, 78, 981–987. [Google Scholar] [CrossRef]
- Francis, R.M.; Nielsen, S.L.; Kryger, P. Varroa-virus interaction in collapsing honey bee colonies. PLoS ONE 2013, 8, e57540. [Google Scholar] [CrossRef]
- Genersch, E.; Aubert, M. Emerging and re-emerging viruses of the honey bee (Apis mellifera L.). Vet. Res. 2010, 41, 41:54. [Google Scholar] [CrossRef] [PubMed]
- Kielmanowicz, M.G.; Inberg, A.; Lerner, I.M.; Golani, Y.; Brown, N.; Turner, C.L.; Hayes, G.J.R.; Ballam, J.M. Prospective large-scale field study generates predictive model identifying major contributors to colony losses. PLoS Pathog. 2015, 11, e1004816. [Google Scholar] [CrossRef] [PubMed]
- Mordecai, G.J.; Wilfert, L.; Martin, S.J.; Jones, I.M.; Schroeder, D.C. Diversity in a honey bee pathogen: First report of a third master variant of the deformed wing virus quasispecies. ISME J. 2015, 10, 1264–1273. [Google Scholar] [CrossRef]
- Mordecai, G.J.; Brettell, L.E.; Martin, S.J.; Dixon, D.; Jones, I.M.; Schroeder, D.C. Superinfection exclusion and the long-term survival of honey bees in Varroa-infested colonies. ISME J. 2015. [Google Scholar] [CrossRef]
- De Miranda, J.R.; Cordoni, G.; Budge, G. The Acute bee paralysis virus—Kashmir bee virus—Israeli acute paralysis virus complex. J. Invertbr. Pathol. 2010, 103, S30–S47. [Google Scholar] [CrossRef]
- Ribière, M.; Olivier, V.; Blanchard, P. Chronic bee paralysis: A disease and a virus like no other? J. Invertbr. Pathol. 2010, 103, S120–S131. [Google Scholar] [CrossRef]
- Celle, O.; Blanchard, P.; Olivier, V.; Schuff, F.; Cougoule, N.; Faucon, J.P.; Ribiere, M. Detection of chronic bee paralysis virus (CBPV) genome and its replicative RNA form in various hosts and possible ways of spread. Virus Res. 2008, 133, 280–284. [Google Scholar] [CrossRef]
- Pedler, R.D.; Brandle, R.; Read, J.L.; Southgate, R.; Bird, P.; Moseby, K.E. Rabbit biocontrol and landscape-scale recovery of threatened desert mammals. Conserv. Biol. 2016, 30, 774–782. [Google Scholar] [CrossRef]
- Bardin, M.; Ajouz, S.; Comby, M.; Lopez-Ferber, M.; Graillot, B.; Siegwart, M.; Nicot, P.C. Is the efficacy of biological control against plant diseases likely to be more durable than that of chemical pesticides? Front. Plant Sci. 2015, 6. [Google Scholar] [CrossRef]
- Murray, E.A.; Burand, J.; Trikoz, N.; Schnabel, J.; Grab, H.; Danforth, B.N. Viral transmission in honey bees and native bees, supported by a global black queen cell virus phylogeny. Environ. Microbiol. 2019, 21, 972–983. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 3. [Google Scholar] [CrossRef]
- Schulz, M.H.; Zerbino, D.R.; Vingron, M.; Birney, E. Oases: Robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics 2012, 28, 1086–1092. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Bigot, D.; Atyame, C.M.; Weill, M.; Justy, F.; Herniou, E.A.; Gayral, P. Discovery of Culex pipiens associated Tunisia virus: A new ssRNA(+) virus representing a new insect associated virus family. Virus Evol. 2018, 4. [Google Scholar] [CrossRef]
- Bigot, D.; Dalmon, A.; Roy, B.; Hou, C.; Germain, M.; Romary, M.; Deng, S.; Diao, Q.; Weinert, L.A.; Cook, J.M.; et al. The discovery of HalictiVirus Resolves the Sinaivirus phylogeny. J. General Virol. 2017, 98, 2864–2875. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinf. 2010, 11, 119. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinf. 2009, 10, 421. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Meth. 2012, 9, 357. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed]
- Kofler, R.; Orozco-terWengel, P.; De Maio, N.; Pandey, R.V.; Nolte, V.; Futschik, A.; Kosiol, C.; Schlotterer, C. PoPoolation: A toolbox for population genetic analysis of next generation sequencing data from pooled individuals. PLoS ONE 2011, 6, e15925. [Google Scholar] [CrossRef]
- Hammer, Ø.; Harper, D.A.T.; Ryan, P.D. PAST: Paleontological statistics software package for education and data analysis. Palaeontol. Electron. 2001, 4, 9. [Google Scholar]
- Edgar, R.C. Muscle: Multiple sequence alignment with high accuracy and high throughput. Nucl. Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef]
- Gouy, M.; Guindon, S.; Gascuel, O. SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 2010, 27, 221–224. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef]
- Lefort, V.; Longueville, J.E.; Gascuel, O. SMS: Smart model selection in PhyML. Mol. Biol. Evol. 2017, 34, 2422–2424. [Google Scholar] [CrossRef] [PubMed]
- Reynaldi, F.J.; Sguazza, G.H.; Albicoro, F.J.; Pecoraro, M.R.; Galosi, C.M. First molecular detection of co-infection of honey bee viruses in asymptomatic Bombus atratus in South America. Braz. J. Biol. 2014, 73, 797–800. [Google Scholar] [CrossRef] [PubMed]
- Sguazza, G.H.; Reynaldi, F.J.; Galosi, C.M.; Pecoraro, M.R. Simultaneous detection of bee viruses by multiplex PCR. J. Virol. Meth. 2013, 194, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Dalmon, A. Evidence for positive selection and recombination hotspots in deformed wing virus (DWV). Sci. Rep. 2017, 7, 41045. [Google Scholar] [CrossRef]
- Van der Wilk, F.; Dullemans, A.M.; Verbeek, M.; van den Heuvel, J.F. Nucleotide sequence and genomic organization of Acyrthosiphon pisum virus. Virology 1997, 238, 353–362. [Google Scholar] [CrossRef][Green Version]
- Shi, M.; Lin, X.D.; Tian, J.H.; Chen, L.J.; Chen, X.; Li, C.X.; Qin, X.C.; Li, J.; Cao, J.P.; Eden, J.S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef]
- Ekstrom, J.O.; Habayeb, M.S.; Srivastava, V.; Kieselbach, T.; Wingsle, G.; Hultmark, D. Drosophila Nora virus capsid proteins differ from those of other picorna-like viruses. Virus Res. 2011, 160, 51–58. [Google Scholar] [CrossRef]
- Habayeb, M.S.; Ekengren, S.K.; Hultmark, D. Nora virus, a persistent virus in Drosophila, defines a new picorna-like virus family. J. General Virol. 2006, 87, 3045–3051. [Google Scholar] [CrossRef]
- Medd, N.C.; Fellous, S.; Waldron, F.M.; Xuereb, A.; Nakai, M.; Cross, J.V.; Obbard, D.J. The virome of Drosophila suzukii, an invasive pest of soft fruit. Virus Evol. 2018, 4, vey009. [Google Scholar] [CrossRef]
- Webster, C.L.; Longdon, B.; Lewis, S.H.; Obbard, D.J. Twenty-Five new viruses associated with the Drosophilidae (Diptera). Evol. Bioinf. Online 2016, 12, 13–25. [Google Scholar] [CrossRef]
- Webster, C.L.; Waldron, F.M.; Robertson, S.; Crowson, D.; Ferrari, G.; Quintana, J.F.; Brouqui, J.M.; Bayne, E.H.; Longdon, B.; Buck, A.H.; et al. The discovery, distribution, and evolution of viruses associated with Drosophila melanogaster. PLoS Biol. 2015, 13, e1002210. [Google Scholar] [CrossRef] [PubMed]
- De Miranda, J.R.; Genersch, E. Deformed wing virus. J. Invertbr. Pathol. 2010, 103, S48–S61. [Google Scholar] [CrossRef] [PubMed]
- McMahon, D.P.; Natsopoulou, M.E.; Doublet, V.; Fürst, M.; Weging, S.; Brown, M.J.F.; Gogol-Döring, A.; Paxton, R.J. Elevated virulence of an emerging viral genotype as a driver of honeybee loss. Proc. R. Soc. B Biol. Sci. 2016, 283. [Google Scholar] [CrossRef] [PubMed]
- Forsgren, E.; Shi, W.; Ding, G.L.; Liu, Z.G.; Tran, T.V.; Tang, P.T.; Truong, T.A.; Dinh, T.Q.; Fries, I. Preliminary observations on possible pathogen spill-over from Apis mellifera to Apis cerana. Apidologie 2015, 46, 265–275. [Google Scholar] [CrossRef]
- McMahon, D.P.; Furst, M.A.; Caspar, J.; Theodorou, P.; Brown, M.J.F.; Paxton, R.J. A sting in the spit: Widespread cross-infection of multiple RNA viruses across wild and managed bees. J. Anim. Ecol. 2015, 84, 615–624. [Google Scholar] [CrossRef]
- Van den Heuvel, J.F.; Hummelen, H.; Verbeek, M.; Dullemans, A.M.; van der Wilk, F. Characteristics of acyrthosiphon pisum virus, a newly identified virus infecting the pea aphid. J. Invertebr. Pathol. 1997, 70, 169–176. [Google Scholar] [CrossRef]
- Dacheux, L.; Cervantes-Gonzalez, M.; Guigon, G.; Thiberge, J.M.; Vandenbogaert, M.; Maufrais, C.; Caro, V.; Bourhy, H. A preliminary study of viral metagenomics of french bat species in contact with humans: Identification of new mammalian viruses. PLoS ONE 2014, 9, e87194. [Google Scholar] [CrossRef]
- Reuter, G.; Pankovics, P.; Gyongyi, Z.; Delwart, E.; Boros, A. Novel dicistrovirus from bat guano. Arch. Virol. 2014, 159, 3453–3456. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Number of Read Pairs | Number of Contigs | Total Size of Contigs (in Nucleotides) | Read Alignment Rate (*) | Pair Alignment Rate (*) |
|---|---|---|---|---|---|
| 159M | 41,650,349 | 85,968 | 44,138,196 | 99.35% | 71.34% |
| 144I | 50,537,821 | 468,927 | 276,819,205 | 98.44% | 41.19% |
| 140M | 49,181,345 | 420,974 | 246,505,091 | 98.81% | 51.54% |
| 140C | 49,526,919 | 820,766 | 374,354,293 | 99.08% | 62.55% |
| Library | Virus Acronym | Virus Name | Family | Genus | Genome | Number of. Contig (s) Assembled | Contig(s) size | Expected Full-Length Genome Size (kb) | % Missing Genome | Coverage of Assembled Contigs * | Coverage of a Full-Length Expected Genome |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 159M | DWV-B | Deformed wing virus type B | Iflaviridae | Iflavirus | Nearly full-length | 1 | 9434 | 10.1 | 6.7 | 11.1 | 10.4 |
| 140C | na | Vespa velutina associated acypi-like virus | unclassified | unclassified | Full-length | 1 | 9872 | 10 | 0 | 535.1 | 535.1 |
| DWV-B | Deformed wing virus type B | Iflaviridae | Iflavirus | Nearly full-length | 4 | 9665 | 10.1 | 4.1 | 8.5 | 8.2 | |
| KBV | Kashmir bee virus | Dicistroviridae | Aparavirus | Partial | 16 | 3536 | 9.5 | 62.9 | 1.9 | 0.7 | |
| 140M | na | Vespa velutina associated acypi-like virus | unclassified | unclassified | Full-length | 1 | 9962 | 10 | 0 | 1186 | 1186 |
| DWV-B | Deformed wing virus type B | Iflaviridae | Iflavirus | Nearly full-length | 2 | 10000 | 10.1 | 0.8 | 13.2 | 13.2 | |
| na | Vespa velutina associated triato-like virus | Dicistroviridae | Triatovirus | Partial | 4 | 2380 | 9.3 | 74.3 | 4.5 | 1.2 | |
| 144I | DWV-B | Deformed wing virus type B | Iflaviridae | Iflavirus | Full-length | 1 | 10113 | 10.1 | 0 | 110,316.3 | 110,316.3 |
| na | Vespa velutina associated acypi-like virus | unclassified | unclassified | Full-length | 1 | 9748 | 10 | 0 | 122 | 122 | |
| ABPV | Acute bee paralysis virus | Dicistroviridae | Aparavirus | Full-length | 1 | 9474 | 9.5 | 0 | 1794.6 | 1794.6 | |
| BQCV | Black queen cell virus | Dicistroviridae | Triatovirus | Full-length | 1 | 8464 | 8.6 | 0 | 268.4 | 268.4 | |
| na | Vespa velutina associated Menton virus | unclassified | unclassified | Full-length segment 1 | 1 | 3031 | 2.8 (seg.1) | 0 (Seg 2 missing: 1.5kb) | 316.7 | 316.7 | |
| na | Vespa velutina associated ifla-like virus | unclassified | unclassified | Full-length | 1 | 10023 | 9.9 | 0 | 19.5 | 19.5 | |
| na | Vespa velutina associated permutotetra-like virus 1 | unclassified | unclassified | Full-length | 1 | 4851 | 4.8 | 0 | 34.7 | 34.7 | |
| na | Vespa velutina associated permutotetra-like virus 2 | unclassified | unclassified | Full-length | 1 | 4742 | 4.8 | 0 | 11.7 | 11.7 | |
| na | Vespa velutina associated partiti-like virus 1 | Partitiviridae | unclassified | Full-length segment 1 | 1 | 1779 | 1.7 | 0 (Seg 2 missing: 1.7 kb) | 1082.5 | 1082.5 | |
| na | Vespa velutina associated partiti-like virus 2 | Partitiviridae | unclassified | Full-length segment 1 | 1 | 1415 | 1.4 | 0 (Seg 2 missing: 1.7 kb) | 44.6 | 44.6 | |
| na | Vespa velutina associated partiti-like virus 3 | Partitiviridae | unclassified | Full-length segment 1 | 1 | 1417 | 1.4 | 0 (Seg 2 missing: 1.7 kb) | 100.3 | 100.3 | |
| na | Vespa velutina associated partiti-like virus 4 | Partitiviridae | unclassified | Partial segment 1 | 1 | 467 | 1.5 | Seg1: 63.2 (Seg 2 missing: 1.7 kb) | 6.2 | 1.9 | |
| na | Vespa velutina associated nora-like virus | unclassified | unclassified | Partial | 1 | 1406 | 8 | 17.5 | 6.6 | 1.2 | |
| na | Vespa velutina associated triato-like virus | Dicistroviridae | Triatovirus | Partial | 3 | 3149 | 9.3 | 66 | 4.9 | 1.6 | |
| ALPV | Aphid lethal paralysis virus | Dicistroviridae | Cripavirus | Partial | 20 | 4186 | 9.8 | 57.4 | 2 | 0.9 | |
| na | Moku virus | Iflaviridae | Iflavirus | Partial | 23 | 6232 | 10.1 | 38 | 2 | 1.3 | |
| BMLV | Bee Macula-like virus | Tymoviridae | na | Partial | 13 | 3102 | 6.3 | 50.3 | 2.4 | 1.2 | |
| DWV-C | Deformed wing virus type C | Iflaviridae | Iflavirus | Partial | 15 | 3292 | 10.1 | 64.9 | 9,634.2 | 3,377 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dalmon, A.; Gayral, P.; Decante, D.; Klopp, C.; Bigot, D.; Thomasson, M.; Herniou, E.A.; Alaux, C.; Le Conte, Y. Viruses in the Invasive Hornet Vespa velutina. Viruses 2019, 11, 1041. https://doi.org/10.3390/v11111041
Dalmon A, Gayral P, Decante D, Klopp C, Bigot D, Thomasson M, Herniou EA, Alaux C, Le Conte Y. Viruses in the Invasive Hornet Vespa velutina. Viruses. 2019; 11(11):1041. https://doi.org/10.3390/v11111041
Chicago/Turabian StyleDalmon, Anne, Philippe Gayral, Damien Decante, Christophe Klopp, Diane Bigot, Maxime Thomasson, Elisabeth A Herniou, Cédric Alaux, and Yves Le Conte. 2019. "Viruses in the Invasive Hornet Vespa velutina" Viruses 11, no. 11: 1041. https://doi.org/10.3390/v11111041
APA StyleDalmon, A., Gayral, P., Decante, D., Klopp, C., Bigot, D., Thomasson, M., Herniou, E. A., Alaux, C., & Le Conte, Y. (2019). Viruses in the Invasive Hornet Vespa velutina. Viruses, 11(11), 1041. https://doi.org/10.3390/v11111041