Visualization of Positive and Negative Sense Viral RNA for Probing the Mechanism of Direct-Acting Antivirals against Hepatitis C Virus

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Compounds and Antibodies
2.3. Strand-Specific Quantitative RT-PCR
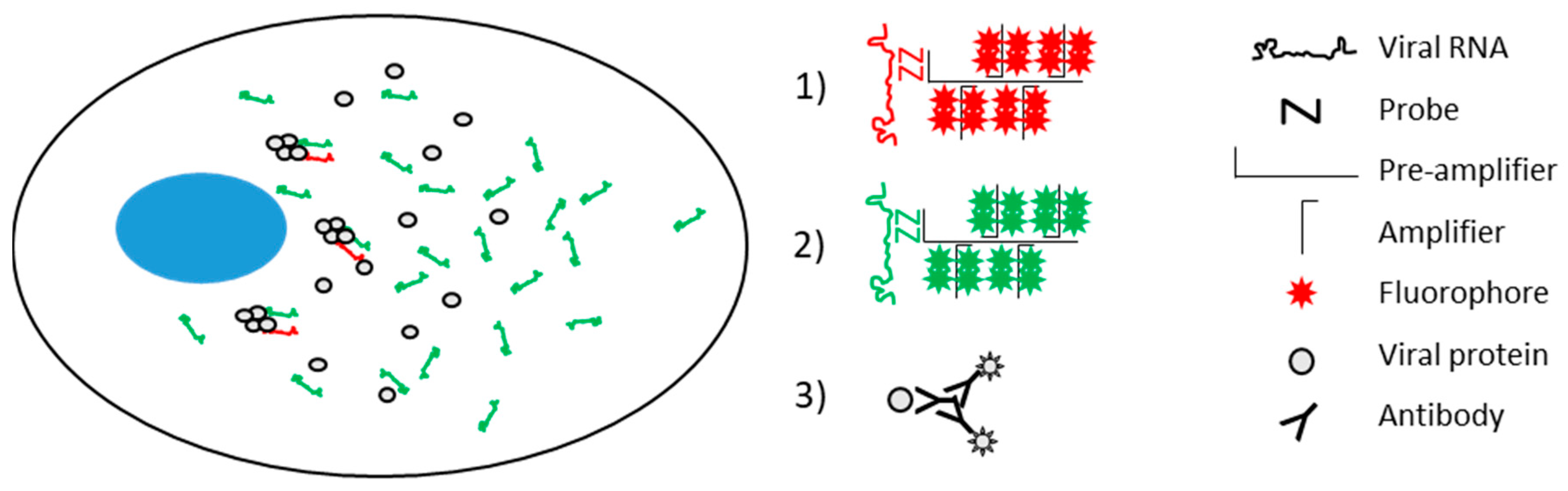
2.4. Branched DNA In Situ Hybridization (bDNA FISH) for Strand-Specific Nucleic Acid Visualization
2.5. Statistical Analyses
3. Results
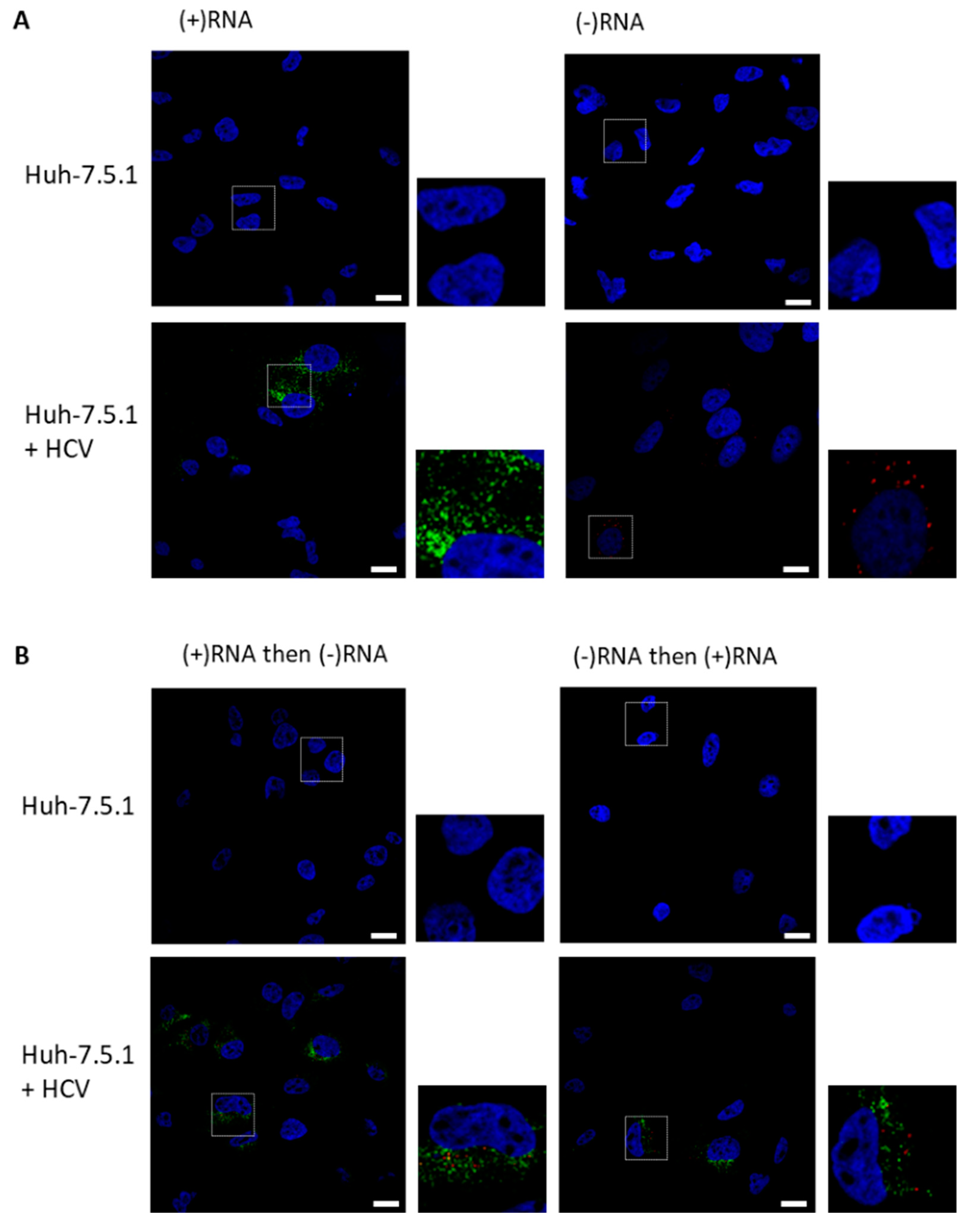
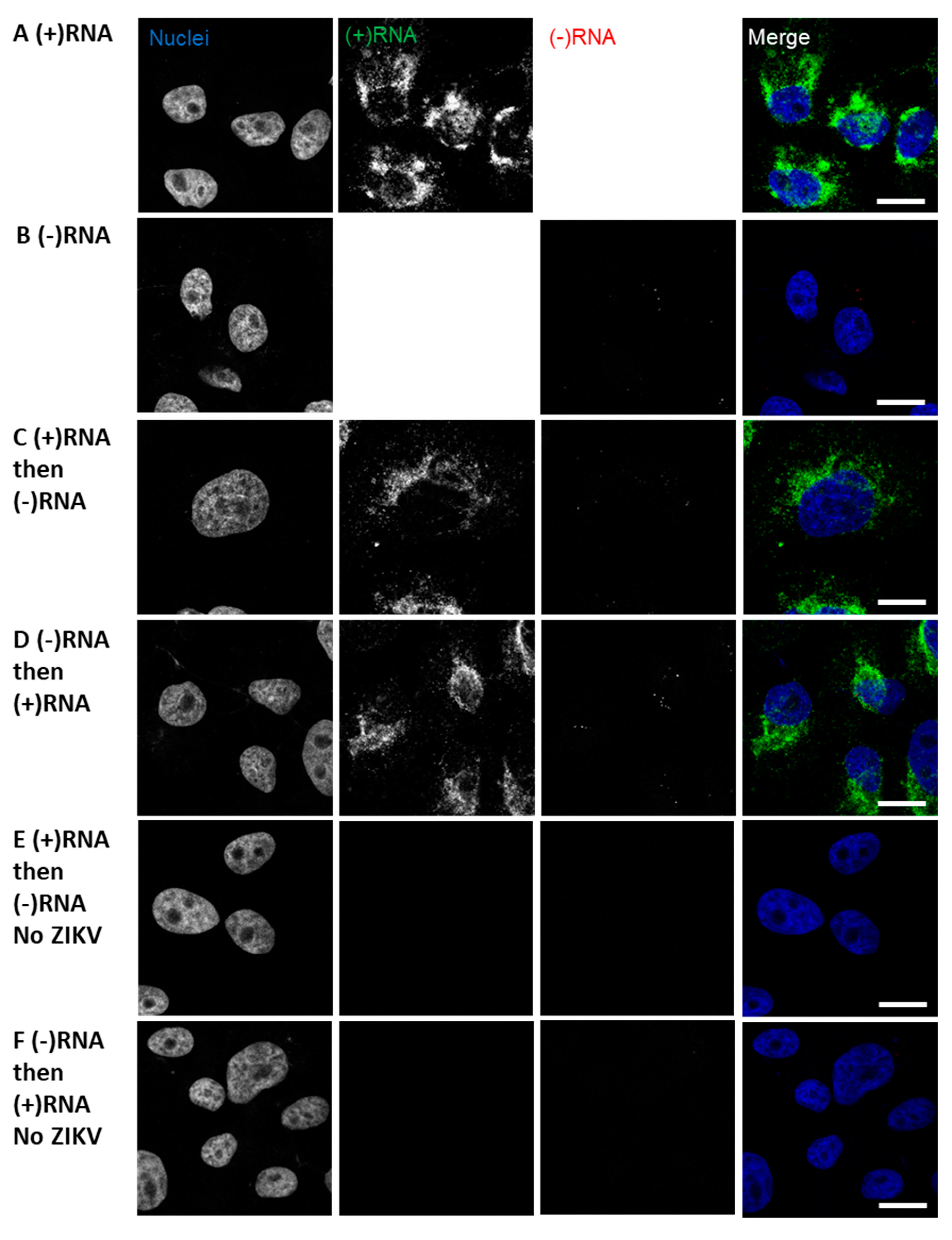
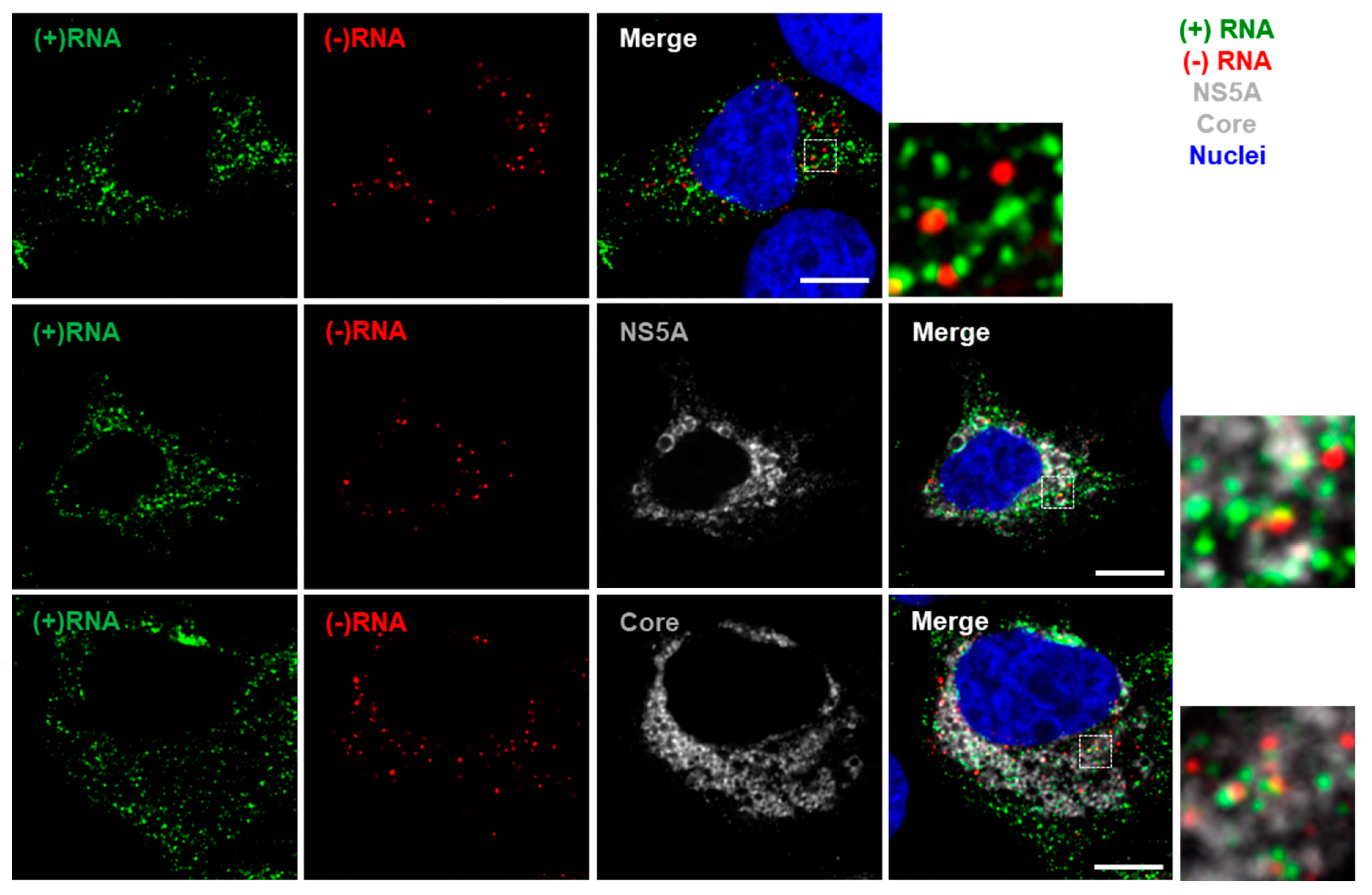
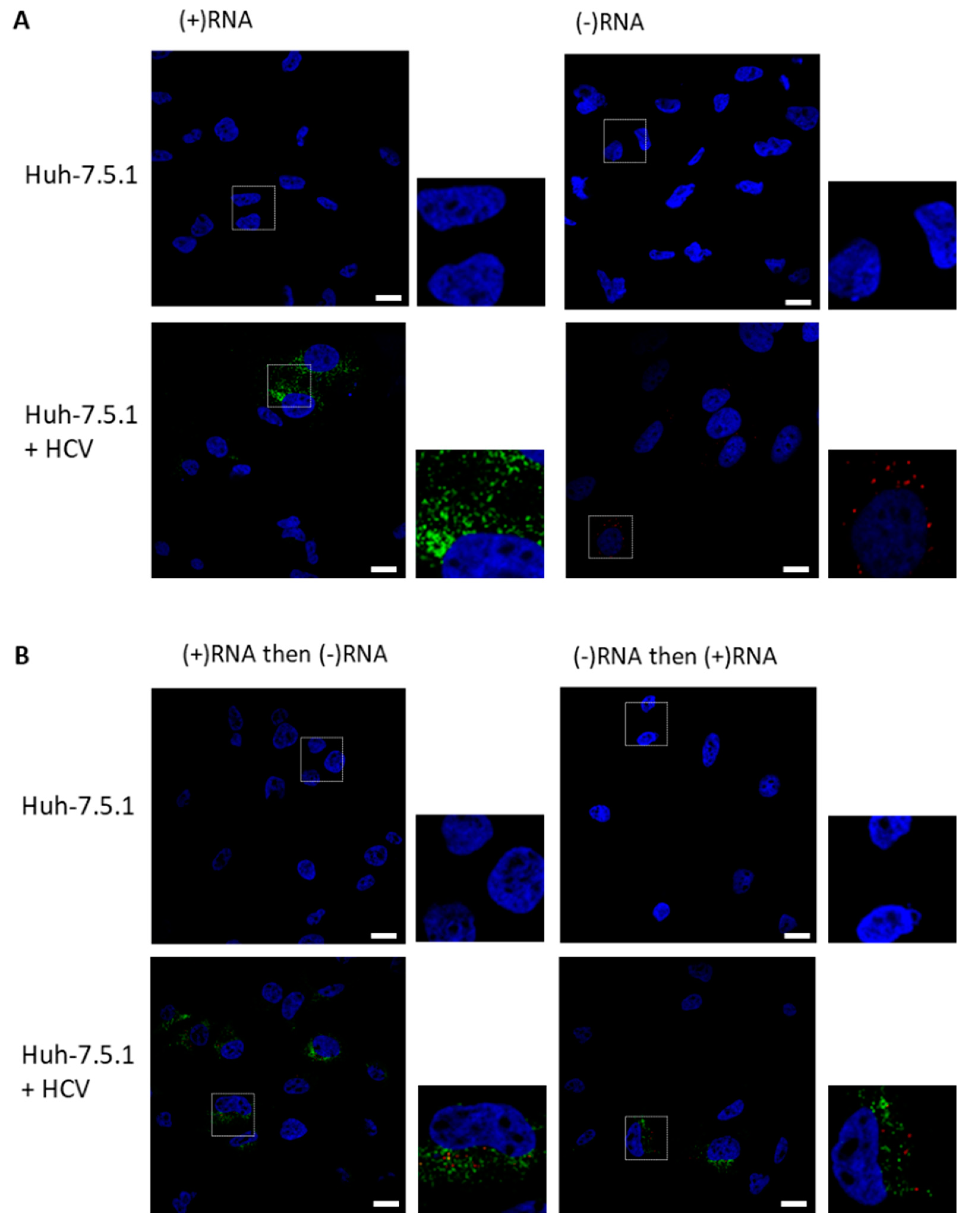
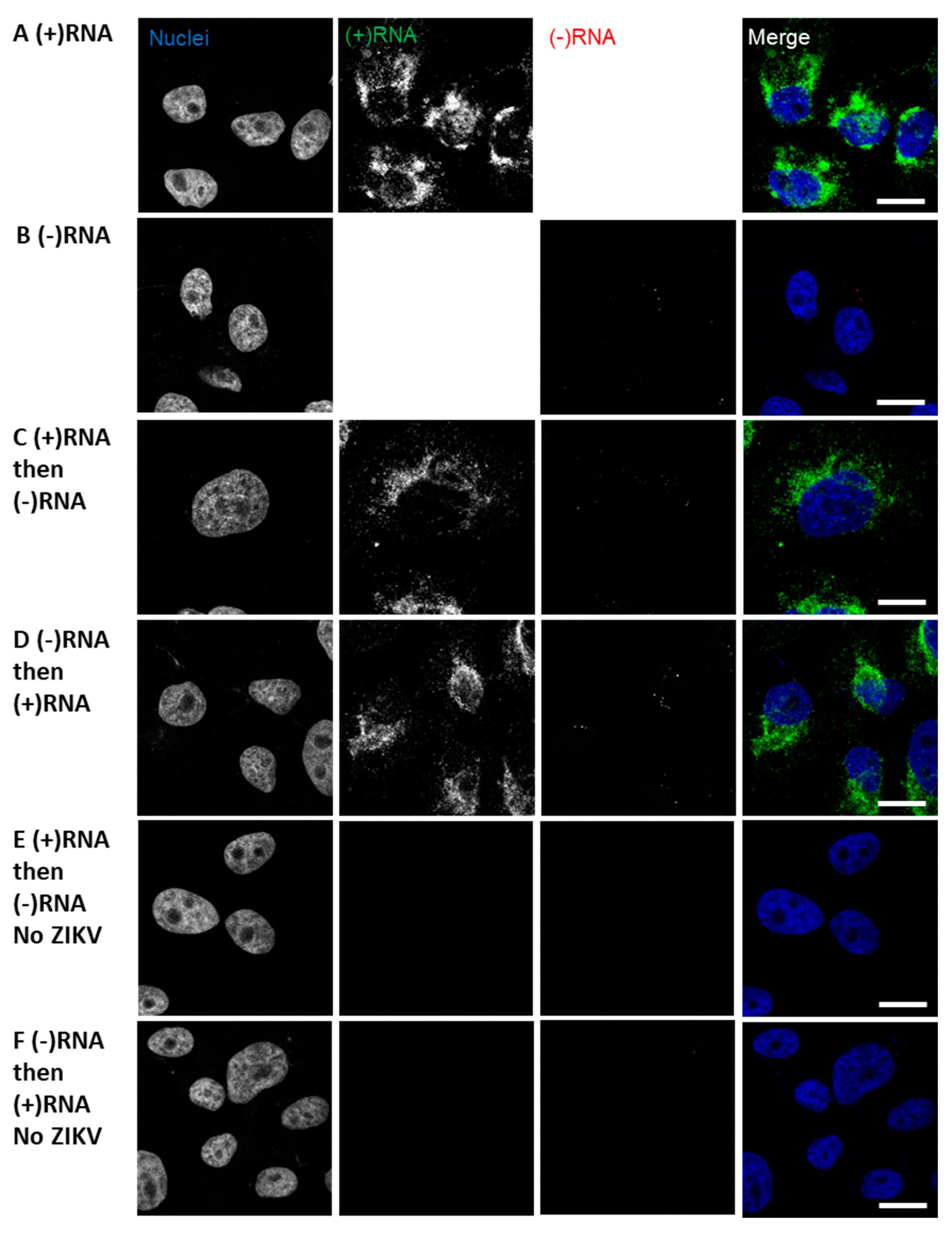
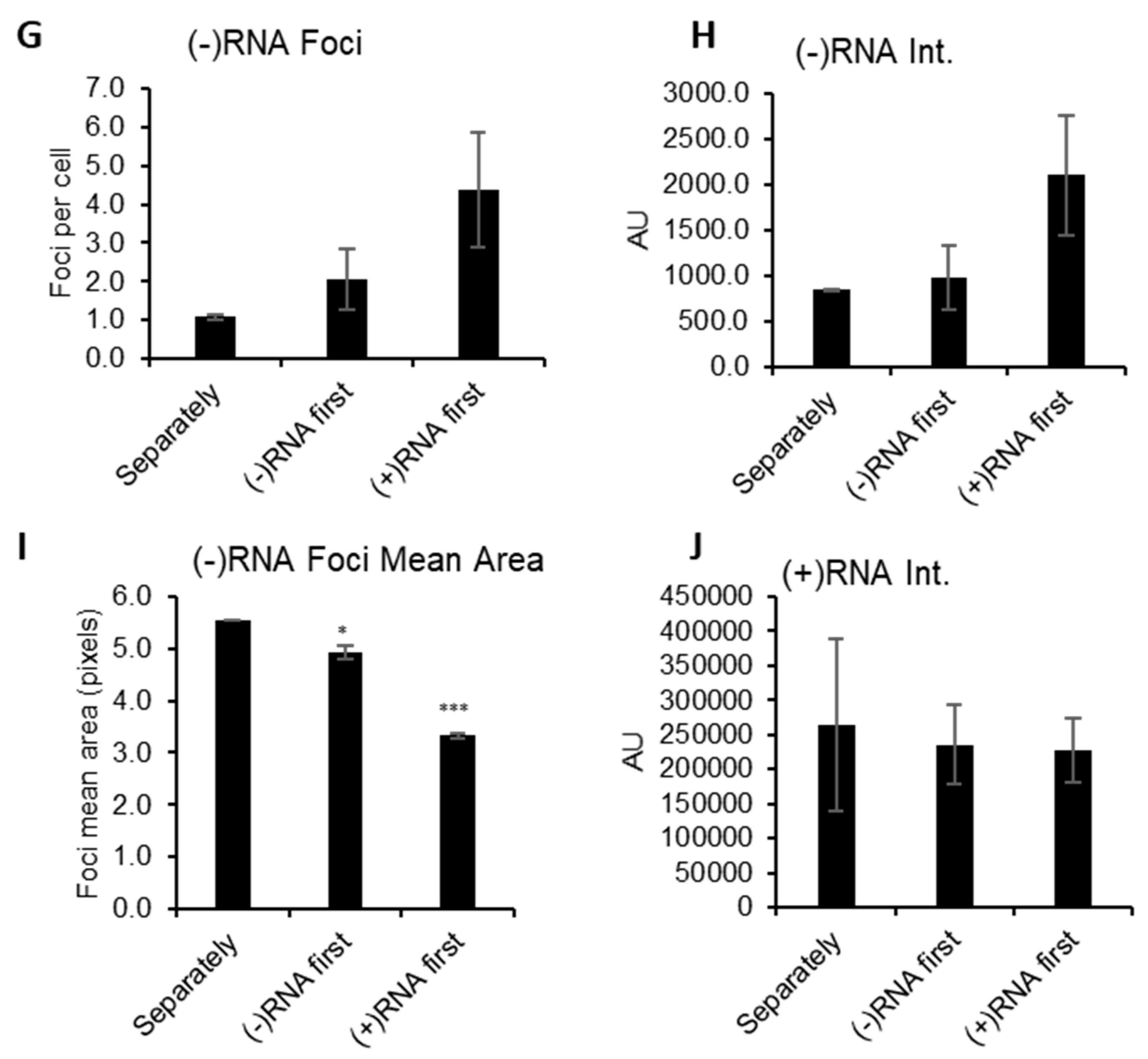
3.1. RNAscope Permits Simultaneous Imaging of (+) and (−)RNA Strands
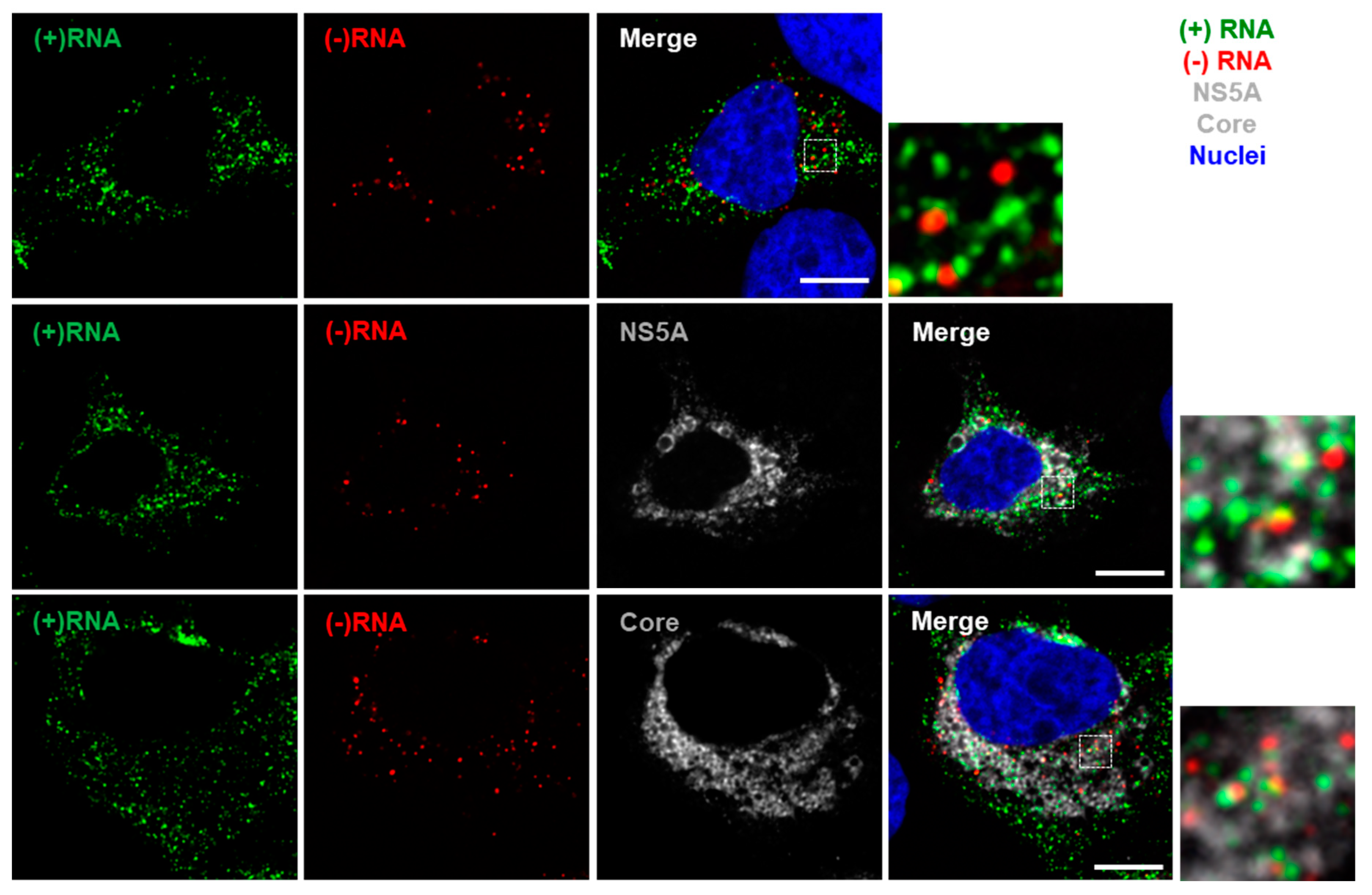
3.2. Staining of Nucleic Acids and Protein in HCV-Infected Cells
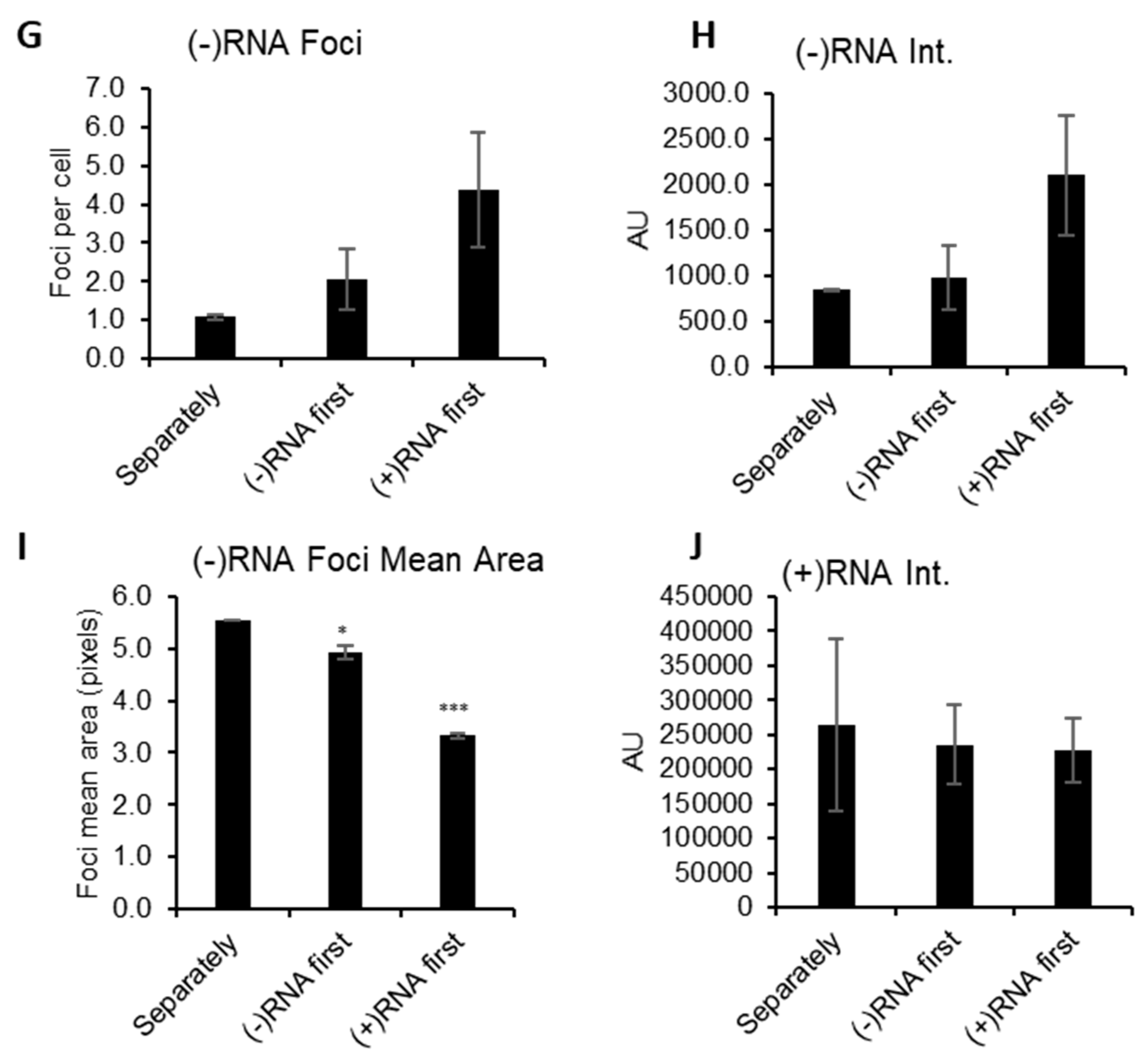
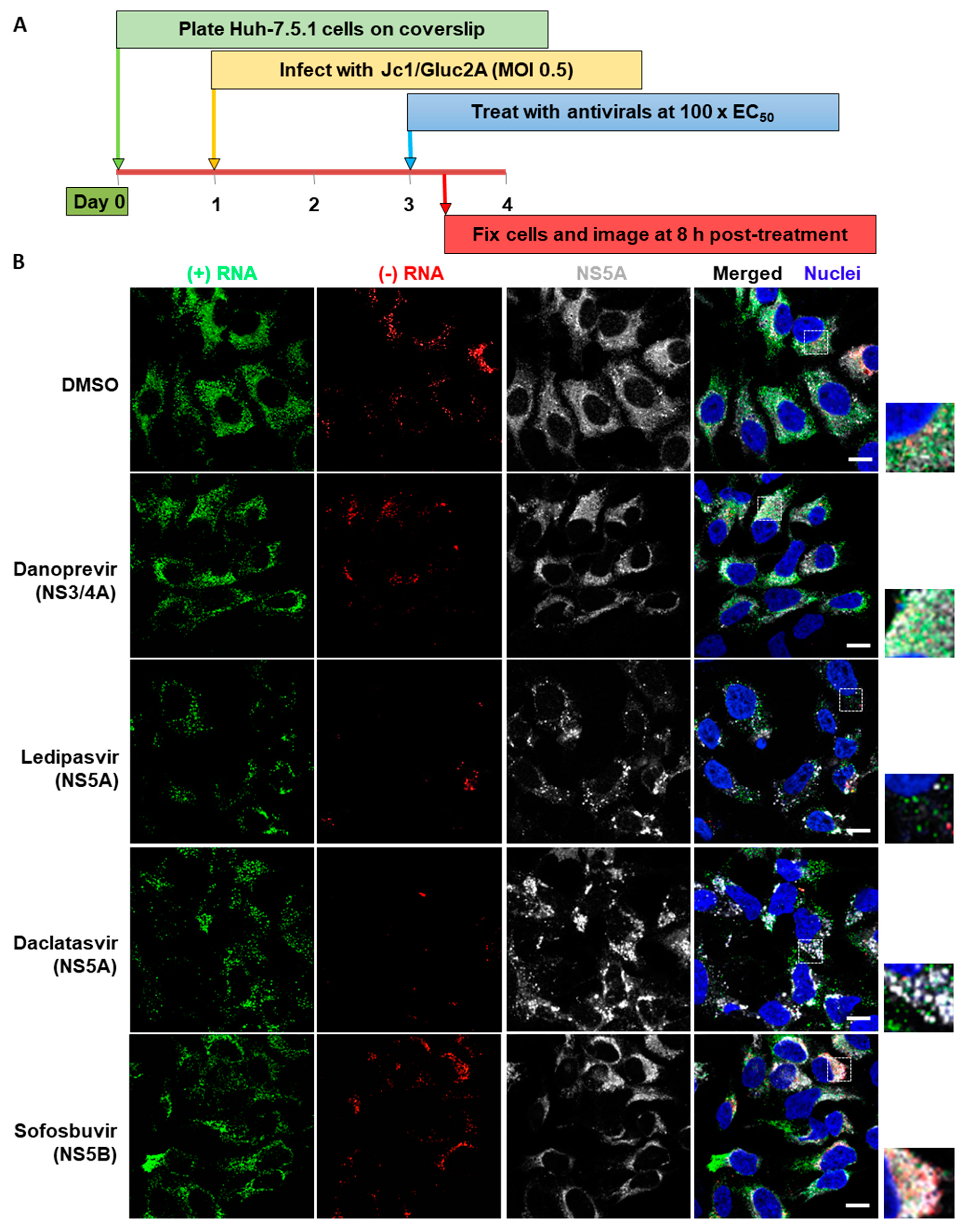
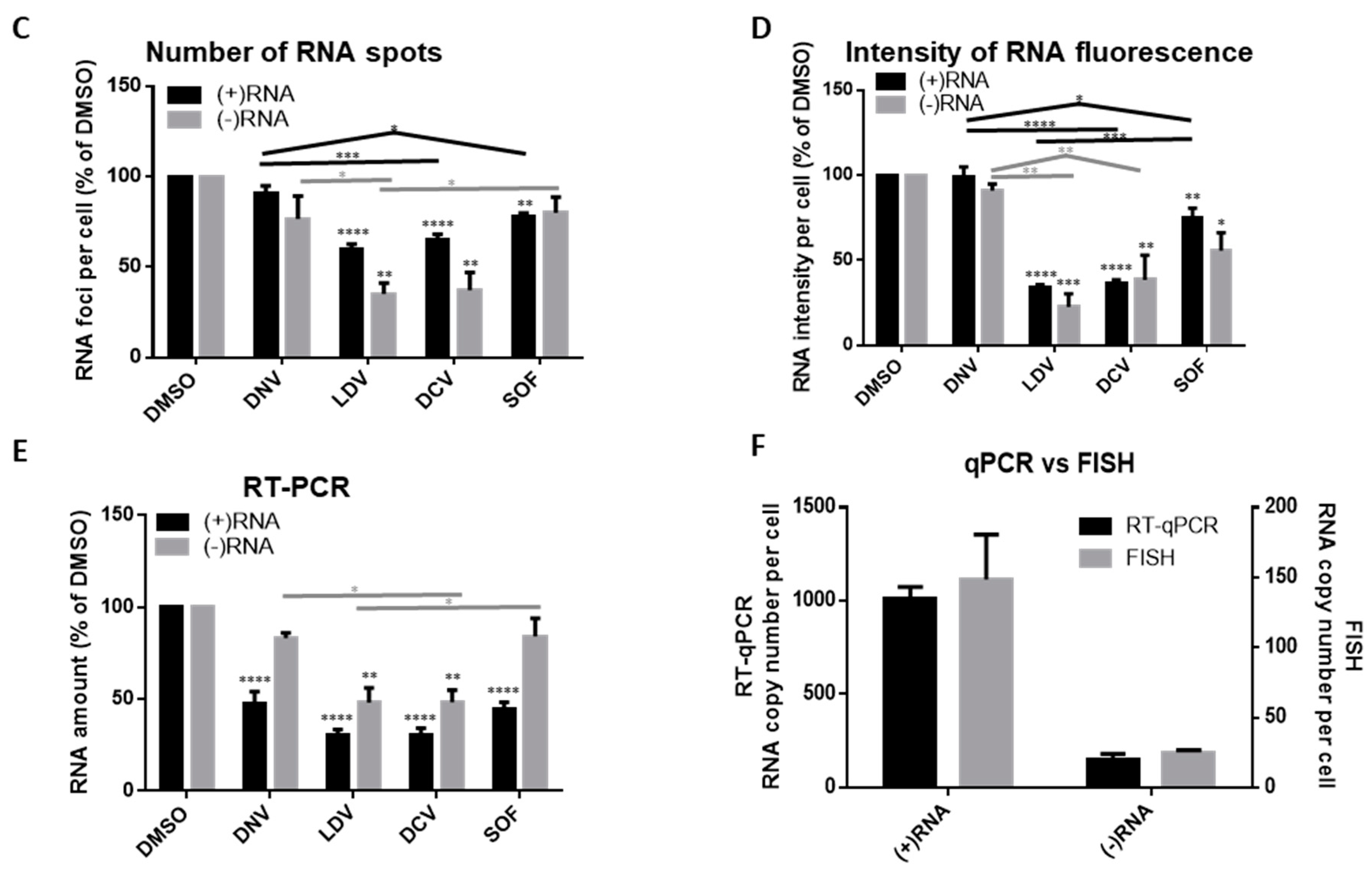
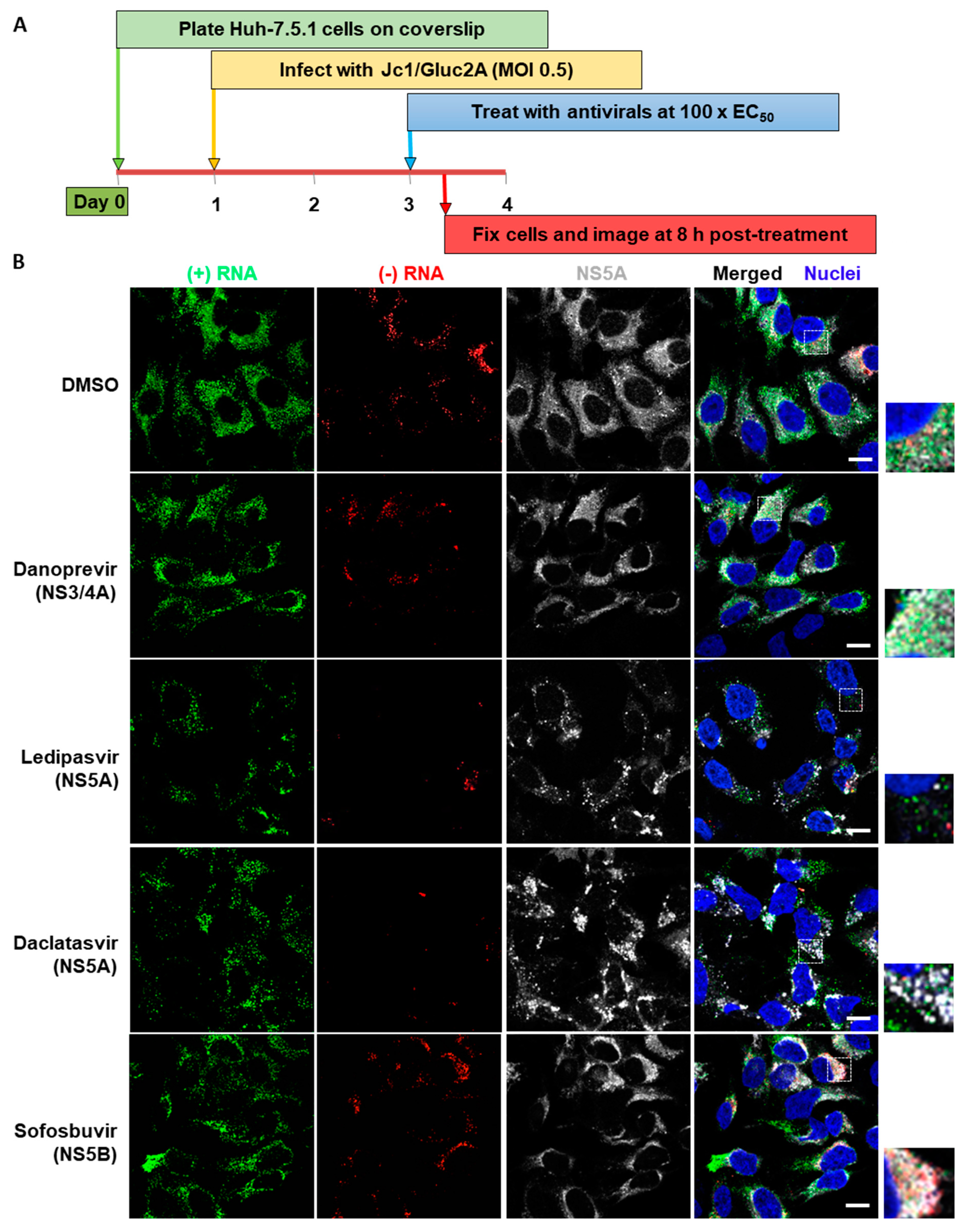
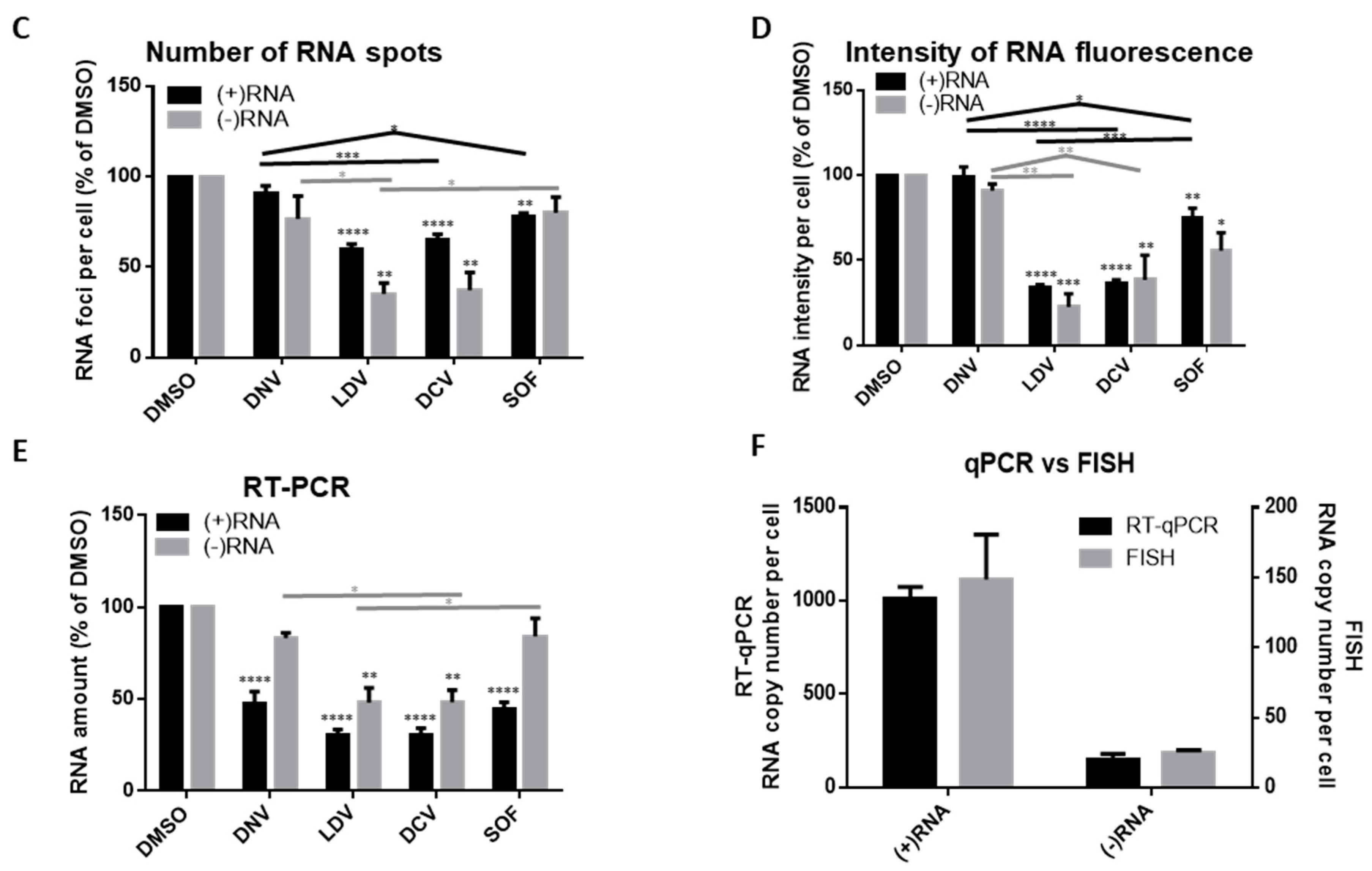
3.3. Analysis of Differential Phenotypes of HCV Inhibitors
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lévêque, N.; Semler, B.L. A 21st century perspective of poliovirus replication. PLoS Pathog. 2015, 11, e1004825. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Rubella Virus. In Epidemiology and Prevention of Vaccine-Preventable Diseases, 13th ed.; Hamborsky, J., Kroger, A., Wolfe, S., Eds.; Public Health Foundation: Washington, DC, USA, 2015. [Google Scholar]
- Pierson, T.C.; Diamond, M.S. Flaviviruses. In Field’s Virology; Knipe, D.M., Howley, P., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 747–794. ISBN 978-1451105636. [Google Scholar]
- Petersen, L.R.; Jamieson, D.J.; Powers, A.M.; Honein, M.A. Zika virus. N. Engl. J. Med. 2016, 374, 1552–1563. [Google Scholar] [CrossRef] [PubMed]
- Kim, A. Hepatitis C virus. In Annals of Internal Medicine; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2016; Volume 165, p. ITC33. ISBN 978-4-431-56099-9. [Google Scholar]
- Chatel-Chaix, L.; Bartenschlager, R. Dengue virus-and hepatitis c virus-induced replication and assembly compartments: The enemy inside—Caught in the Web. J. Virol. 2014, 88, 5907–5911. [Google Scholar] [CrossRef] [PubMed]
- Player, A.N.; Shen, L.P.; Kenny, D.; Antao, V.P.; Kolberg, J.A. Single-copy gene detection using branched DNA (bDNA) in situ hybridization. J. Histochem. Cytochem. 2001, 49, 603–611. [Google Scholar] [CrossRef]
- Wang, F.; Flanagan, J.; Su, N.; Wang, L.C.; Bui, S.; Nielson, A.; Wu, X.; Vo, H.T.; Ma, X.J.; Luo, Y. RNAscope: A novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J. Mol. Diagnostics 2012, 14, 22–29. [Google Scholar] [CrossRef]
- Porichis, F.; Hart, M.G.; Griesbeck, M.; Everett, H.L.; Hassan, M.; Baxter, A.E.; Lindqvist, M.; Miller, S.M.; Soghoian, D.Z.; Kavanagh, D.G.; et al. High-throughput detection of miRNAs and gene-specific mRNA at the single-cell level by flow cytometry. Nat. Commun. 2014, 5, 5641. [Google Scholar] [CrossRef]
- Battich, N.; Stoeger, T.; Pelkmans, L. Image-based transcriptomics in thousands of single human cells at single-molecule resolution. Nat. Methods 2013, 10, 1127–1136. [Google Scholar] [CrossRef]
- Baxter, A.E.; Niessl, J.; Fromentin, R.; Richard, J.; Porichis, F.; Charlebois, R.; Massanella, M.; Brassard, N.; Alsahafi, N.; Delgado, G.G.; et al. Single-cell characterization of viral translation-competent reservoirs in hiv-infected individuals. Cell Host Microbe 2016, 20, 368–380. [Google Scholar] [CrossRef]
- Martrus, G.; Niehrs, A.; Cornelis, R.; Rechtien, A.; García-Beltran, W.; Lütgehetmann, M.; Hoffmann, C.; Altfeld, M. Kinetics of HIV-1 latency reversal quantified on the single—Cell level using a novel flow-based technique. J. Virol. 2016, 90, 9018–9028. [Google Scholar] [CrossRef]
- Grau-Expósito, J.; Serra-Peinado, C.; Miguel, L.; Navarro, J.; Curran, A.; Burgos, J.; Ocaña, I.; Ribera, E.; Torrella, A.; Planas, B.; et al. A novel single-cell FISH-flow assay identifies effector memory CD4+T cells as a major niche for HIV-1 transcription in HIV-infected patients. MBio 2017, 8, e00876-17. [Google Scholar] [CrossRef]
- Chang, S.Y.; Keeney, M.; Law, M.; Donovan, J.; Aubry, M.C.; Garcia, J. Detection of human papillomavirus in non-small cell carcinoma of the lung. Hum. Pathol. 2015, 46, 1592–1597. [Google Scholar] [CrossRef] [PubMed]
- Ukpo, O.C.; Flanagan, J.J.; Ma, X.J.; Luo, Y.; Thorstad, W.L.; Lewis, J.S. High-risk human papillomavirus E6/E7 mRNA detection by a novel in situ hybridization assay strongly correlates with p16 expression and patient outcomes in oropharyngeal squamous cell carcinoma. Am. J. Surg. Pathol. 2011, 35, 1343–1350. [Google Scholar] [CrossRef] [PubMed]
- Bishop, J.A.; Ma, X.J.; Wang, H.; Luo, Y.; Illei, P.B.; Begum, S.; Taube, J.M.; Koch, W.M.; Westra, W.H. Detection of transcriptionally active high-risk HPV in patients with head and neck squamous cell carcinoma as visualized by a novel E6/E7 mRNA in situ hybridization method. Am. J. Surg. Pathol. 2012, 36, 1874–1882. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, M.X.-M.; Su, N.; Wang, L.; Wu, X.; Bui, S.; Nielsen, A.; Vo, H.-T.; Nguyen, N.; Luo, Y.; et al. RNAscope for In situ detection of transcriptionally active human papillomavirus in head and neck squamous cell carcinoma. J. Vis. Exp. 2014, 85, 1–5. [Google Scholar] [CrossRef]
- Carossino, M.; Loynachan, A.T.; James MacLachlan, N.; Drew, C.; Shuck, K.M.; Timoney, P.J.; Del Piero, F.; Balasuriya, U.B.R. Detection of equine arteritis virus by two chromogenic RNA in situ hybridization assays (conventional and RNAscope®) and assessment of their performance in tissues from aborted equine fetuses. Arch. Virol. 2016, 161, 3125–3136. [Google Scholar] [CrossRef]
- Honeycutt, J.B.; Wahl, A.; Baker, C.; Spagnuolo, R.A.; Foster, J.; Zakharova, O.; Wietgrefe, S.; Caro-Vegas, C.; Madden, V.; Sharpe, G.; et al. Macrophages sustain HIV replication in vivo independently of T cells. J. Clin. Investig. 2016, 126, 1353–1366. [Google Scholar] [CrossRef]
- Feeley, E.M.; Sims, J.S.; John, S.P.; Chin, C.R.; Pertel, T.; Chen, L.M.; Gaiha, G.D.; Ryan, B.J.; Donis, R.O.; Elledge, S.J.; et al. IFITM3 inhibits influenza a virus infection by preventing cytosolic entry. PLoS Pathog. 2011, 7, e1002337. [Google Scholar] [CrossRef]
- Wieland, S.; Makowska, Z.; Campana, B.; Calabrese, D.; Dill, M.T.; Chung, J.; Chisari, F.V.; Heim, M.H. Simultaneous detection of hepatitis C virus and interferon stimulated gene expression in infected human liver. Hepatology 2014, 59, 2121–2130. [Google Scholar] [CrossRef]
- Puray-Chavez, M.; Tedbury, P.R.; Huber, A.D.; Ukah, O.B.; Yapo, V.; Liu, D.; Ji, J.; Wolf, J.J.; Engelman, A.N.; Sarafianos, S.G. Multiplex single-cell visualization of nucleic acids and protein during HIV infection. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Deleage, C.; Wietgrefe, S.W.; Del Prete, G.; Morcock, D.R.; Hao, X.-P.; Piatak, M., Jr.; Bess, J.; Anderson, J.L.; Perkey, K.; Reilly, C.; et al. Defining HIV and SIV Reservoirs in Lymphoid Tissues. Pathog. Immun. 2016, 1, 68. [Google Scholar] [CrossRef]
- Chin, C.R.; Perreira, J.M.; Savidis, G.; Portmann, J.M.; Aker, A.M.; Feeley, E.M.; Smith, M.C.; Brass, A.L. Direct visualization of HIV-1 replication intermediates shows that capsid and CPSF6 modulate HIV-1 intra-nuclear invasion and integration. Cell Rep. 2015, 13, 1717–1731. [Google Scholar] [CrossRef] [PubMed]
- Marini, B.; Kertesz-Farkas, A.; Ali, H.; Lucic, B.; Lisek, K.; Manganaro, L.; Pongor, S.; Luzzati, R.; Recchia, A.; Mavilio, F.; et al. Nuclear architecture dictates HIV-1 integration site selection. Nature 2015, 521, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Josefsson, L.; King, M.S.; Makitalo, B.; Brannstrom, J.; Shao, W.; Maldarelli, F.; Kearney, M.F.; Hu, W.-S.; Chen, J.; Gaines, H.; et al. Majority of CD4+ T cells from peripheral blood of HIV-1-infected individuals contain only one HIV DNA molecule. Proc. Natl. Acad. Sci. USA 2011, 108, 11199–11204. [Google Scholar] [CrossRef] [PubMed]
- Quinkert, D.; Bartenschlager, R.; Lohmann, V. Quantitative analysis of the hepatitis c virus replication complex. J. Virol. 2005, 79, 13594–13605. [Google Scholar] [CrossRef] [PubMed]
- Andino, R.; Rieckhof, G.E.; Baltimore, D. A functional ribonucleoprotein complex forms around the 5′ end of poliovirus RNA. Cell 1990, 63, 369–380. [Google Scholar] [CrossRef]
- Novak, J.E.; Kirkegaard, K. Improved method for detecting poliovirus negative strands used to demonstrate specificity of positive-strand encapsidation and the ratio of positive to negative strands in infected cells. J. Virol. 1991, 65, 3384–3387. [Google Scholar] [Green Version]
- Cleaves, G.R.; Ryan, T.E.; Walter Schlesinger, R. Identification and characterization of type 2 dengue virus replicative intermediate and replicative form RNAs. Virology 1981, 111, 73–83. [Google Scholar] [CrossRef]
- Liu, D.; Ji, J.; Ndongwe, T.P.; Michailidis, E.; Rice, C.M.; Ralston, R.; Sarafianos, S.G. Fast hepatitis C virus RNA elimination and NS5A redistribution by NS5A inhibitors studied by a multiplex assay approach. Antimicrob. Agents Chemother. 2015, 59, 3482–3492. [Google Scholar] [CrossRef]
- Gao, M.; Nettles, R.E.; Belema, M.; Snyder, L.B.; Nguyen, V.N.; Fridell, R.A.; Serrano-Wu, M.H.; Langley, D.R.; Sun, J.H.; O’Boyle, D.R.; et al. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature 2010, 465, 96–100. [Google Scholar] [CrossRef]
- Guedj, J.; Dahari, H.; Rong, L.; Sansone, N.D.; Nettles, R.E.; Cotler, S.J.; Layden, T.J.; Uprichard, S.L.; Perelson, A.S. Modeling shows that the NS5A inhibitor daclatasvir has two modes of action and yields a shorter estimate of the hepatitis C virus half-life. Proc. Natl. Acad. Sci. USA 2013, 110, 3991–3996. [Google Scholar] [CrossRef] [Green Version]
- Zhong, J.; Gastaminza, P.; Cheng, G.; Kapadia, S.; Kato, T.; Burton, D.R.; Wieland, S.F.; Uprichard, S.L.; Wakita, T.; Chisari, F.V. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. USA 2005, 102, 9294–9299. [Google Scholar] [CrossRef] [PubMed]
- Naoki, O.; Arihiro, K.; Toshiyuki, Y.; Noriko, H.; Fumio, K.; Suyoshi, S.; Makoto, K.; Kentaro, H.; Hattori, M. The genome landscape of the African Green Monkey kidney-derived vero cell line. DNA Res. 2014, 21, 673–683. [Google Scholar]
- Emeny, J.M.; Morgan, M.J. Regulation of the interferon system: Evidence that vero cells have a genetic defect in interferon production. J. Gen. Virol. 1979, 43, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Diaz, M.O.; Ziemin, S.; Le Beau, M.M.; Pitha, P.; Smith, S.D.; Chilcote, R.R.; Rowley, J.D. Homozygous deletion of the alpha- and beta 1-interferon genes in human leukemia and derived cell lines. Proc. Natl. Acad. Sci. USA 1988, 85, 5259–5263. [Google Scholar] [CrossRef]
- Marukian, S.; Jones, C.T.; Andrus, L.; Evans, M.J.; Ritola, K.D.; Charles, E.D.; Rice, C.M.; Dustin, L.B. Cell culture-produced hepatitis C virus does not infect peripheral blood mononuclear cells. Hepatology 2008, 48, 1843–1850. [Google Scholar] [CrossRef] [Green Version]
- Sakkas, H.; Economou, V.; Papadopoulou, C. Zika virus infection: Past and present of another emerging vector-borne disease. J. Vector Borne Dis. 2016, 53, 305–311. [Google Scholar]
- Dick, G.W.A. Zika virus (II). Pathogenicity and physical properties. Trans. R. Soc. Trop. Med. Hyg. 1952, 46, 521–534. [Google Scholar] [CrossRef]
- Lindenbach, B.D. Complete Replication of Hepatitis C Virus in Cell Culture. Science. 2005, 309, 623–626. [Google Scholar] [CrossRef] [Green Version]
- Komurian-Pradel, F.; Perret, M.; Deiman, B.; Sodoyer, M.; Lotteau, V.; Paranhos-Baccalà, G.; André, P. Strand specific quantitative real-time PCR to study replication of hepatitis C virus genome. J. Virol. Methods 2004, 116, 103–106. [Google Scholar] [CrossRef]
- Schwarz, M.C.; Sourisseau, M.; Espino, M.M.; Gray, E.S.; Chambers, M.T.; Tortorella, D.; Evans, M.J. Rescue of the 1947 Zika virus prototype strain with a cytomegalovirus promoter-driven cDNA clone. mSphere 2016, 1, e00246-16. [Google Scholar] [CrossRef]
- McGivern, D.R.; Masaki, T.; Williford, S.; Ingravallo, P.; Feng, Z.; Lahser, F.; Asante-Appiah, E.; Neddermann, P.; De Francesco, R.; Howe, A.Y.; et al. Kinetic analyses reveal potent and early blockade of hepatitis C virus assembly by NS5A inhibitors. Gastroenterology 2014, 147, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zeng, Y.; Li, J.; Guo, Y.; Fu, Y.; He, J.; Sun, S.; Zhou, Y. A novel helper-dependent adenovirus-based cell culture model for Hepatitis C virus replication and production. Virol. J. 2013, 10, 273. [Google Scholar] [CrossRef] [PubMed]
- Pietschmann, T.; Lohmann, V.; Kaul, A.; Krieger, N.; Rinck, G.; Rutter, G.; Strand, D.; Bartenschlager, R. Persistent and transient replication of full-length hepatitis C virus genomes in cell culture. J. Virol. 2002, 76, 4008–4021. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, R.; Mason, I. In-situ hybridization of radioactive riboprobes to RNA in tissue sections. Methods Mol. Biol. 2008, 461, 675–686. [Google Scholar] [PubMed]
- Jensen, S.; Thomsen, A.R. Sensing of RNA viruses: A Review of innate immune receptors involved in recognizing RNA virus invasion. J. Virol. 2012, 86, 2900–2910. [Google Scholar] [CrossRef]
- Hornung, V.; Ellegast, J.; Kim, S.; Brzózka, K.; Jung, A.; Kato, H.; Poeck, H.; Akira, S.; Conzelmann, K.K.; Schlee, M.; et al. 5′-Triphosphate RNA is the ligand for RIG-I. Science 2006, 314, 994–997. [Google Scholar] [CrossRef]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef]
- Lim, S.M.; Koraka, P.; Osterhaus, A.D.M.E.; Martina, B.E.E. Development of a strand-specific real-time qRT-PCR for the accurate detection and quantitation of West Nile virus RNA. J. Virol. Methods 2013, 194, 146–153. [Google Scholar] [CrossRef]
- Steger, G.; Müller, H.; Riesner, D. Helix-coil transitions in double-stranded viral RNA. Fine resolution melting and ionic strength dependence. BBA Sect. Nucleic Acids Protein Synth. 1980, 606, 274–284. [Google Scholar] [CrossRef]
- Gomatos, P.J.; Tamm, I. The secondary structure of reovirus rna. Proc. Natl. Acad. Sci. USA 1963, 49, 707–714. [Google Scholar] [CrossRef]
- Klepper, A.; Eng, F.J.; Doyle, E.H.; El-Shamy, A.; Rahman, A.H.; Fiel, M.I.; Avino, G.C.; Lee, M.; Ye, F.; Roayaie, S.; et al. Hepatitis C virus double-stranded RNA is the predominant form in human liver and in interferon-treated cells. Hepatology 2017, 66, 357–370. [Google Scholar] [CrossRef] [PubMed]
- Forestier, N.; Larrey, D.; Guyader, D.; Marcellin, P.; Rouzier, R.; Patat, A.; Smith, P.; Bradford, W.; Porter, S.; Blatt, L.; et al. Treatment of chronic hepatitis C patients with the NS3/4A protease inhibitor danoprevir (ITMN-191/RG7227) leads to robust reductions in viral RNA: A phase 1b multiple ascending dose study. J. Hepatol. 2011, 54, 1130–1136. [Google Scholar] [CrossRef] [PubMed]
- Seiwert, S.D.; Andrews, S.W.; Jiang, Y.; Serebryany, V.; Tan, H.; Kossen, K.; Ravi Rajagopalan, P.T.; Misialek, S.; Stevens, S.K.; Stoycheva, A.; et al. Preclinical characteristics of the hepatitis C virus NS3/4A protease inhibitor ITMN-191 (R7227). Antimicrob. Agents Chemother. 2008, 52, 4432–4441. [Google Scholar] [CrossRef] [PubMed]
- Murakami, E.; Tolstykh, T.; Bao, H.; Niu, C.; Micolochick Steuer, H.M.; Bao, D.; Chang, W.; Espiritu, C.; Bansal, S.; Lam, A.M.; et al. Mechanism of activation of PSI-7851 and its diastereoisomer PSI-7977. J. Biol. Chem. 2010, 285, 34337–34347. [Google Scholar] [CrossRef] [PubMed]
- Lawitz, E.J.; Gruener, D.; Hill, J.M.; Marbury, T.; Moorehead, L.; Mathias, A.; Cheng, G.; Link, J.O.; Wong, K.A.; Mo, H.; et al. A phase 1, randomized, placebo-controlled, 3-day, dose-ranging study of GS-5885, an NS5A inhibitor, in patients with genotype 1 hepatitis C. J. Hepatol. 2012, 57, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Pevear, D.C.; Collett, M.S.; Chunduru, S.; Young, D.C.; Benetatos, C.; Jordan, R. Newly synthesized hepatitis c virus replicon rna is protected from nuclease activity by a protease-sensitive factor(s). J. Virol. 2004, 78, 10202–10205. [Google Scholar] [CrossRef] [PubMed]
- Berger, C.; Romero-Brey, I.; Radujkovic, D.; Terreux, R.; Zayas, M.; Paul, D.; Harak, C.; Hoppe, S.; Gao, M.; Penin, F.; et al. Daclatasvir-like inhibitors of NS5A block early biogenesis of hepatitis C virus-induced membranous replication factories, independent of RNA replication. Gastroenterology 2014, 147, 1094–1105.e5. [Google Scholar] [CrossRef]
- Ramanan, V.; Trehan, K.; Ong, M.L.; Luna, J.M.; Hoffmann, H.H.; Espiritu, C.; Sheahan, T.P.; Chandrasekar, H.; Schwartz, R.E.; Christine, K.S.; et al. Viral genome imaging of hepatitis C virus to probe heterogeneous viral infection and responses to antiviral therapies. Virology 2016, 494, 236–247. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nucleic Acid Target | Probe Name | Catalog# | Target Region of Genome (Coded Protein) |
|---|---|---|---|
| HCV (+)RNA | V-HCV-GT2a | 441361 | 4268–5505 (NS3-NS4B) |
| HCV (−)RNA | HCV-GT2a-sense-C2 | 441371 | 4268–5505 (NS3-NS4B) |
| ZIKV (+)RNA | V-ZIKA-pp-O2 | 464531 | 866–1763 (M-E) |
| ZIKV (−)RNA | V-ZIKA-pp-O2-sense-C2 | 478731-C2 | 866–1763 (M-E) |
| Copies per Cell | S.E.M. | Fold Difference | S.E.M. | |
|---|---|---|---|---|
| (+)RNA | 3.0 × 106 | 5.7 × 105 | 33 | 2.4 |
| (−)RNA | 9.0 × 104 | 2.2 × 104 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, D.; Tedbury, P.R.; Lan, S.; Huber, A.D.; Puray-Chavez, M.N.; Ji, J.; Michailidis, E.; Saeed, M.; Ndongwe, T.P.; Bassit, L.C.; et al. Visualization of Positive and Negative Sense Viral RNA for Probing the Mechanism of Direct-Acting Antivirals against Hepatitis C Virus. Viruses 2019, 11, 1039. https://doi.org/10.3390/v11111039
Liu D, Tedbury PR, Lan S, Huber AD, Puray-Chavez MN, Ji J, Michailidis E, Saeed M, Ndongwe TP, Bassit LC, et al. Visualization of Positive and Negative Sense Viral RNA for Probing the Mechanism of Direct-Acting Antivirals against Hepatitis C Virus. Viruses. 2019; 11(11):1039. https://doi.org/10.3390/v11111039
Chicago/Turabian StyleLiu, Dandan, Philip R. Tedbury, Shuiyun Lan, Andrew D. Huber, Maritza N. Puray-Chavez, Juan Ji, Eleftherios Michailidis, Mohsan Saeed, Tanyaradzwa P. Ndongwe, Leda C. Bassit, and et al. 2019. "Visualization of Positive and Negative Sense Viral RNA for Probing the Mechanism of Direct-Acting Antivirals against Hepatitis C Virus" Viruses 11, no. 11: 1039. https://doi.org/10.3390/v11111039
APA StyleLiu, D., Tedbury, P. R., Lan, S., Huber, A. D., Puray-Chavez, M. N., Ji, J., Michailidis, E., Saeed, M., Ndongwe, T. P., Bassit, L. C., Schinazi, R. F., Ralston, R., Rice, C. M., & Sarafianos, S. G. (2019). Visualization of Positive and Negative Sense Viral RNA for Probing the Mechanism of Direct-Acting Antivirals against Hepatitis C Virus. Viruses, 11(11), 1039. https://doi.org/10.3390/v11111039