Aptamer Profiling of A549 Cells Infected with Low-Pathogenicity and High-Pathogenicity Influenza Viruses

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.1.1. Cells
2.1.2. Viruses
2.1.3. Infections
2.2. Quantitative SOMAscan® Analyses
2.3. Statistical and Bioinformatics Analyses
3. Results
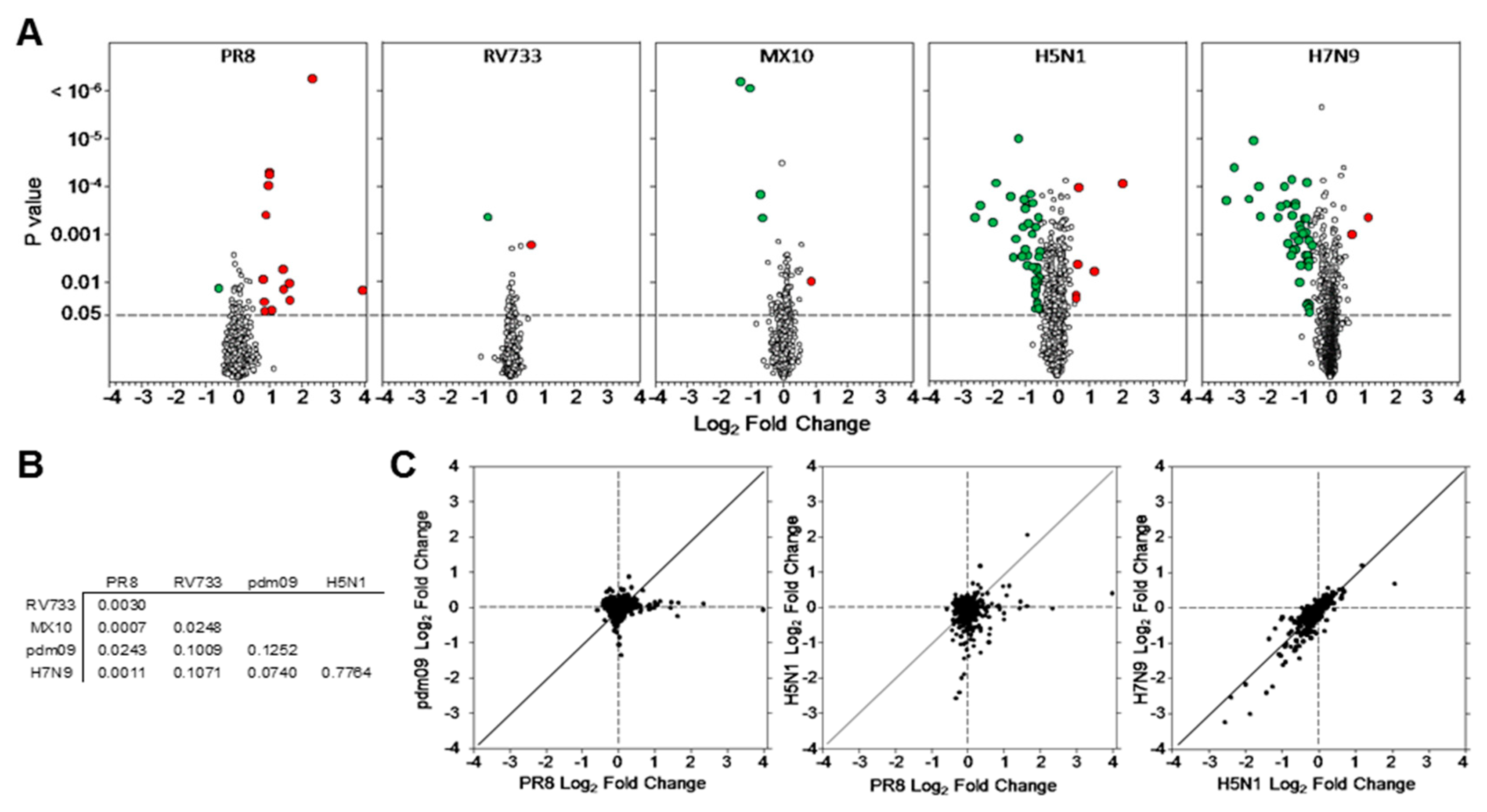
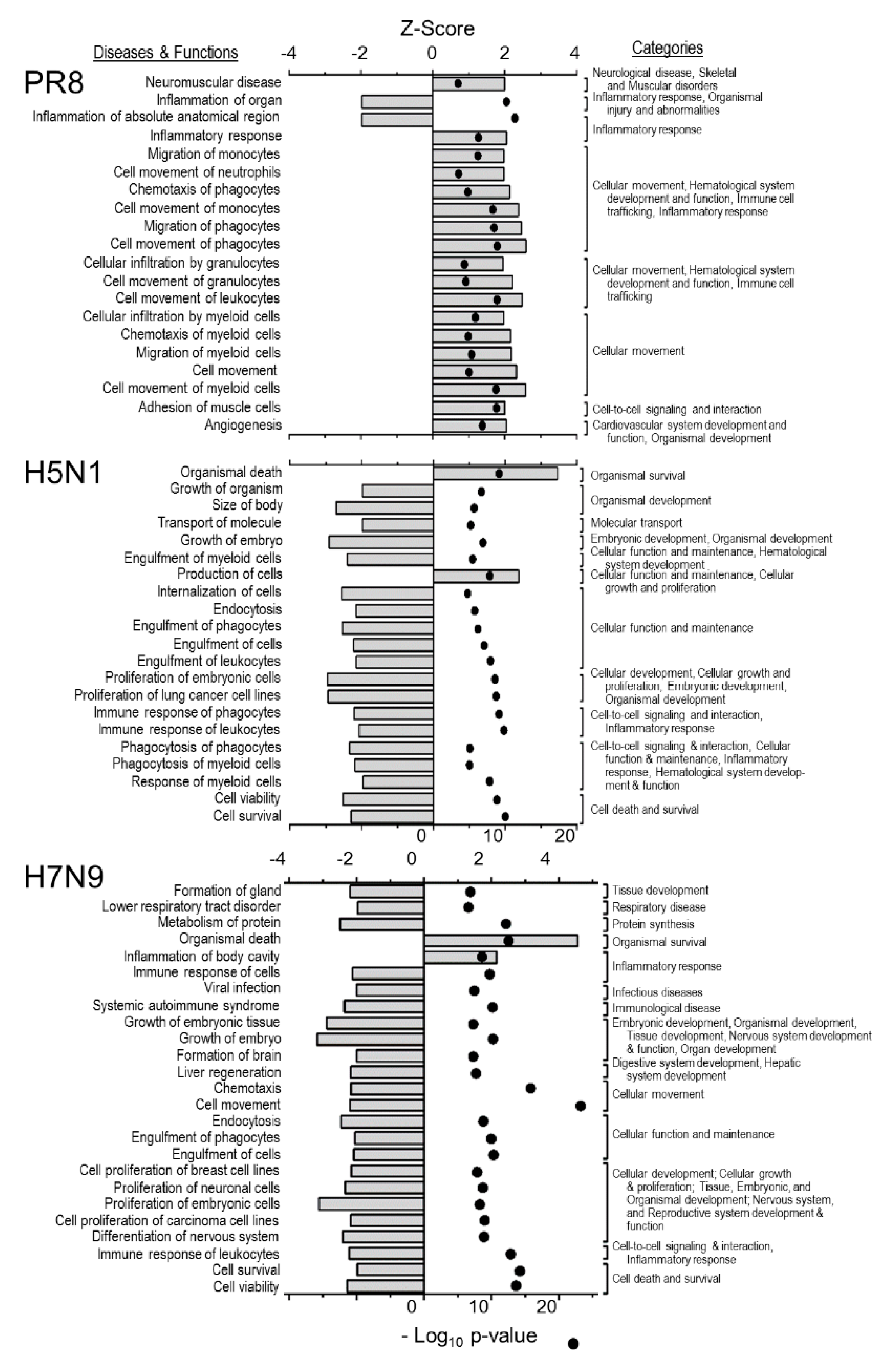
3.1. Dysregulation of A549 Proteins Determined Using SOMAmers
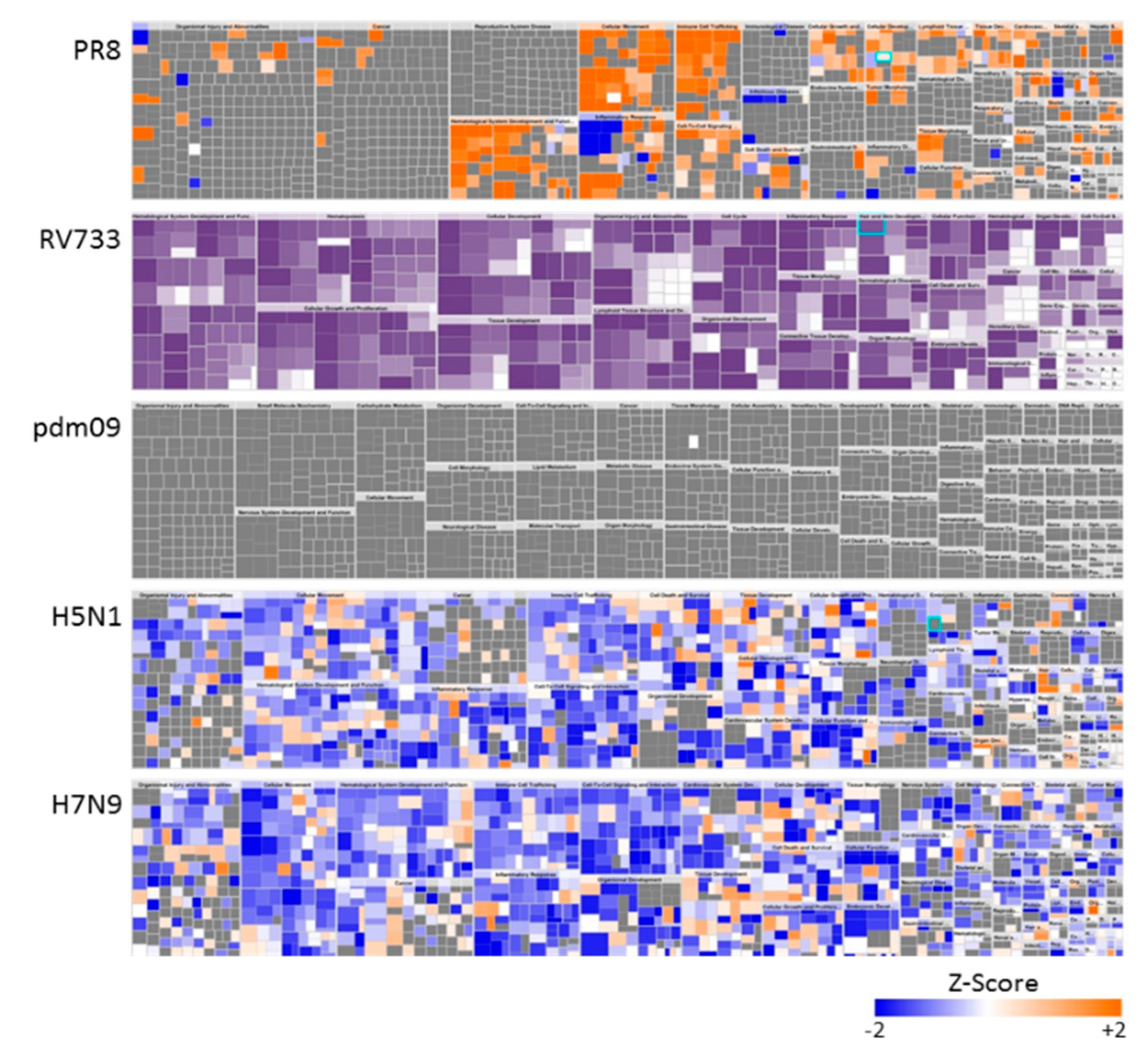
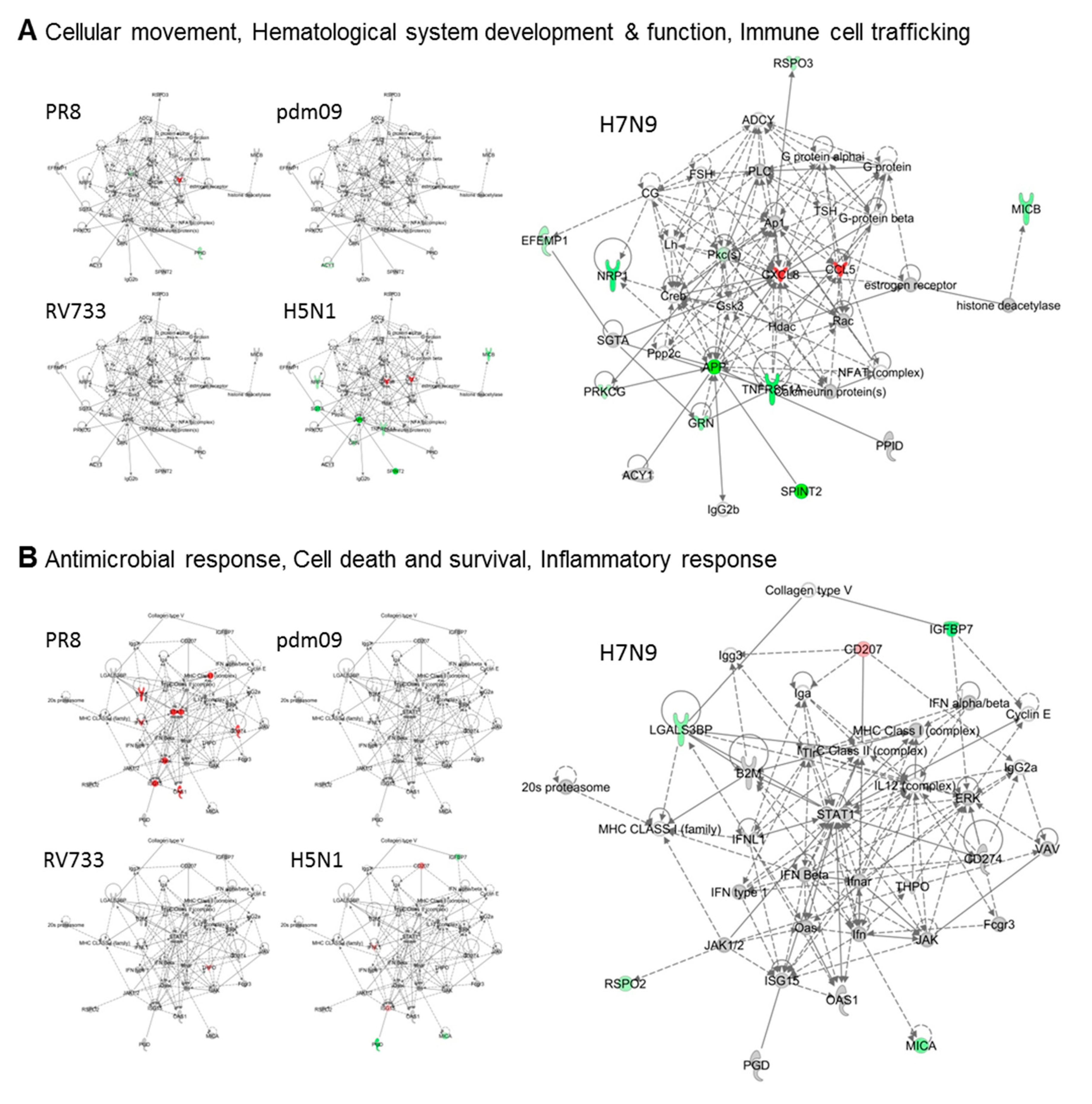
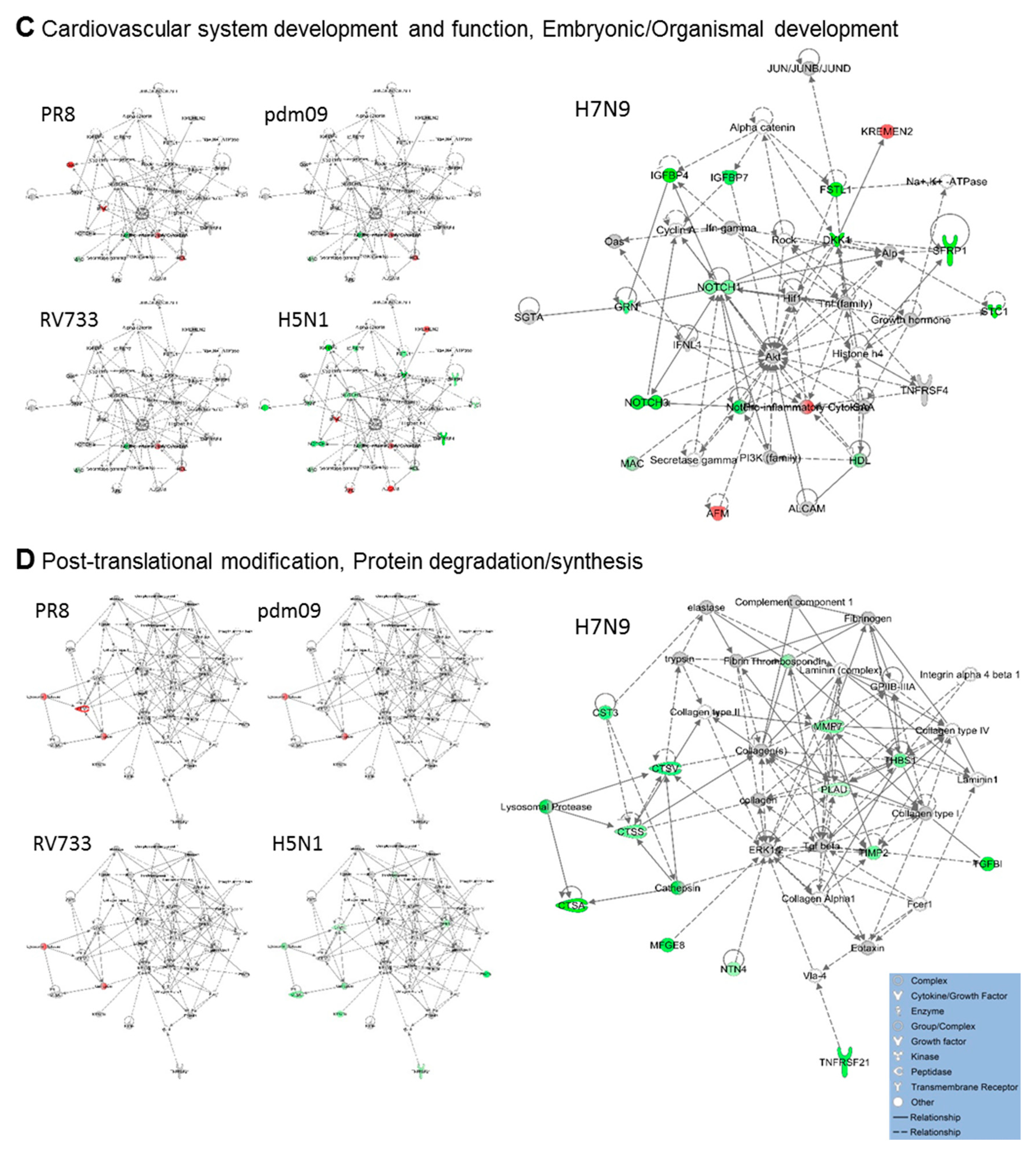
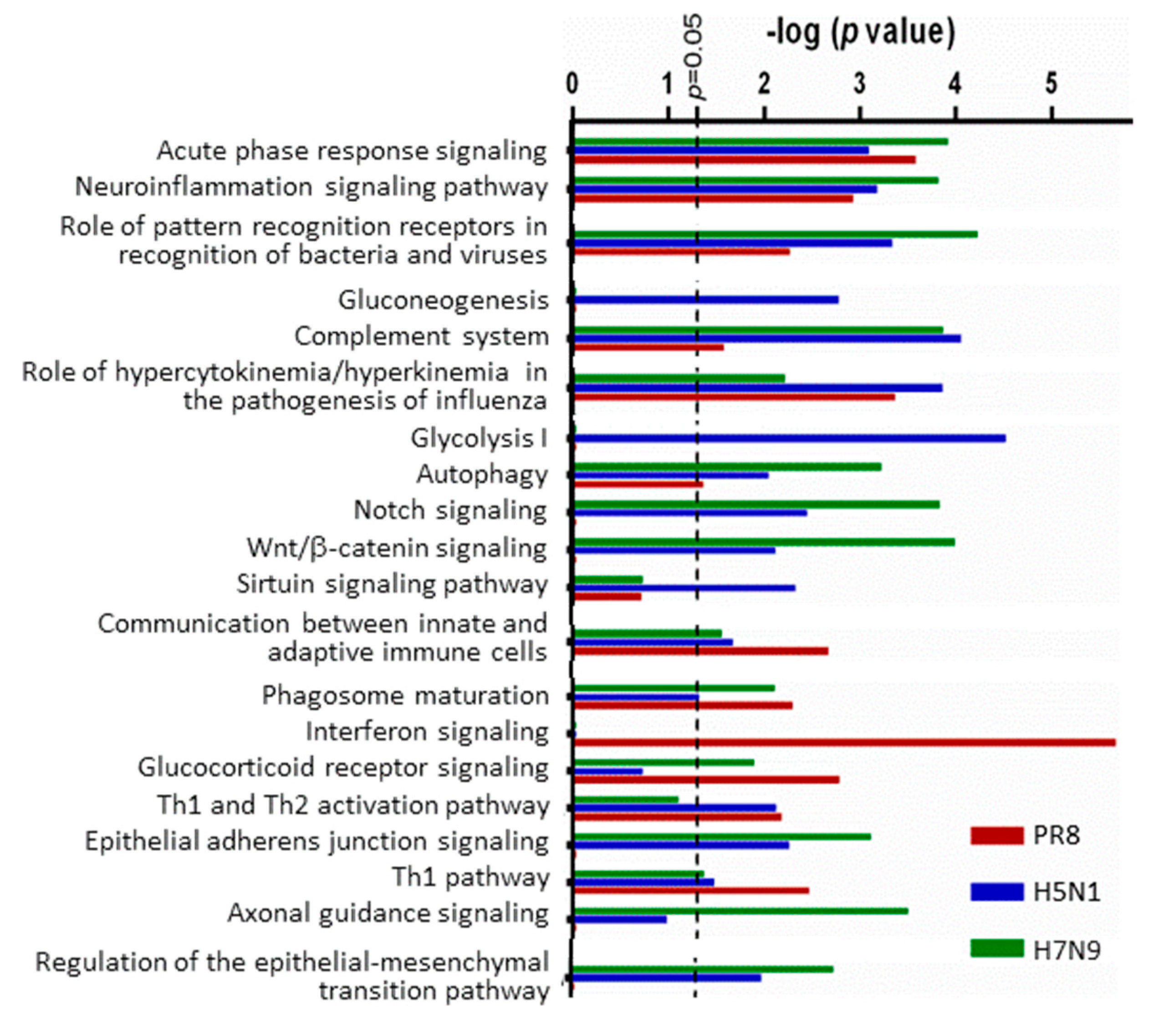
3.2. H5N1 and H7N9 Induce More Profound Proteomic Dysregulation
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Murray, C.J.L.; Lopez, A.D. The Global Burden of Disease. In A Comprehensive Assessment of Mortality and Disability from Diseases, Injuries, and Risk Factors in 1990 and prOjected to 2020; Harvard School of Public Health: Boston, MA, USA, 1996; p. 990. [Google Scholar]
- Taubenberger, J.K.; Hultin, J.V.; Morens, D.M. Discovery and characterization of the 1918 pandemic influenza virus in historical context. Antivir. Ther. 2007, 12, 581–591. [Google Scholar] [PubMed]
- Palese, P.; Shaw, M.L. Orthomyxoviridae: The viruses and their replication. In Fields Virology, 5th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007; pp. 1647–1689. [Google Scholar]
- Jagger, B.W.; Wise, H.M.; Kash, J.C.; Walters, K.A.; Wills, N.M.; Xiao, Y.L.; Dunfee, R.L.; Schwartzman, L.M.; Ozinsky, A.; Bell, G.L.; et al. An overlapping protein-coding region in influenza A virus segment 3 modulates the host response. Science 2012, 337, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Rahim, M.N.; Selman, M.; Sauder, P.J.; Forbes, N.E.; Stecho, W.; Xu, W.; Lebar, M.; Brown, E.G.; Coombs, K.M. Generation and characterization of a new panel of broadly-reactive monoclonal anti-NS1 antibodies for detection of Influenza A virus. J. Gen. Virol. 2013, 94, 592–604. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.X.; Li, Y.; Rivailler, P.; Conrardy, C.; Castillo, D.A.A.; Chen, L.M.; Recuenco, S.; Ellison, J.A.; Davis, C.T.; York, I.A.; et al. A distinct lineage of influenza A virus from bats. Proc. Natl. Acad. Sci. USA 2012, 109, 4269–4274. [Google Scholar] [CrossRef] [Green Version]
- Alexander, D.J. An overview of the epidemiology of avian influenza. Vaccine 2007, 25, 5637–5644. [Google Scholar] [CrossRef]
- Krauss, S.; Obert, C.A.; Franks, J.; Walker, D.; Jones, K.; Seiler, P.; Niles, L.; Pryor, S.P.; Obenauer, J.C.; Naeve, C.W.; et al. Influenza in migratory birds and evidence of limited intercontinental virus exchange. PLoS Path. 2007, 3, 1684–1693. [Google Scholar] [CrossRef]
- Richard, M.; de Graaf, M.; Herfst, S. Avian influenza A viruses: From zoonosis to pandemic. Future Virol. 2014, 9, 513–524. [Google Scholar] [CrossRef]
- Coombs, K.M.; Berard, A.; Xu, W.; Krokhin, O.; Meng, X.; Cortens, J.P.; Kobasa, D.; Wilkins, J.; Brown, E.G. Quantitative proteomic analyses of influenza virus-infected cultured human lung cells. J. Virol. 2010, 84, 10888–10906. [Google Scholar] [CrossRef]
- Kroeker, A.L.; Ezzati, P.; Coombs, K.M.; Halayko, A.J. Influenza A infection of primary human airway epithelial cells up-regulates proteins related to purine metabolism and ubiquitin-related signaling. J. Proteome Res. 2013, 12, 3139–3151. [Google Scholar] [CrossRef]
- Kroeker, A.L.; Ezzati, P.; Halayko, A.J.; Coombs, K.M. Response of primary human airway epithelial cells to Influenza infection – A quantitative proteomic study. J. Proteome Res. 2012, 11, 4132–4136. [Google Scholar] [CrossRef]
- Simon, P.F.; McCorrister, S.; Hu, P.Z.; Chong, P.; Silaghi, A.; Westmacott, G.; Coombs, K.M.; Kobasa, D. Highly pathogenic H5N1 and novel H7N9 influenza A viruses induce more profound proteomic host responses than seasonal and pandemic H1N1 strains. J. Proteome Res. 2015, 14, 4511–4523. [Google Scholar] [CrossRef]
- Vester, D.; Rapp, E.; Gade, D.; Genzel, Y.; Reichl, U. Quantitative analysis of cellular proteome alterations in human influenza A virus-infected mammalian cell lines. Proteomics 2009, 9, 3316–3327. [Google Scholar] [CrossRef] [PubMed]
- Dove, B.K.; Surtees, R.; Bean, T.J.; Munday, D.; Wise, H.M.; Digard, P.; Carroll, M.W.; Ajuh, P.; Barr, J.N.; Hiscox, J.A. A quantitative proteomic analysis of lung epithelial (A549) cells infected with 2009 pandemic influenza A virus using stable isotope labelling with amino acids in cell culture. Proteomics 2012, 12, 1431–1436. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhou, J.; Wang, Y.; Mason, R.J.; Funk, C.J.; Du, Y. Proteome alterations in primary human alveolar macrophages in response to influenza a virus infection. J. Proteome Res. 2012, 11, 4091–4101. [Google Scholar] [CrossRef] [PubMed]
- Cheung, C.Y.; Chan, E.Y.; Krasnoselsky, A.; Purdy, D.; Navare, A.T.; Bryan, J.T.; Leung, C.K.L.; Hui, K.P.Y.; Peiris, J.S.M.; Katze, M.G. H5N1 virus causes significant perturbations in host proteome very early in Influenza virus-infected primary human monocyte-derived macrophages. J. Infect. Dis. 2012, 206, 640–645. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, S.; Vaught, J.D.; Bock, C.; Gold, L.; Katilius, E.; Keeney, T.R.; Kim, N.; Saccomano, N.A.; Wilcox, S.K.; Zichi, D.; et al. From SOMAmer-based biomarker discovery to diagnostic and clinical applications: A SOMAmer-based, streamlined multiplex proteomic assay. PLoS ONE 2011, 6, e26332. [Google Scholar] [CrossRef] [PubMed]
- Gold, L.; Walker, J.J.; Wilcox, S.K.; Williams, S. Advances in human proteomics at high scale with the SOMAscan proteomics platform. New Bio. 2012, 29, 543–549. [Google Scholar] [CrossRef]
- Webber, J.; Stone, T.C.; Katilius, E.; Smith, B.C.; Gordon, B.; Mason, M.D.; Tabi, Z.; Brewis, I.A.; Clayton, A. Proteomics analysis of cancer exosomes using a novel modified aptamer-based array (SOMAscan(TM)) Platform. Mol. Cell. Proteom. 2014, 13, 1050–1064. [Google Scholar] [CrossRef]
- Brown, E.G. Increased virulence of a mouse-adapted variant of Influenza A/Fm/1/47 virus is controlled by mutations in genome segments 4, 5, 7, and 8. J. Virol. 1990, 64, 4523–4533. [Google Scholar]
- SOMAScan. Proteomic Assay Technical White Paper, SSM-002, Rev. 4; SOMAScan: Venice, Italy, 2017. [Google Scholar]
- Glover, K.K.M.; Gao, A.; Coombs, K.M. Vero cell proteomic changes induced by Zika virus infection. Proteomics 2019, 19, e1800309. [Google Scholar] [CrossRef]
- Xie, H.; Wan, X.F.; Ye, Z.P.; Plant, E.P.; Zhao, Y.Q.; Xu, Y.F.; Li, X.; Finch, C.; Zhao, N.; Kawano, T.; et al. H3N2 mismatch of 2014–2015 Northern hemisphere influenza vaccines and head-to-head comparison between human and ferret antisera derived antigenic maps. Sci. Rep. 2015, 5, 15279. [Google Scholar] [CrossRef] [PubMed]
- Lewnard, J.A.; Cobey, S. Immune history and influenza vaccine effectiveness. Vaccines 2018, 6, 28. [Google Scholar] [CrossRef] [PubMed]
- Lackenby, A.; Thompson, C.I.; Democratis, J. The potential impact of neuraminidase inhibitor resistant influenza. Curr. Opin. Inf. Dis. 2008, 21, 626–638. [Google Scholar] [CrossRef] [PubMed]
- Hurt, A.C.; Besselaar, T.G.; Daniels, R.S.; Ermetal, B.; Fry, A.; Gubareva, L.; Huang, W.J.; Lackenby, A.; Lee, R.T.C.; Lo, J.; et al. Global update on the susceptibility of human influenza viruses to neuraminidase inhibitors, 2014–2015. Antivir. Res. 2016, 132, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Karlas, A.; Machuy, N.; Shin, Y.; Pleissner, K.P.; Artarini, A.; Heuer, D.; Becker, D.; Khalil, H.; Ogilvie, L.A.; Hess, S.; et al. Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature 2010, 463, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Konig, R.; Stertz, S.; Zhou, Y.; Inoue, A.; Hoffmann, H.H.; Bhattacharyya, S.; Alamares, J.G.; Tscherne, D.M.; Ortigoza, M.B.; Liang, Y.; et al. Human host factors required for influenza virus replication. Nature 2010, 463, 813–817. [Google Scholar] [CrossRef]
- Watanabe, T.; Watanabe, S.; Kawaoka, Y. Cellular networks involved in the Influenza virus life cycle. Cell Host Microbe 2010, 7, 427–439. [Google Scholar] [CrossRef]
- Tran, A.T.; Rahim, M.N.; Ranadheera, C.; Kroeker, A.; Cortens, J.P.; Opanubi, K.J.; Wilkins, J.A.; Coombs, K.M. Knockdown of specific host factors protects against influenza virus-induced cell death. Cell Death Dis. 2013, 4, e769. [Google Scholar] [CrossRef]
- Dhingra, V.; Li, Q.; Allison, A.B.; Stallknecht, D.E.; Fu, Z.F. Proteomic profiling and neurodegeneration in West-Nile-virus-infected neurons. J. Biomed. Biotechnol. 2005, 2005, 271–279. [Google Scholar] [CrossRef]
- Zhang, M.C.; Zheng, X.Y.; Wu, Y.; Gan, M.; He, A.; Li, Z.Y.; Zhang, D.J.; Wu, X.S.; Zhan, X.M. Differential proteomics of Aedes albopictus salivary gland, midgut and C6/36 cell induced by dengue virus infection. Virology 2013, 444, 109–118. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.K.; Chai, F.; Li, H.Y.; Xiao, G.F.; Guo, L. Identification of host proteins involved in Japanese encephalitis virus infection by quantitative proteomics analysis. J. Proteome Res. 2013, 12, 2666–2678. [Google Scholar] [CrossRef]
- Yang, R.W.; Klimentova, J.; Gockel-Krzikalla, E.; Ly, R.; Gmelin, N.; Hotz-Wagenblatt, A.; Rehulkova, H.; Stulik, J.; Rosl, F.; Niebler, M. Combined transcriptome and proteome analysis of immortalized human keratinocytes expressing human papillomavirus 16 (HPV16) oncogenes reveals novel key factors and networks in HPV-induced carcinogenesis. Msphere 2019, 4, e00129-19. [Google Scholar] [CrossRef] [PubMed]
- Niu, D.D.; Sui, J.J.; Zhang, J.H.; Feng, H.X.; Chen, W.N. iTRAQ-coupled 2-D LC-MS/MS analysis of protein profile associated with HBV-modulated DNA methylation. Proteomics 2009, 9, 3856–3868. [Google Scholar] [CrossRef] [PubMed]
- Shobahah, J.; Xue, S.J.; Hu, D.B.; Zhao, C.; Wei, M.; Quan, Y.P.; Yu, W. Quantitative phosphoproteome on the silkworm (Bombyx mori) cells infected with baculovirus. Virol. J. 2017, 14, 117. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.K.; Wang, B.; Xin, Q.L.; Shang, W.J.; Shen, S.; Xiao, G.F.; Deng, F.; Wang, H.L.; Hu, Z.H.; Wang, M.L. Quantitative proteomic analysis reveals unfolded-protein response involved in severe fever with thrombocytopenia syndrome virus infection. J. Virol. 2019, 93, e00308–e00319. [Google Scholar] [CrossRef]
- Luber, C.A.; Cox, J.; Lauterbach, H.; Fancke, B.; Selbach, M.; Tschopp, J.; Akira, S.; Wiegand, M.; Hochrein, H.; O’Keeffe, M.; et al. Quantitative proteomics reveals subset-specific viral recognition in dendritic cells. Immunity 2010, 32, 279–289. [Google Scholar] [CrossRef]
- Romas, L.M.; Hasselrot, K.; Aboud, L.G.; Birse, K.D.; Ball, T.B.; Broliden, K.; Burgener, A.D. A comparative proteomic analysis of the soluble immune factor environment of rectal and oral mucosa. PLoS ONE 2014, 9, e100820. [Google Scholar] [CrossRef]
- Lemay, M.L.; Otto, A.; Maass, S.; Plate, K.; Becher, D.; Moineau, S. Investigating Lactococcus lactis MG1363 response to phage p2 infection at the proteome level. Mol. Cell. Proteom. 2019, 18, 704–714. [Google Scholar] [CrossRef]
- Yates, J.R.; Ruse, C.I.; Nakorchevsky, A. Proteomics by mass spectrometry: Approaches, advances, and applications. Ann. Rev. Biomed. Eng. 2009, 11, 49–79. [Google Scholar] [CrossRef]
- Coombs, K.M. Quantitative proteomics of complex mixtures. Expert Rev. Proteom. 2011, 8, 659–677. [Google Scholar] [CrossRef]
- Vidova, V.; Spacil, Z. A review on mass spectrometry-based quantitative proteomics: Targeted and data independent acquisition. Anal. Chim. Acta 2017, 964, 7–23. [Google Scholar] [CrossRef] [PubMed]
- Mehan, M.R.; Ayers, D.; Thirstrup, D.; Xiong, W.; Ostroff, R.M.; Brody, E.N.; Walker, J.J.; Gold, L.; Jarvis, T.C.; Janjic, N.; et al. Protein signature of lung cancer tissues. PLoS ONE 2012, 7, e35157. [Google Scholar] [CrossRef] [PubMed]
- Sattlecker, M.; Kiddle, S.J.; Newhouse, S.; Proitsi, P.; Nelson, S.; Williams, S.; Johnston, C.; Killick, R.; Simmons, A.; Westman, E.; et al. Alzheimer’s disease biomarker discovery using SOMAscan multiplexed protein technology. Alzheimers Dement. 2014, 10, 724–734. [Google Scholar] [CrossRef] [PubMed]
- Marion, T.; Elbahesh, H.; Thomas, P.G.; DeVincenzo, J.P.; Webby, R.; Schughart, K. Respiratory mucosal proteome quantification in human influenza infections. PLoS ONE 2016, 11, e0153674. [Google Scholar] [CrossRef]
- Sher, A.A.; Glover, K.K.M.; Coombs, K.M. Zika virus infection disrupts astrocytic proteins involved in synapse control and axon guidance. Front. Microbiol. 2019, 10, 596. [Google Scholar] [CrossRef]
- Menachery, V.D.; Eisfeld, A.J.; Schafer, A.; Josset, L.; Sims, A.C.; Proll, S.; Fan, S.F.; Li, C.J.; Neumann, G.; Tilton, S.C.; et al. Pathogenic influenza viruses and coronaviruses utilize similar and contrasting approaches to control interferon-stimulated gene responses. Mbio 2014, 5, e01174-14. [Google Scholar] [CrossRef]
- Bavagnoli, L.; Maga, G. Identification of host cell factors involved in influenza A virus infection. Future Virol. 2013, 8, 195–208. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number That Are Significant | Total Unique | PR8 | RV733 | pdm09 | H5N1 | H7N9 |
|---|---|---|---|---|---|---|
| and fold-change > 1.000 | 510 | 33 | 20 | 67 | 194 | 133 |
| and fold-change < 0.9999 | 15 | 7 | 38 | 166 | 168 | |
| and fold-change > 1.250 | 128 | 17 | 1 | 7 | 15 | 10 |
| and fold-change < 0.8000 | 1 | 1 | 14 | 57 | 65 | |
| and fold-change > 1.333 | 98 | 15 | 1 | 6 | 8 | 6 |
| and fold-change < 0.7500 | 1 | 1 | 6 | 45 | 56 | |
| and fold-change > 1.500 | 76 | 14 | 1 | 1 | 6 | 2 |
| and fold-change < 0.6667 | 1 | 1 | 4 | 32 | 45 | |
| and fold-change > 2.000 | 33 | 8 | 0 | 0 | 2 | 1 |
| and fold-change < 0.5000 | 0 | 0 | 2 | 11 | 19 | |
| and fold-change > 3.000 | 11 | 4 | 0 | 0 | 1 | 0 |
| and fold-change < 0.3333 | 0 | 0 | 0 | 4 | 7 | |
| and fold-change > 5.000 | 6 | 2 | 0 | 0 | 0 | 0 |
| and fold-change < 0.2000 | 0 | 0 | 0 | 2 | 4 |
| EntrezGene Symbol | Protein | Fold-Change Compared to Sham-Infected | ||||
|---|---|---|---|---|---|---|
| H1N1 Viruses | ||||||
| PR8 | RV733 | pdm09 | H5N1 | H7N9 | ||
| Up-Regulated Proteins | ||||||
| ISG15 | Ubiquitin-like protein ISG15 | 15.9 | 1.03 | 0.96 | 1.33 | 1.10 |
| OAS1 | 2′-5′-oligoadenylate synthase 1 | 5.06 | 0.99 | 1.08 | 0.97 | 0.96 |
| CCL5 | C-C motif chemokine 5 | 3.11 | 1.06 | 1.09 | 4.18 | 1.62 |
| STAT1 | Signal transducer and activator of transcription 1-alpha/beta | 3.09 | 1.26 | 0.85 | 1.02 | 1.00 |
| B2M | Beta-2-microglobulin | 2.72 | 0.97 | 1.11 | 1.12 | 0.99 |
| APOL1 | Apolipoprotein L1 | 2.68 | 0.98 | 0.99 | 0.99 | 0.92 |
| CD274 | Programmed cell death 1 ligand 1 | 2.09 | 1.06 | 1.05 | 1.27 | 1.13 |
| CTSS | Cathepsin S | 2.00 | 0.99 | 1.05 | 0.79 | 0.59 |
| SERPINE1 | Plasminogen activator inhibitor 1 | 1.99 | 1.00 | 1.10 | 0.89 | 0.70 |
| IFNL1 | Interferon lambda-1 | 1.97 | 1.00 | 0.99 | 1.52 | 1.21 |
| F2 | Thrombin | 1.84 | 1.03 | 1.00 | 0.97 | 0.89 |
| PLAUR | Urokinase plasminogen activator surface receptor | 1.80 | 0.98 | 1.05 | 1.05 | 0.85 |
| MDK | Midkine | 1.80 | 1.01 | 0.97 | 1.02 | 0.93 |
| CFB | Complement factor B | 1.75 | 1.04 | 1.03 | 1.03 | 0.96 |
| THPO | Thrombopoietin | 1.01 | 1.58 | 1.01 | 1.00 | 1.10 |
| L1CAM | Neural cell adhesion molecule L1 | 1.21 | 1.28 | 1.83 | 1.57 | 1.47 |
| CXCL8 | Interleukin-8 | 1.26 | 1.00 | 0.99 | 2.26 | 2.30 |
| CD207 | C-type lectin domain family 4 member K | 1.10 | 1.05 | 1.06 | 1.61 | 1.38 |
| F9 | Coagulation factor IX | 2.21 | 1.03 | 1.14 | 1.53 | 1.29 |
| Down-regulated proteins | ||||||
| PPID | Peptidyl-prolyl cis-trans isomerase D | 0.66 | 1.15 | 0.95 | 0.97 | 1.03 |
| TGM3 | Protein-glutamine gamma-glutamyltransferase E | 0.88 | 0.61 | 1.21 | 0.75 | 0.90 |
| PGAM1 | Phosphoglycerate mutase 1 | 1.05 | 0.83 | 0.39 | 0.54 | 0.50 |
| MDH1 | Malate dehydrogenase, cytoplasmic | 1.01 | 1.02 | 0.48 | 0.59 | 0.83 |
| LDHB | L-lactate dehydrogenase B chain | 0.97 | 1.19 | 0.61 | 0.84 | 1.02 |
| ENO1 | Alpha-enolase | 0.91 | 0.96 | 0.63 | 0.77 | 0.82 |
| PCSK9 | Proprotein convertase subtilisin/kexin type 9 | 0.80 | 0.92 | 1.07 | 0.17 | 0.11 |
| DKK1 | Dickkopf-related protein 1 | 0.85 | 0.87 | 0.92 | 0.19 | 0.17 |
| DKK4 | Dickkopf-related protein 4 | 0.88 | 0.91 | 0.97 | 0.25 | 0.22 |
| APP | Amyloid beta A4 protein | 0.93 | 0.94 | 0.99 | 0.27 | 0.12 |
| SPINT2 | Kunitz-type protease inhibitor 2 | 0.93 | 0.87 | 0.95 | 0.37 | 0.19 |
| TNFRSF4 | Tumor necrosis factor receptor superfamily member 4 | 1.01 | 0.53 | 0.56 | 0.39 | 0.54 |
| IGFBP4 | Insulin-like growth factor-binding protein 4 | 1.28 | 0.95 | 1.06 | 0.41 | 0.21 |
| PGD | 6-phosphogluconate dehydrogenase, decarboxylating | 0.98 | 1.12 | 0.80 | 0.44 | 0.66 |
| FN1 | Fibronectin | 1.12 | 0.94 | 0.95 | 0.47 | 0.40 |
| TGFBI | Transforming growth factor-beta-induced protein ig-h3 | 0.87 | 0.95 | 0.95 | 0.48 | 0.39 |
| SGTA | Small glutamine-rich tetratricopeptide repeat-containing protein alpha | 0.93 | 1.01 | 0.65 | 0.49 | 0.80 |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase | 0.97 | 1.00 | 0.70 | 0.50 | 0.83 |
| FSTL3 | Follistatin-related protein 3 | 1.49 | 0.92 | 1.04 | 0.50 | 0.33 |
| FN1 | Fibronectin Fragment 3 | 1.12 | 0.95 | 0.94 | 0.53 | 0.47 |
| CTSA | Lysosomal protective protein | 0.75 | 0.94 | 1.04 | 0.53 | 0.35 |
| MICB | MHC class I polypeptide-related sequence B | 0.88 | 0.93 | 1.00 | 0.57 | 0.60 |
| NOTCH3 | Neurogenic locus notch homolog protein 3 | 1.06 | 0.90 | 0.95 | 0.59 | 0.47 |
| PKM2 | Pyruvate kinase PKM | 0.80 | 1.10 | 1.38 | 0.59 | 0.81 |
| LRIG3 | Leucine-rich repeats and immunoglobulin-like domains protein 3 | 1.50 | 0.87 | 0.92 | 0.59 | 0.63 |
| MFGE8 | Lactadherin | 0.91 | 0.94 | 1.05 | 0.63 | 0.43 |
| PEX5 | Peroxisomal targeting signal 1 receptor | 0.98 | 0.71 | 0.73 | 0.63 | 0.75 |
| WNK3 | Serine/threonine-protein kinase WNK3 | 1.10 | 0.93 | 0.86 | 0.63 | 0.77 |
| TNFRSF21 | Tumor necrosis factor receptor superfamily member 21 | 0.96 | 0.92 | 0.96 | 0.63 | 0.43 |
| SFRP1 | Secreted frizzled-related protein 1 | 1.27 | 1.00 | 0.98 | 0.64 | 0.45 |
| TNFRSF1A | Tumor necrosis factor receptor superfamily member 1A | 0.74 | 0.97 | 1.09 | 0.64 | 0.46 |
| FSTL1 | Follistatin-related protein 1 | 1.28 | 0.92 | 1.02 | 0.65 | 0.44 |
| IGFBP7 | Insulin-like growth factor-binding protein 7 | 1.06 | 0.96 | 1.00 | 0.65 | 0.53 |
| NRP1 | Neuropilin-1 | 1.09 | 0.94 | 0.89 | 0.65 | 0.53 |
| CSF3R | Granulocyte colony-stimulating factor receptor | 1.03 | 0.75 | 0.83 | 0.66 | 0.75 |
| C3 | C3a anaphylatoxin des Arginine | 1.12 | 0.96 | 1.00 | 0.66 | 0.61 |
| CFH | Complement factor H | 1.54 | 1.00 | 0.96 | 0.66 | 0.52 |
| STC1 | Stanniocalcin-1 | 1.19 | 0.89 | 1.05 | 0.69 | 0.37 |
| FGFR1 | Fibroblast growth factor receptor 1 | 1.02 | 0.94 | 1.02 | 0.69 | 0.47 |
| CTSV | Cathepsin L2 | 0.83 | 0.98 | 1.08 | 0.74 | 0.52 |
| CST3 | Cystatin-C | 1.39 | 0.94 | 1.10 | 0.86 | 0.55 |
| PLXNB2 | Plexin-B2 | 1.05 | 1.02 | 1.14 | 0.71 | 0.58 |
| LGALS8 | Galectin-8 | 1.18 | 0.92 | 0.92 | 0.78 | 0.59 |
| NRG1 | Neuregulin-1 | 1.21 | 1.03 | 1.24 | 0.94 | 0.60 |
| GNS | N-acetylglucosamine-6-sulfatase | 0.95 | 1.04 | 1.09 | 0.75 | 0.60 |
| MICA | MHC class I polypeptide-related sequence A | 0.85 | 1.04 | 1.18 | 0.68 | 0.61 |
| LAMA1 LAMB1 LAMC1 | Laminin | 0.76 | 1.09 | 1.14 | 0.79 | 0.62 |
| THBS1 | Thrombospondin-1 | 1.05 | 1.01 | 1.04 | 0.68 | 0.62 |
| TIMP2 | Metalloproteinase inhibitor 2 | 0.97 | 0.93 | 1.03 | 0.85 | 0.62 |
| MMP7 | Matrilysin | 0.93 | 0.99 | 1.03 | 0.93 | 0.63 |
| LCN2 | Neutrophil gelatinase-associated lipocalin | 0.78 | 0.96 | 1.03 | 0.87 | 0.63 |
| GRN | Granulins | 1.04 | 0.96 | 1.04 | 0.67 | 0.63 |
| TFPI | Tissue factor pathway inhibitor | 0.80 | 1.03 | 1.03 | 1.10 | 0.63 |
| GFRA1 | GDNF family receptor alpha-1 | 0.77 | 0.97 | 0.99 | 0.75 | 0.63 |
| MET | Hepatocyte growth factor receptor | 1.23 | 1.03 | 1.11 | 0.89 | 0.64 |
| KIR2DL4 | Killer cell immunoglobulin-like receptor 2DL4 | 0.92 | 1.01 | 0.92 | 0.77 | 0.65 |
| LGALS3BP | Galectin-3-binding protein | 1.49 | 0.97 | 1.01 | 0.83 | 0.65 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coombs, K.M.; Simon, P.F.; McLeish, N.J.; Zahedi-Amiri, A.; Kobasa, D. Aptamer Profiling of A549 Cells Infected with Low-Pathogenicity and High-Pathogenicity Influenza Viruses. Viruses 2019, 11, 1028. https://doi.org/10.3390/v11111028
Coombs KM, Simon PF, McLeish NJ, Zahedi-Amiri A, Kobasa D. Aptamer Profiling of A549 Cells Infected with Low-Pathogenicity and High-Pathogenicity Influenza Viruses. Viruses. 2019; 11(11):1028. https://doi.org/10.3390/v11111028
Chicago/Turabian StyleCoombs, Kevin M., Philippe F. Simon, Nigel J. McLeish, Ali Zahedi-Amiri, and Darwyn Kobasa. 2019. "Aptamer Profiling of A549 Cells Infected with Low-Pathogenicity and High-Pathogenicity Influenza Viruses" Viruses 11, no. 11: 1028. https://doi.org/10.3390/v11111028
APA StyleCoombs, K. M., Simon, P. F., McLeish, N. J., Zahedi-Amiri, A., & Kobasa, D. (2019). Aptamer Profiling of A549 Cells Infected with Low-Pathogenicity and High-Pathogenicity Influenza Viruses. Viruses, 11(11), 1028. https://doi.org/10.3390/v11111028