Gut DNA Virome Diversity and Its Association with Host Bacteria Regulate Inflammatory Phenotype and Neuronal Immunotoxicity in Experimental Gulf War Illness

 ,
,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Animals
2.3. Rodent Model of Gulf War Illness (GWI)
2.4. Microbiome Analysis
Virus-Like Particle (VLP) Enrichment and Total Nucleic Acid Extraction
2.5. Virome Analysis
2.6. Bacteriome Analysis
2.7. Availability of Data and Materials
2.8. Immunohistochemistry
2.9. Immunofluorescence Staining
2.10. Real-Time Quantitative PCR
2.11. Western Blot
2.12. Serum ELISA
2.13. Statistical Analysis
3. Results
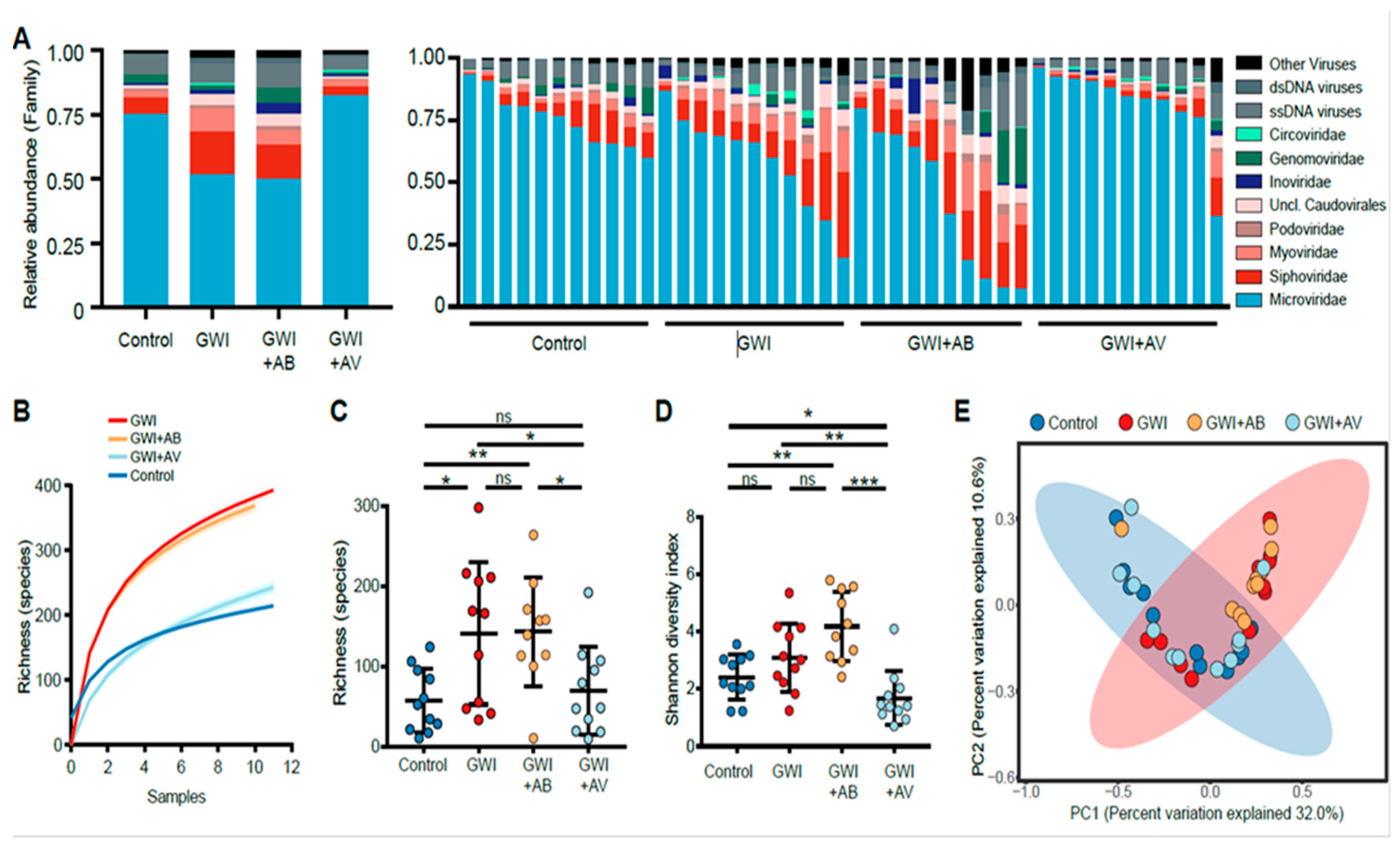
3.1. Gulf War Chemical Exposure Alters the Gut Virome
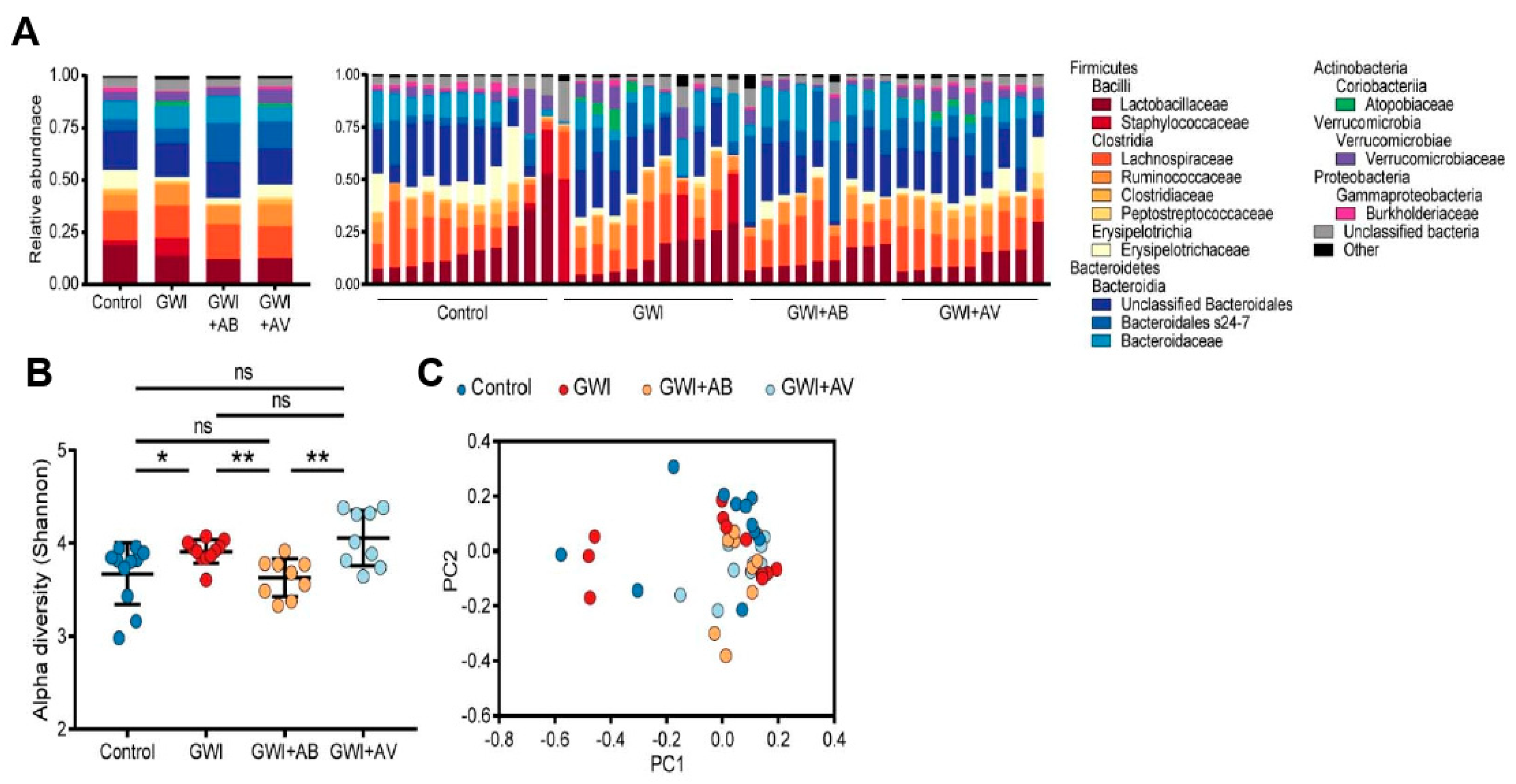
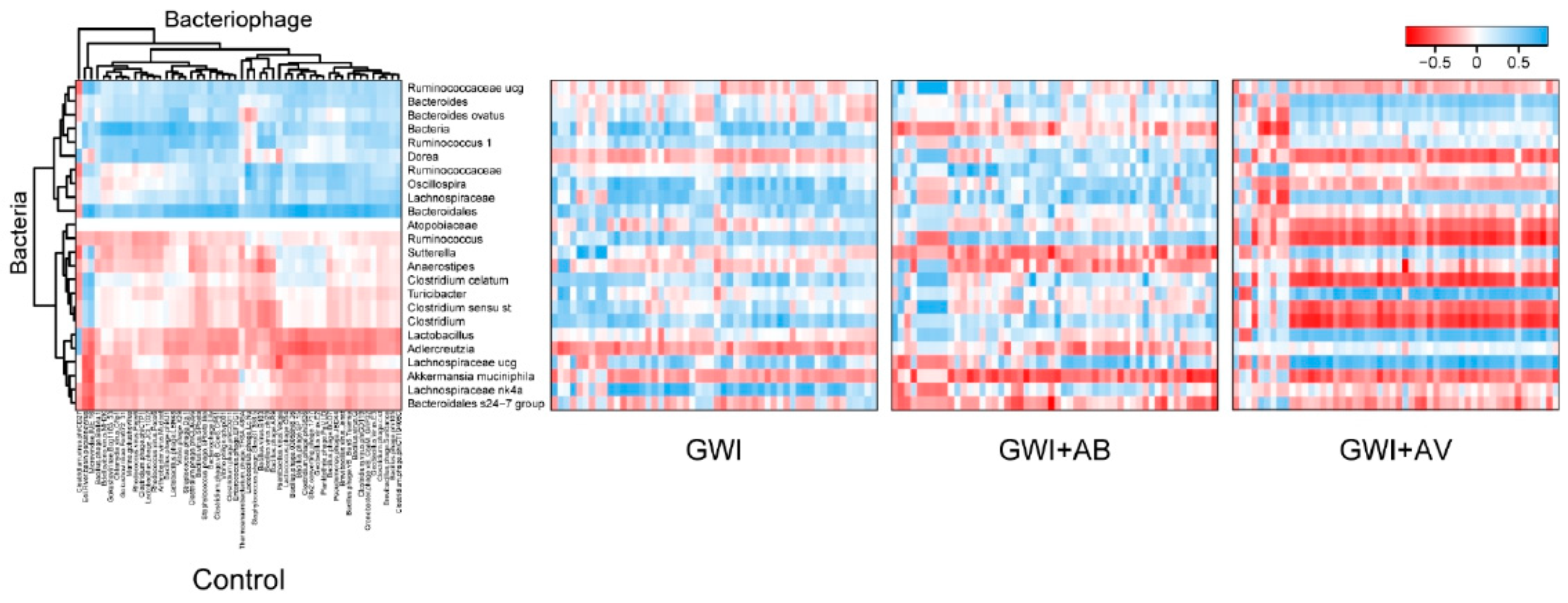
3.2. Gut Bacterial Dysbiosis Induced by Gulf War Chemical Exposure Is Altered by Antibiotics but Not by Antiviral Treatment
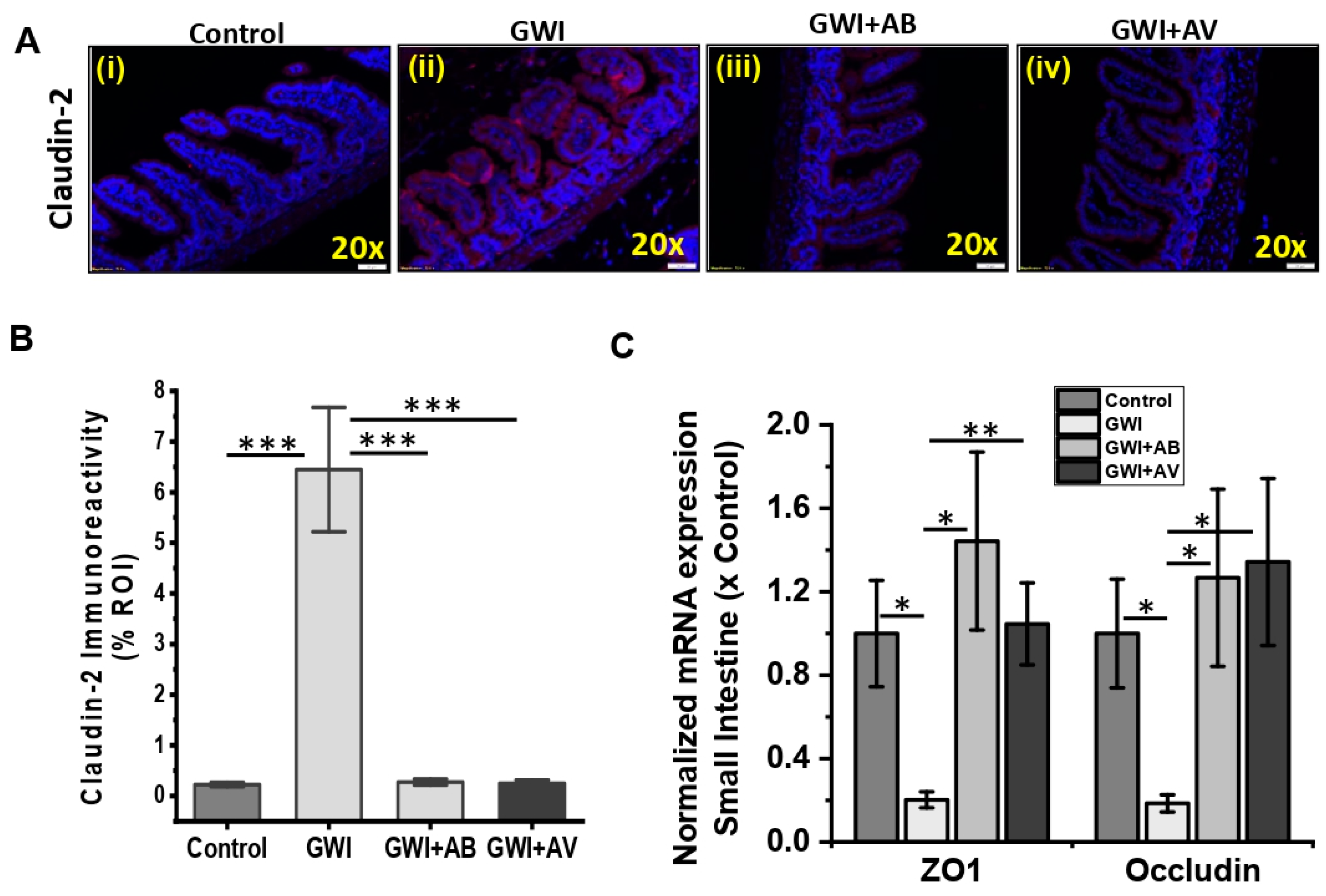
3.3. Gut Bacterial and Viral Dysbiosis Induced Alteration of Intestinal Epithelial Tight Junction Proteins Is Restored by Antibiotics and Antiviral Treatment
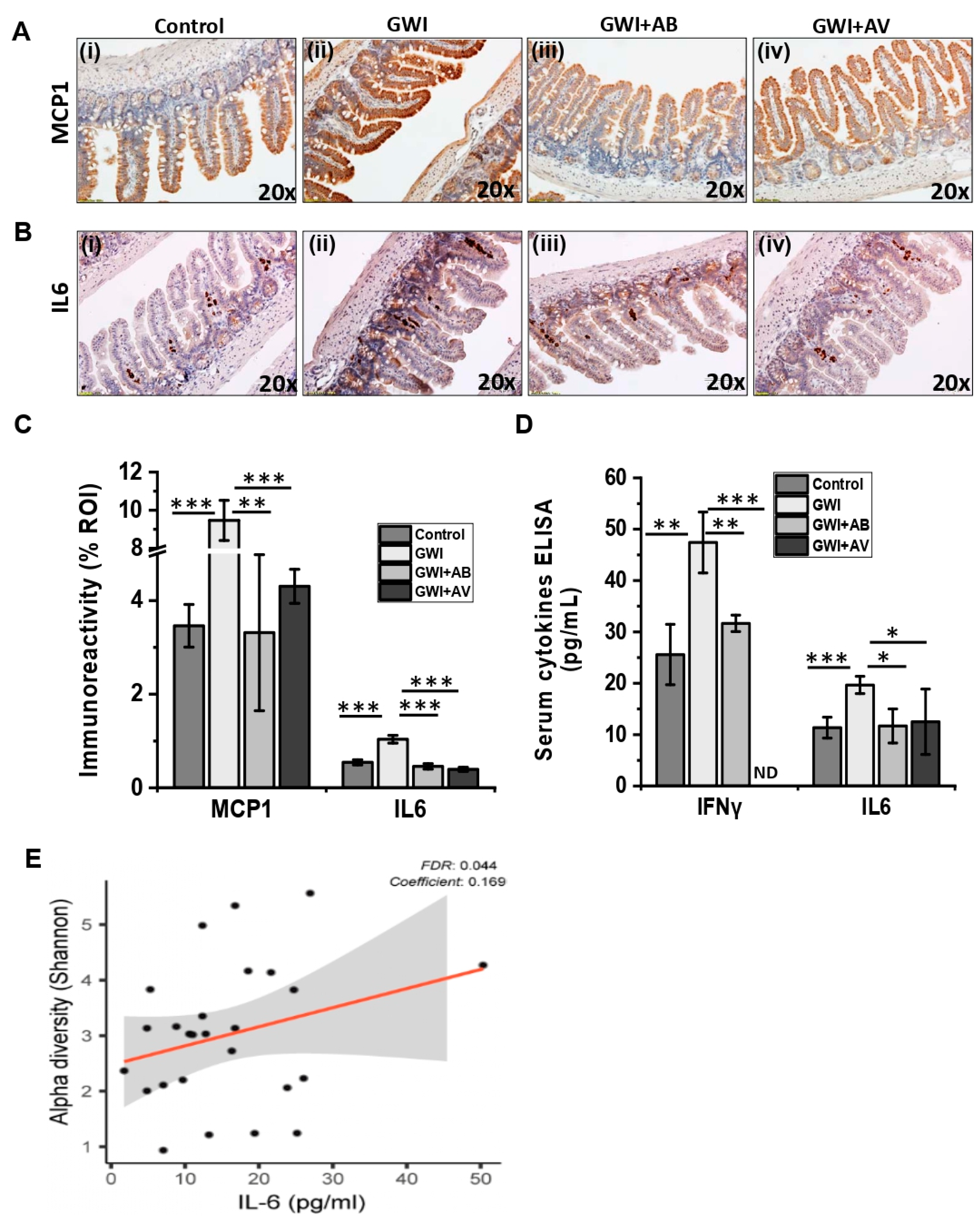
3.4. Viral Dysbiosis in GWI Mice Was Accompanied by Intestinal Inflammation in GWI
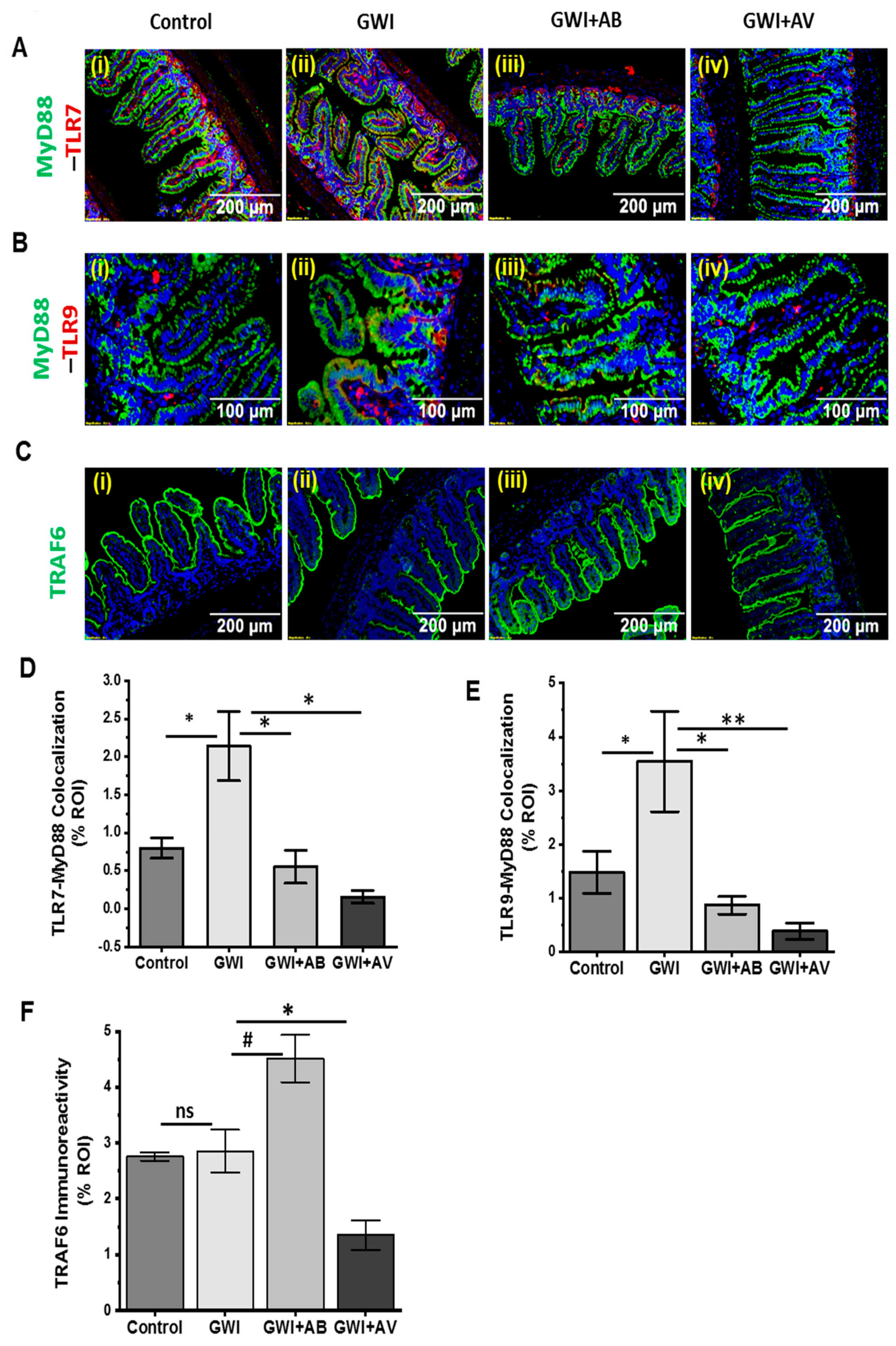
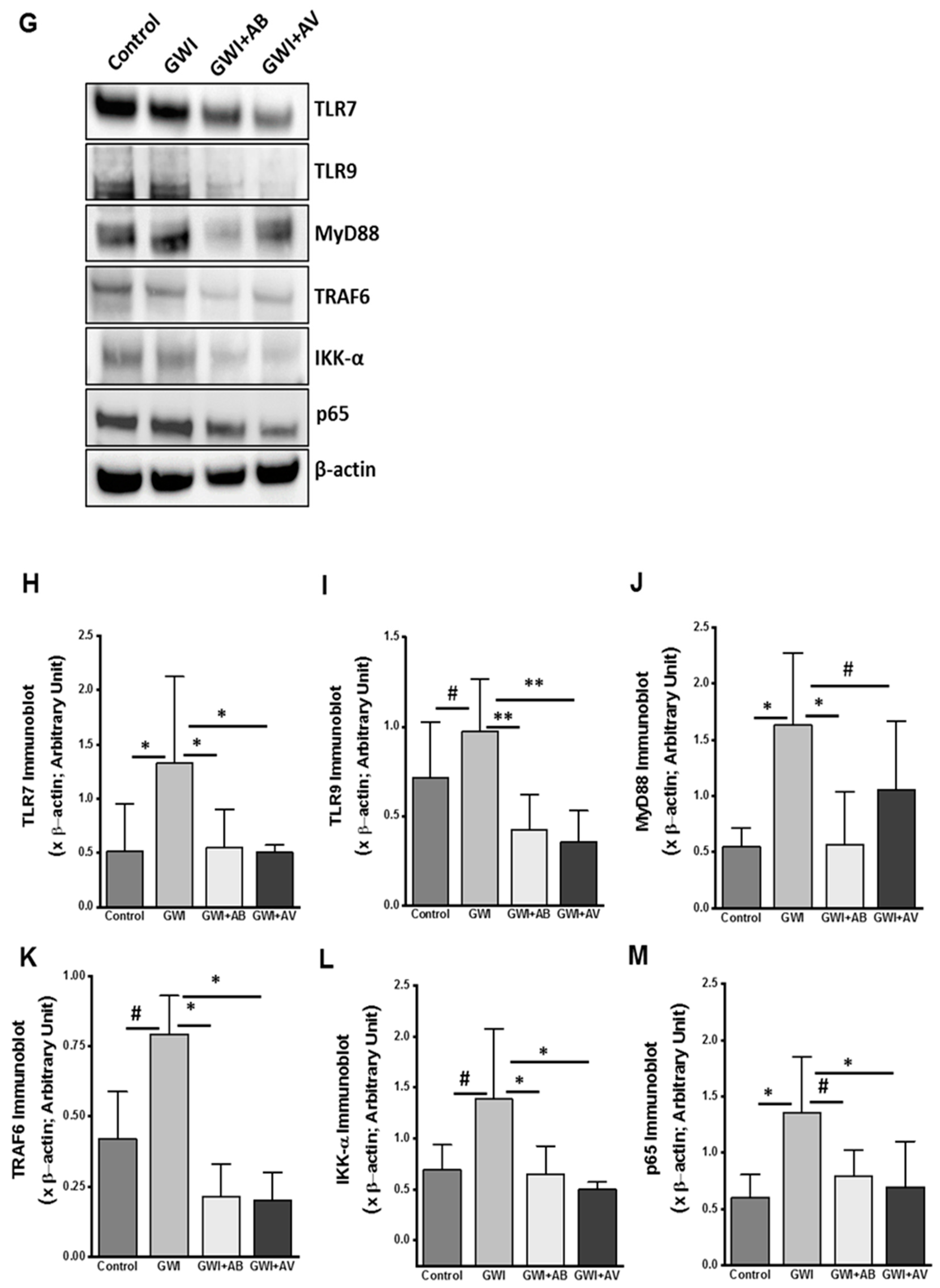
3.5. Altered Resident Enteric Virome Signature Was Accompanied by Activated TLR Signaling in Intestinal Inflammatory Phenotype of GWI
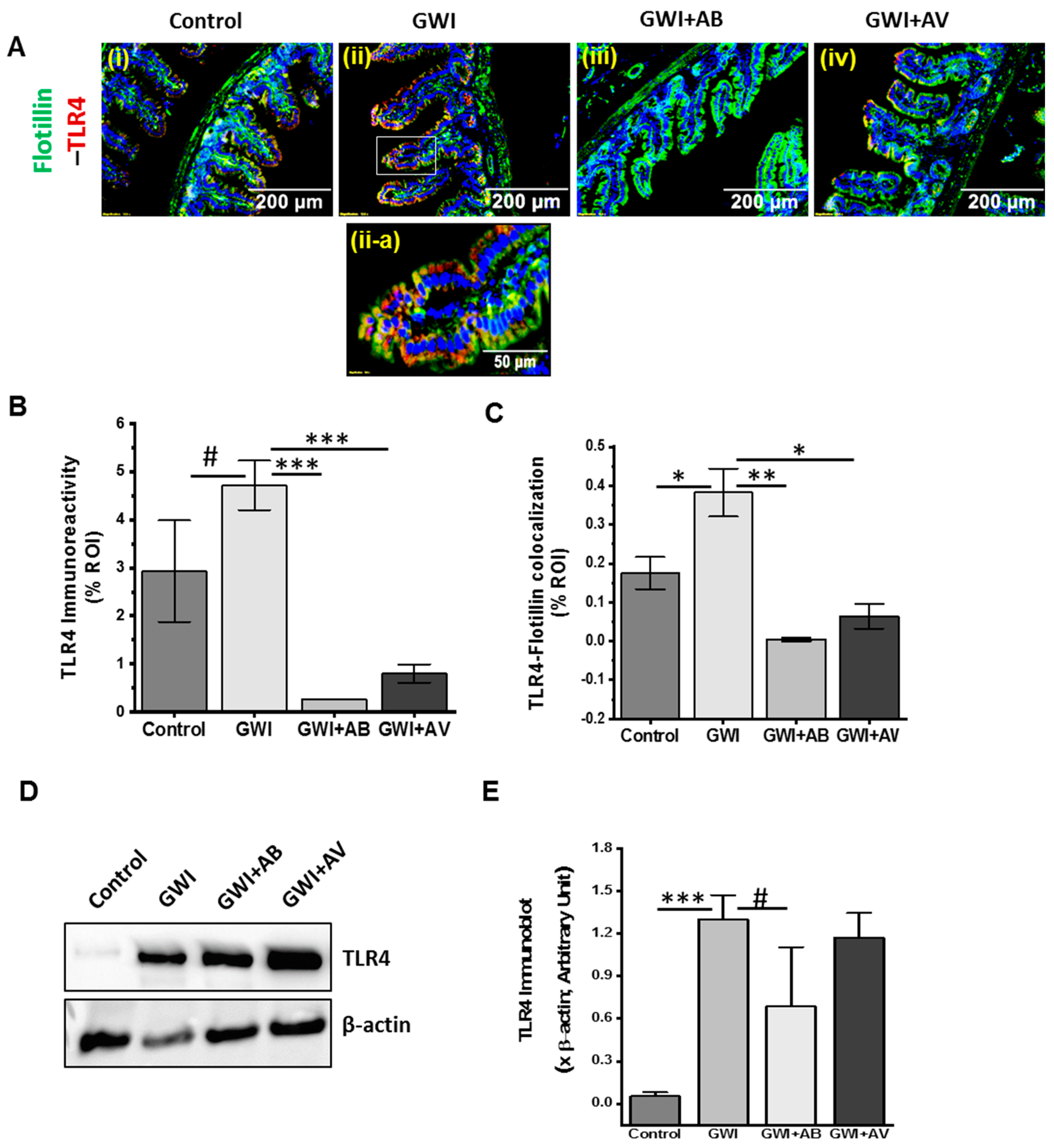
3.6. Inhibition of Virome Alteration by Ribavirin in GWI Decreased TLR4 Activation but Had Limited Impact on TLR4 Protein Levels
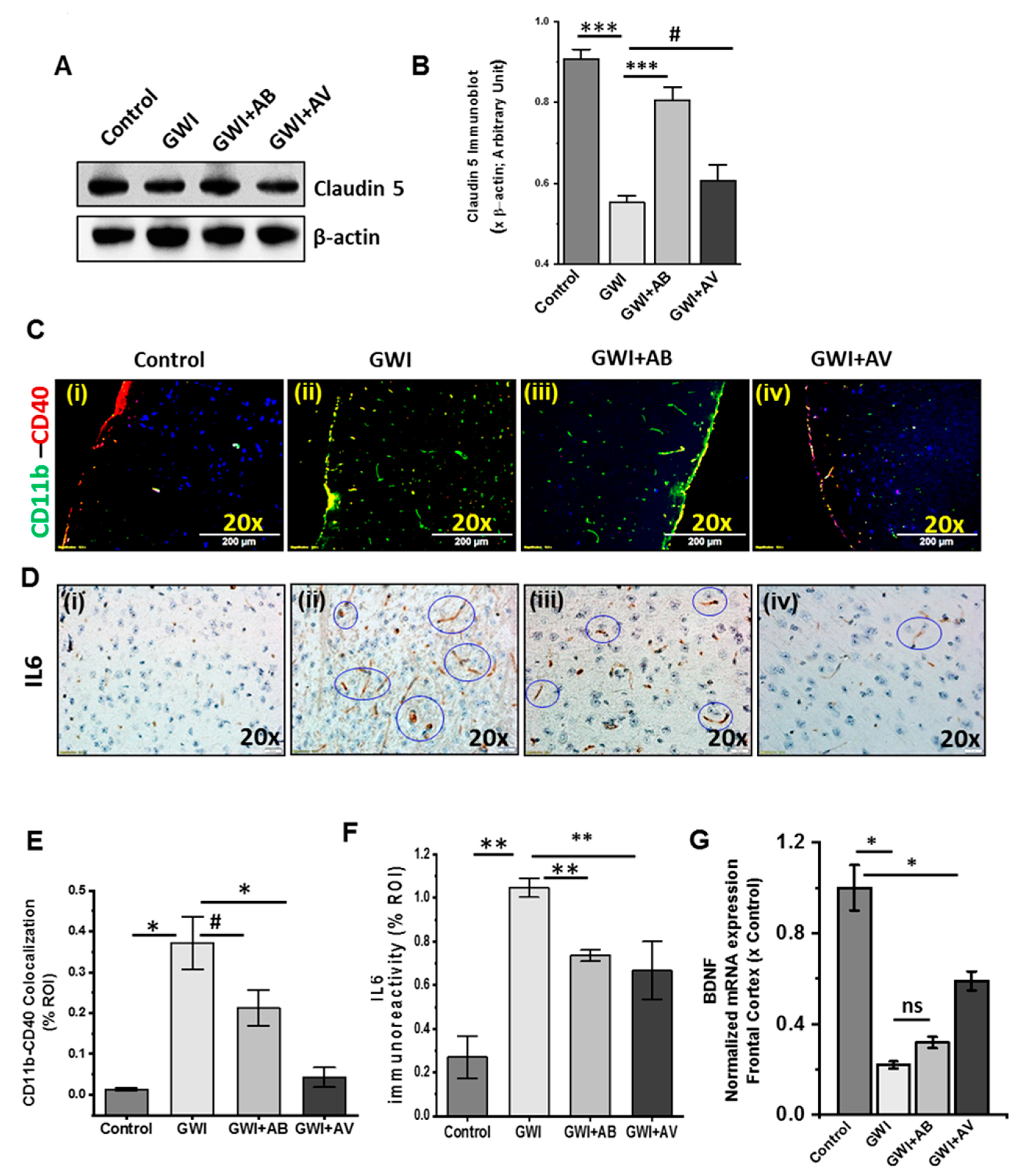
3.7. Altered Gut Virome and Microbiome in Gwi Mice Was Associated with Blood–Brain Barrier (BBB) Integrity Loss and Neuroinflammation While Antiviral Ribavirin Reversed Outcomes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- White, R.F.; Steele, L.; O’Callaghan, J.P.; Sullivan, K.; Binns, J.H.; Golomb, B.A.; Bloom, F.E.; Bunker, J.A.; Crawford, F.; Graves, J.C. Recent research on Gulf War illness and other health problems in veterans of the 1991 Gulf War: Effects of toxicant exposures during deployment. Cortex 2016, 74, 449–475. [Google Scholar] [CrossRef] [PubMed]
- Nettleman, M. Gulf War Illness: Challenges Persist. Trans. Am. Clin. Climatol. Assoc. 2015, 126, 237–247. [Google Scholar] [PubMed]
- Pierce, L.M.; Kurata, W.E.; Matsumoto, K.W.; Clark, M.E.; Farmer, D.M. Long-term epigenetic alterations in a rat model of Gulf War Illness. Neurotoxicology. 2016, 55, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Alhasson, F.; Das, S.; Seth, R.; Dattaroy, D.; Chandrashekaran, V.; Ryan, C.N.; Chan, L.S.; Testerman, T.; Burch, J.; Hofseth, L.J.; et al. Altered gut microbiome in a mouse model of Gulf War Illness causes neuroinflammation and intestinal injury via leaky gut and TLR4 activation. PLoS ONE 2017, 12, e0172914. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.N.; Wang, J.M.; Vogt, D.; Vickers, K.; King, D.W.; King, L.A. Gulf War Illness. J. Occup. Environ. Med. 2013, 55, 104–110. [Google Scholar] [CrossRef] [PubMed]
- The 1990–1991 Gulf War. Available online: http://cdmrp.army.mil/gwirp/pdfs/GWIRP_Landscape.pdf (accessed on 19 October 2019).
- Koo, B.B.; Michalovicz, L.T.; Calderazzo, S.; Kelly, K.A.; Sullivan, K.; Killiany, R.J.; O’Callaghan, J.P. Corticosterone potentiates DFP-induced neuroinflammation and affects high-order diffusion imaging in a rat model of Gulf War Illness. Brain Behav. Immun. 2018, 67, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, L.; Evans, J.E.; Joshi, U.; Crynen, G.; Reed, J.; Mouzon, B.; Baumann, S.; Montague, H.; Zakirova, Z.; Emmerich, T.; et al. Translational potential of long-term decreases in mitochondrial lipids in a mouse model of Gulf War Illness. Toxicology. 2016, 372, 22–33. [Google Scholar] [CrossRef]
- Parihar, V.K.; Hattiangady, B.; Shuai, B.; Shetty, A.K. Mood and Memory Deficits in a Model of Gulf War Illness Are Linked with Reduced Neurogenesis, Partial Neuron Loss and Mild Inflammation in the Hippocampus. Neuropsychopharmacology 2013, 38, 2348–2362. [Google Scholar] [CrossRef]
- Seth, R.K.; Kimono, D.; Alhasson, F.; Sarkar, S.; Albadrani, M.; Lasley, S.K.; Horner, R.; Janulewicz, P.; Nagarkatti, M.; Sullivan, K.; et al. Increased butyrate priming in the gut stalls microbiome associated-gastrointestinal inflammation and hepatic metabolic reprogramming in a mouse model of Gulf War Illness. Toxicol. Appl. Pharmacol. 2018, 350, 64–77. [Google Scholar] [CrossRef]
- Mukhopadhya, I.; Segal, J.P.; Carding, S.R.; Hart, A.L.; Hold, G.L. The gut virome: the ‘missing link’ between gut bacteria and host immunity? Therap. Adv. Gastroenterol. 2019, 12, 1756284819836620. [Google Scholar] [CrossRef]
- Belkaid, Y.; Harrison, O.J. Homeostatic Immunity and the Microbiota. Immunity. 2017, 46, 562–576. [Google Scholar] [CrossRef] [PubMed]
- Kohl, K.D.; Carey, H.V. A place for host-microbe symbiosis in the comparative physiologist’s toolbox. J. Exp. Biol. 2016, 219, 3496–3504. [Google Scholar] [CrossRef] [PubMed]
- Rieder, R.; Wisniewski, P.J.; Alderman, B.L.; Campbell, S.C. Microbes and mental health: A review. Brain Behav. Immun. 2017, 66, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Quigley, E.M.M. Microbiota-Brain-Gut Axis and Neurodegenerative Diseases. Curr. Neurol. Neurosci. Rep. 2017, 17, 94. [Google Scholar] [CrossRef] [PubMed]
- Serban, D.E. Microbiota in inflammatory bowel disease pathogenesis and therapy: Is it all about diet? Nutr. Clin. Pract. 2015, 30, 760–779. [Google Scholar] [CrossRef]
- Metzger, R.N.; Krug, A.B.; Eisenächer, K. Enteric virome sensing—its role in intestinal homeostasis and immunity. Viruses. 2018, 10, 146. [Google Scholar] [CrossRef]
- Brestoff, J.R.; Artis, D. Commensal bacteria at the interface of host metabolism and the immune system. Nat. Immunol. 2013, 14, 676–684. [Google Scholar] [CrossRef]
- Ding, R.X.; Goh, W.R.; Wu, R.N.; Yue, X.Q.; Luo, X.; Khine, W.W.T.; Wu, J.R.; Lee, Y.K. Revisit gut microbiota and its impact on human health and disease. J. Food Drug Anal. 2019, 27, 623–631. [Google Scholar] [CrossRef]
- Sarkar, A.; Lehto, S.M.; Harty, S.; Dinan, T.G.; Cryan, J.F.; Burnet, P.W.J. Psychobiotics and the Manipulation of Bacteria–Gut–Brain Signals. Trends Neurosci. 2016, 39, 763–781. [Google Scholar] [CrossRef]
- Hills, R.D.; Pontefract, B.A.; Mishcon, H.R.; Black, C.A.; Sutton, S.C.; Theberge, C.R. Gut Microbiome: Profound Implications for Diet and Disease. Nutrients 2019, 11, 1613. [Google Scholar] [CrossRef]
- Carding, S.; Verbeke, K.; Vipond, D.T.; Corfe, B.M.; Owen, L.J. Dysbiosis of the gut microbiota in disease. Microb. Ecol. Heal Dis. 2015, 2, 26. [Google Scholar] [CrossRef] [PubMed]
- Lopetuso, L.R.; Ianiro, G.; Scaldaferri, F.; Cammarota, G.; Gasbarrini, A. Gut Virome and Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2016, 22, 1708–1712. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Li, H. The role of gut microbiota in atherosclerosis and hypertension. Front. Pharmacol. 2018, 9, 1082. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.Y.; Chan, Y.L.; Tsai, M.H.; Wang, C.J.; Chiang, M.H.; Chiu, C.C. Gut microbial dysbiosis is associated with allergen-specific IgE responses in young children with airway allergies. World Allergy Organ. J. 2019, 12, 100021. [Google Scholar] [CrossRef] [PubMed]
- Focà, A.; Liberto, M.C.; Quirino, A.; Marascio, N.; Zicca, E.; Pavia, G. Gut inflammation and immunity: what is the role of the human gut virome? Mediators Inflamm. 2015, 2015, 326032. [Google Scholar] [CrossRef]
- Zuo, T.; Lu, X.J.; Zhang, Y.; Cheung, C.P.; Lam, S.; Zhang, F.; Tang, W.; Ching, J.Y.L.; Zhao, R.; Chan, P.K.S.; et al. Gut mucosal virome alterations in ulcerative colitis. Gut 2019, 68, 1169–1179. [Google Scholar] [CrossRef]
- Norman, J.M.; Handley, S.A.; Baldridge, M.T.; Droit, L.; Liu, C.Y.; Keller, B.C.; Kambal, A.; Monaco, C.L.; Zhao, G.; Fleshner, P.; et al. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell 2015, 160, 447–460. [Google Scholar] [CrossRef]
- Monaco, C.L.; Gootenberg, D.B.; Zhao, G.; Handley, S.A.; Ghebremichael, M.S.; Lim, E.S.; Lankowski, A.; Baldridge, M.T.; Wilen, C.B.; Flagg, M.; et al. Altered Virome and Bacterial Microbiome in Human Immunodeficiency Virus-Associated Acquired Immunodeficiency Syndrome. Cell Host Microbe 2016, 19, 311–322. [Google Scholar] [CrossRef]
- Morris, G.; Berk, M.; Carvalho, A.; Caso, J.; Sanz, Y.; Maes, M. The Role of Microbiota and Intestinal Permeability in the Pathophysiology of Autoimmune and Neuroimmune Processes with an Emphasis on Inflammatory Bowel Disease Type 1 Diabetes and Chronic Fatigue Syndrome. Curr. Pharm. Des. 2016, 22, 6058–6075. [Google Scholar] [CrossRef]
- Mandarano, A.H.; Giloteaux, L.; Keller, B.A.; Levine, S.M.; Hanson, M.R. Eukaryotes in the gut microbiota in myalgic encephalomyelitis/chronic fatigue syndrome. PeerJ 2018, 6, e4282. [Google Scholar] [CrossRef]
- O’Callaghan, J.P.; Kelly, K.A.; Locker, A.R.; Miller, D.B.; Lasley, S.M. Corticosterone primes the neuroinflammatory response to DFP in mice: potential animal model of Gulf War Illness. J. Neurochem. 2015, 133, 708–721. [Google Scholar] [CrossRef] [PubMed]
- Zakirova, Z.; Crynen, G.; Hassan, S.; Abdullah, L.; Horne, L.; Mathura, V.; Crawford, F.; Ait-Ghezala, G. A Chronic Longitudinal Characterization of Neurobehavioral and Neuropathological Cognitive Impairment in a Mouse Model of Gulf War Agent Exposure. Front. Integr. Neurosci. 2016, 9, 71. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Beier, S.; Flade, I.; Górska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.J.; Tappu, R. MEGAN Community Edition - Interactive Exploration and Analysis of Large-Scale Microbiome Sequencing Data. PLoS Comput. Biol. 2016, 12, e1004957. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Leung, H.C.M.; Yiu, S.M.; Chin, F.Y.L. IDBA-UD: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Sommer, D.D.; Delcher, A.L.; Salzberg, S.L.; Pop, M. Minimus: A fast, lightweight genome assembler. BMC Bioinform. 2007, 8, 64. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Enault, F.; Hurwitz, B.L.; Sullivan, M.B. VirSorter: Mining viral signal from microbial genomic data. PeerJ 2015, 3, e985. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. Available online: http://arxiv.org/abs/1303.3997 (accessed on 19 October 2019).
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Community Ecology Package. R package version 2.5-5. 2019. Available online: https://cran.r-project.org/package=vegan (accessed on 19 October 2019).
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Pfeiffer, J.K.; Virgin, H.W. Viral immunity. Transkingdom control of viral infection and immunity in the mammalian intestine. Science 2016, 351, 6270. [Google Scholar] [CrossRef]
- Ray, K. Gut mucosal virome altered in ulcerative colitis. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 262. [Google Scholar] [CrossRef]
- Tokarz, R.; Hyams, J.S.; Mack, D.R.; Boyle, B.; Griffiths, A.M.; LeLeiko, N.S.; Sauer, C.G.; Shah, S.; Markowitz, J.; Baker, S.S.; et al. Characterization of Stool Virome in Children Newly Diagnosed With Moderate to Severe Ulcerative Colitis. Inflamm. Bowel Dis. 2019. [Google Scholar] [CrossRef] [PubMed]
- Duerkop, B.A.; Hooper, L.V. Resident viruses and their interactions with the immune system. Nat. Immunol. 2013, 14, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Yong, Y.H.; Liu, S.F.; Hua, G.H.; Jia, R.M.; Gooneratne, R.; Zhao, Y.T.; Liao, M.; Ju, X.H. Goose toll-like receptor 3 (TLR3) mediated IFN-γ and IL-6 in anti-H5N1 avian influenza virus response. Vet. Immunol. Immunopathol. 2018, 197, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Mohan, C. Toll-like receptor signaling pathways--therapeutic opportunities. Mediators Inflamm. 2010, 2010, 781235. [Google Scholar] [CrossRef] [PubMed]
- Cakebread, J.A.; Xu, Y.; Grainge, C.; Kehagia, V.; Howarth, P.H.; Holgate, S.T.; Davies, D.E. Exogenous IFN-β has antiviral and anti-inflammatory properties in primary bronchial epithelial cells from asthmatic subjects exposed to rhinovirus. J. Allergy Clin. Immunol. 2011, 127, 1148–1154.e9. [Google Scholar] [CrossRef] [PubMed]
- Janulewicz, P.; Krengel, M.; Quinn, E.; Heeren, T.; Toomey, R.; Killiany, R.; Zundel, C.; Ajama, J.; O’Callaghan, J.; Steele, L.; et al. The multiple hit hypothesis for gulf war illness: Self-reported chemical/biological weapons exposure and mild traumatic brain injury. Brain Sci. 2018, 8, 198. [Google Scholar] [CrossRef]
- Van Belleghem, J.D.; Dąbrowska, K.; Vaneechoutte, M.; Barr, J.J.; Bollyky, P.L. Interactions between bacteriophage, bacteria, and the mammalian immune system. Viruses 2018, 11, 10. [Google Scholar] [CrossRef]
- Park, S.Y.; Baek, S.; Lee, S.O.; Choi, S.H.; Kim, Y.S.; Woo, J.H.; Sung, H.; Kim, M.N.; Kim, D.Y.; Lee, J.H.; et al. Efficacy of oral ribavirin in hematologic disease patients with paramyxovirus infection: Analytic strategy using propensity scores. Antimicrob. Agents Chemother. 2013, 57, 983–989. [Google Scholar] [CrossRef]
- Gavin, P.J.; Katz, B.Z. Intravenous ribavirin treatment for severe adenovirus disease in immunocompromised children. Pediatrics 2002, 110, e9. [Google Scholar] [CrossRef]
- Ventre, K.; Randolph, A.G. Ribavirin for respiratory syncytial virus infection of the lower respiratory tract in infants and young children. Cochrane Database of Syst. Rev. 2007. [Google Scholar] [CrossRef]
- Lin, D.M.; Koskella, B.; Lin, H.C. Phage therapy: An alternative to antibiotics in the age of multi-drug resistance. World J. Gastrointest Pharmacol. Ther. 2017, 8, 162. [Google Scholar] [CrossRef] [PubMed]
- Hetta, H.F.; Mekky, M.A.; Khalil, N.K.; Mohamed, W.A.; El-Feky, M.A.; Ahmed, S.H.; Daef, E.A.; Medhat, A.; Nassar, M.I.; Sherman, K.E.; et al. Extra-hepatic infection of hepatitis C virus in the colon tissue and its relationship with hepatitis C virus pathogenesis. J. Med. Microbiol. 2016, 65, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Udina, M.; Navinés, R.; Egmond, E.; Oriolo, G.; Langohr, K.; Gimenez, D.; Valdés, M.; Gómez-Gil, E.; Grande, I.; Gratacós, M.; et al. Glucocorticoid Receptors, Brain-Derived Neurotrophic Factor, Serotonin and Dopamine Neurotransmission are Associated with Interferon-Induced Depression. Int. J. Neuropsychopharmacol. 2016, 19, pyv135. [Google Scholar] [CrossRef] [PubMed]
- Bozic, I.; Savic, D.; Jovanovic, M.; Bjelobaba, I.; Laketa, D.; Nedeljkovic, N.; Stojiljkovic, M.; Pekovic, S.; Lavrnja, I. Low-Dose Ribavirin Treatments Attenuate Neuroinflammatory Activation of BV-2 Cells by Interfering with Inducible Nitric Oxide Synthase. Anal. Cell Pathol. 2015, 2015, 623614. [Google Scholar] [CrossRef] [PubMed]
- Angelucci, F.; Cechova, K.; Amlerova, J.; Hort, J. Antibiotics, gut microbiota, and Alzheimer’s disease. J. Neuroinflammation. 2019, 16, 108. [Google Scholar] [CrossRef]
- Pagliari, D.; Piccirillo, C.A.; Larbi, A.; Cianci, R. The Interactions between Innate Immunity and Microbiota in Gastrointestinal Diseases. J. Immunol. Res. 2015, 2015, 898297. [Google Scholar] [CrossRef]
- Belkaid, Y.; Hand, T.W. Role of the microbiota in immunity and inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef]
- Lazar, V.; Ditu, L.M.; Pircalabioru, G.G.; Gheorghe, I.; Curutiu, C.; Holban, A.M.; Picu, A.; Petcu, L.; Chifiriuc, M.C. Aspects of gut microbiota and immune system interactions in infectious diseases, immunopathology, and cancer. Front. Immunol. 2018, 9, 1830. [Google Scholar] [CrossRef]
- Karst, S.M. Viral Safeguard: The Enteric Virome Protects against Gut Inflammation. Immunity 2016, 44, 715–718. [Google Scholar] [CrossRef]
- Yang, J.Y.; Kim, M.S.; Kim, E.; Cheon, J.H.; Lee, Y.S.; Kim, Y.; Lee, S.H.; Seo, S.U.; Shin, S.H.; Choi, S.S.; et al. Enteric Viruses Ameliorate Gut Inflammation via Toll-like Receptor 3 and Toll-like Receptor 7-Mediated Interferon-β Production. Immunity 2016, 44, 889–900. [Google Scholar] [CrossRef]
- Perales-Linares, R.; Navas-Martin, S. Toll-like receptor 3 in viral pathogenesis: Friend or foe? Immunology. 2013, 140, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Kordjazy, N.; Haj-Mirzaian, A.; Haj-Mirzaian, A.; Rohani, M.M.; Gelfand, E.W.; Rezaei, N.; Abdolghaffari, A.H. Role of toll-like receptors in inflammatory bowel disease. Pharmacol. Res. 2018, 129, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Macht, V.A.; Woodruff, J.L.; Maissy, E.S.; Grillo, C.A.; Wilson, M.A.; Fadel, J.R.; Reagan, L.P. Pyridostigmine bromide and stress interact to impact immune function, cholinergic neurochemistry and behavior in a rat model of Gulf War Illness. Brain Behav. Immun. 2019, 80, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Phillips, K.F.; Santos, E.; Blair, R.E.; Deshpande, L.S. Targeting intracellular calcium stores alleviates neurological morbidities in a DFP-based rat model of gulf war illness. Toxicol. Sci. 2019, 169, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Banjara, M.; Ghosh, C. Sterile Neuroinflammation and Strategies for Therapeutic Intervention. Int. J. Inflammation. 2017, 2017, 8385961. [Google Scholar] [CrossRef] [PubMed]
- Daniels, B.P.; Holman, D.W.; Cruz-Orengo, L.; Jujjavarapu, H.; Durrant, D.M.; Klein, R.S. Viral pathogen-associated molecular patterns regulate blood-brain barrier integrity via competing innate cytokine signals. MBio 2014, 5, e01476-14. [Google Scholar] [CrossRef] [PubMed]
- Spindler, K.R.; Hsu, T.H. Viral disruption of the blood-brain barrier. Trends Microbiol. 2012, 20, 282–290. [Google Scholar] [CrossRef]
- Elwood, E.; Lim, Z.; Naveed, H.; Galea, I. The effect of systemic inflammation on human brain barrier function. Brain Behav. Immun. 2017, 62, 35–40. [Google Scholar] [CrossRef]
- Varatharaj, A.; Galea, I. The blood-brain barrier in systemic inflammation. Brain Behav. Immun. 2017, 60, 1–12. [Google Scholar] [CrossRef]
- Obrenovich, M. Leaky Gut, Leaky Brain? Microorganisms 2018, 6, 107. [Google Scholar] [CrossRef]
- Zhang, J.; Sadowska, G.B.; Chen, X.; Park, S.Y.; Kim, J.E.; Bodge, C.A.; Cummings, E.; Lim, Y.P.; Makeyev, O.; Besio, W.G. Anti-IL-6 neutralizing antibody modulates blood-brain barrier function in the ovine fetus. FASEB J. 2015, 29, 1739–1753. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sequences (3′-5′) |
|---|---|
| ZO1 | Sense: CCACCTCTGTCCAGCTCTTC Antisense: CACCGGAGTGATGGTTTTCT |
| Occludin | Sense: GTGAGCTGTGATGTGTGTTGAGCT Antisense: GTGGGGAACGTGGCCGATATAATG |
| BDNF | Sense: TGCAGGGGCATAGACAAAAGG Antisense: CTTATGAATCGCCAGCCAATTCTC |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seth, R.K.; Maqsood, R.; Mondal, A.; Bose, D.; Kimono, D.; Holland, L.A.; Janulewicz Lloyd, P.; Klimas, N.; Horner, R.D.; Sullivan, K.; et al. Gut DNA Virome Diversity and Its Association with Host Bacteria Regulate Inflammatory Phenotype and Neuronal Immunotoxicity in Experimental Gulf War Illness. Viruses 2019, 11, 968. https://doi.org/10.3390/v11100968
Seth RK, Maqsood R, Mondal A, Bose D, Kimono D, Holland LA, Janulewicz Lloyd P, Klimas N, Horner RD, Sullivan K, et al. Gut DNA Virome Diversity and Its Association with Host Bacteria Regulate Inflammatory Phenotype and Neuronal Immunotoxicity in Experimental Gulf War Illness. Viruses. 2019; 11(10):968. https://doi.org/10.3390/v11100968
Chicago/Turabian StyleSeth, Ratanesh K., Rabia Maqsood, Ayan Mondal, Dipro Bose, Diana Kimono, LaRinda A. Holland, Patricia Janulewicz Lloyd, Nancy Klimas, Ronnie D. Horner, Kimberly Sullivan, and et al. 2019. "Gut DNA Virome Diversity and Its Association with Host Bacteria Regulate Inflammatory Phenotype and Neuronal Immunotoxicity in Experimental Gulf War Illness" Viruses 11, no. 10: 968. https://doi.org/10.3390/v11100968
APA StyleSeth, R. K., Maqsood, R., Mondal, A., Bose, D., Kimono, D., Holland, L. A., Janulewicz Lloyd, P., Klimas, N., Horner, R. D., Sullivan, K., Lim, E. S., & Chatterjee, S. (2019). Gut DNA Virome Diversity and Its Association with Host Bacteria Regulate Inflammatory Phenotype and Neuronal Immunotoxicity in Experimental Gulf War Illness. Viruses, 11(10), 968. https://doi.org/10.3390/v11100968