An Immortalized Hepatocyte-Like Cell Line (imHC) Accommodated Complete Viral Lifecycle, Viral Persistence Form, cccDNA and Eventual Spreading of a Clinically-Isolated HBV

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Detection of Sodium Taurocholate Cotransporting Polypeptide (NTCP) in Hepatocytes
2.3. Production of HBV from Cultured Cells
2.4. The Infection to Hepatocytes Using HBV Derived from HepG2.2.15 or Clinical Isolate
2.5. Evaluating HBV Progeny Produced from Infected imHC and HepaRG
2.6. Measuring the HBsAg and HBeAg in Supernatant Using ELISA
2.7. Immunofluorescent Staining
2.8. Detection of Intracellular HBV DNA and Viral Load Using Quantitative Real-Time PCR
2.9. Detection of HBV cccDNA in Infected Hepatocytes Using Quantitative Real-Time PCR
2.10. Treatment of HBV Infected Hepatocyte with Entry Inhibitor, Nucleos(t)ide Analogs and Antiviral Cytokines
2.11. Determination of Viral Spreading Using Flow Cytometry
2.12. Ethics Statement
2.13. Statistical Analysis
3. Results
3.1. Expression of Hepatic Phenotypes and NTCP an Entry Receptor for HBV in imHC
3.2. Presenting of Hepatic Maturation Features and Upregulating of NTCP Receptor Expression in imHC after Hepatic Maturation Induction
3.3. The imHCs were Susceptible to HBV Infection, Sustained Viral DNA Replication and Generated Viral Particles after Infection with either HepG2.2.15 Derived HBV or the Clinical Isolates
3.4. Infected imHCs Released HBsAg and HBeAg into the Surrounding Medium
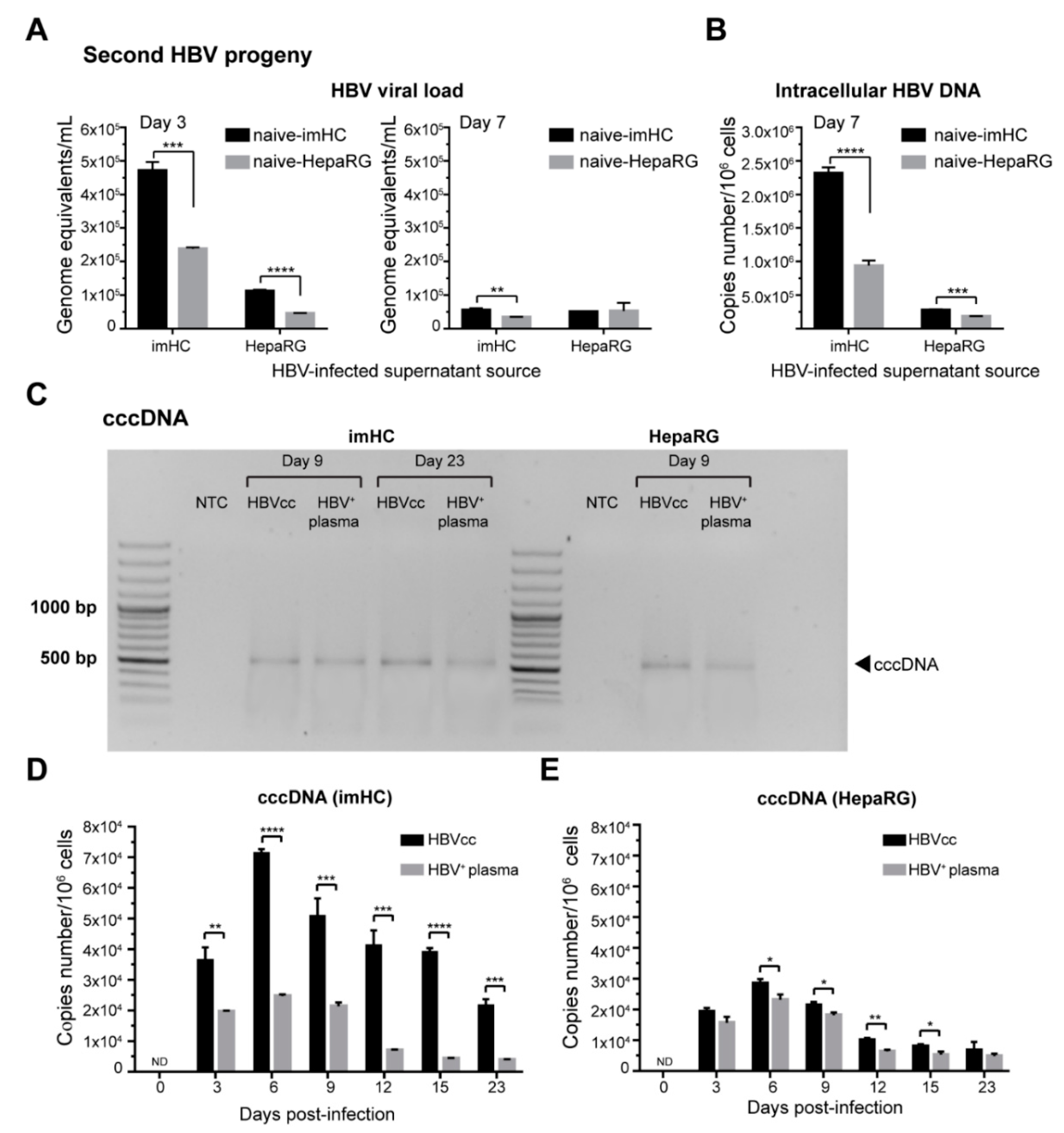
3.5. imHCs Allowed the Production of Secondary HBV Progeny, cccDNA and Viral DNA Replication
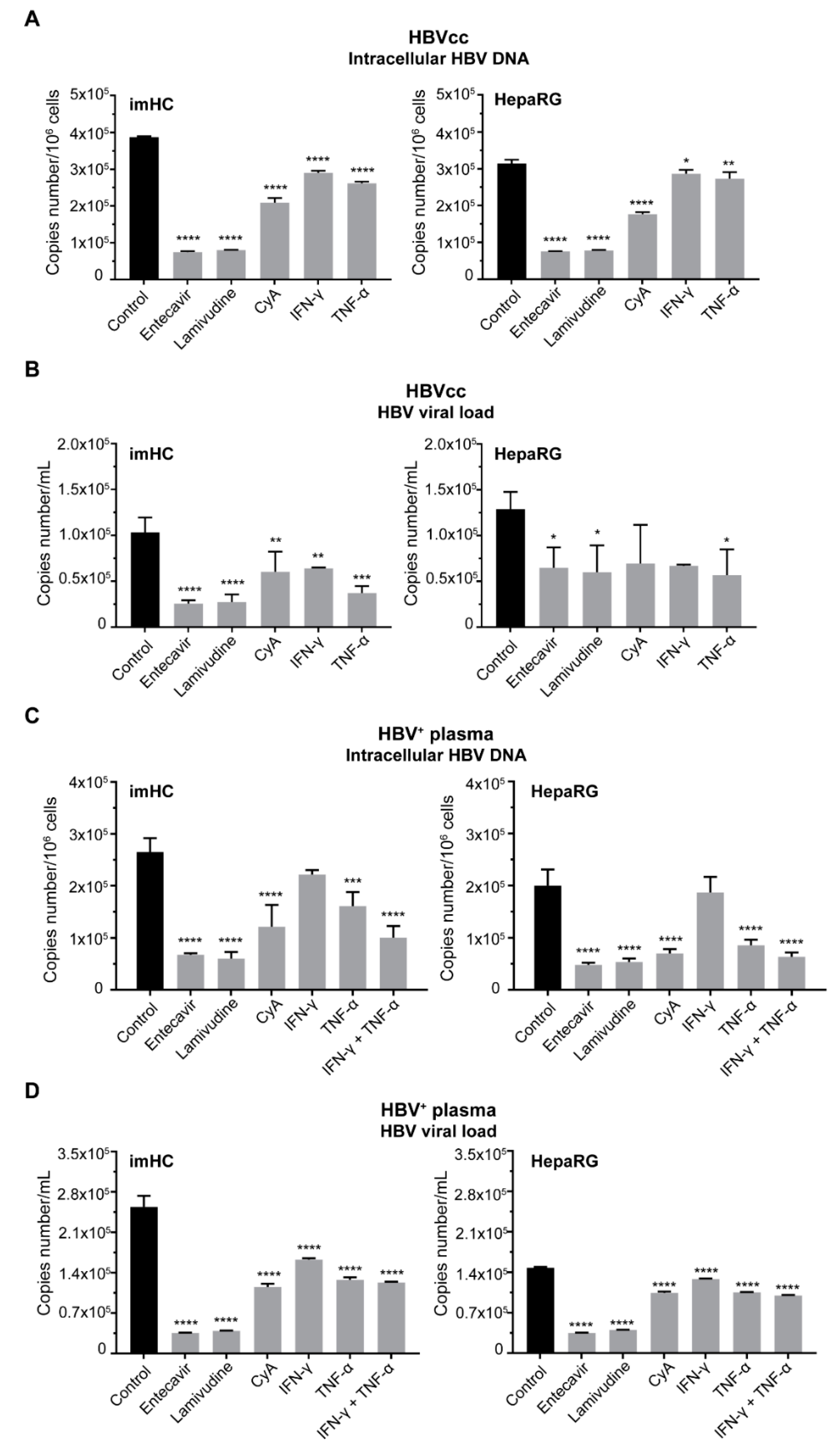
3.6. Treatment of Infected imHCs with Entry Inhibitor, Nucleos(t)ide Analogs and Antiviral Cytokines Decreased Intracellular HBV DNA and HBV Viral Load
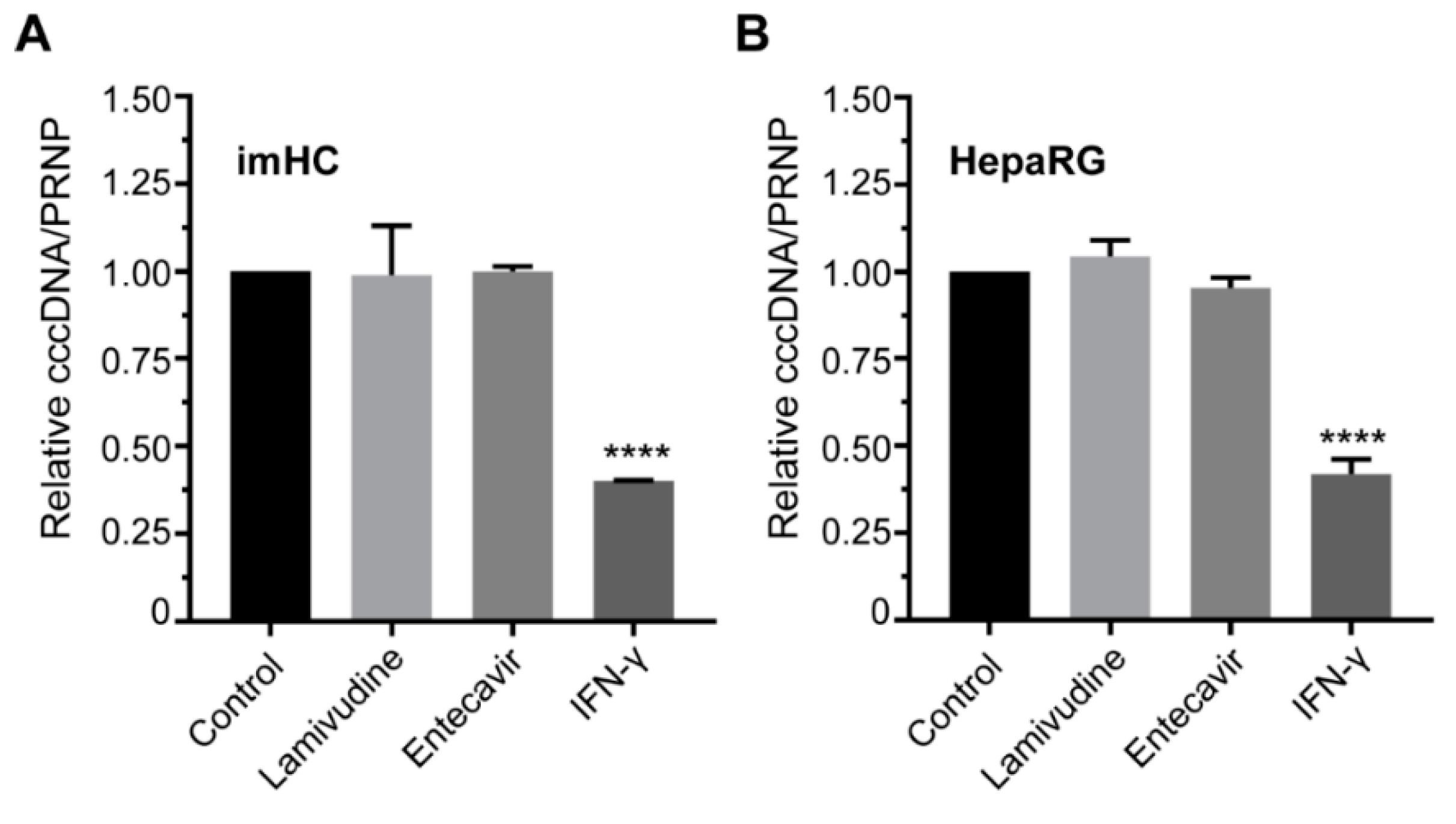
3.7. IFN-γ Treatment Lessened the HBV cccDNA Level in Infected Hepatocyte
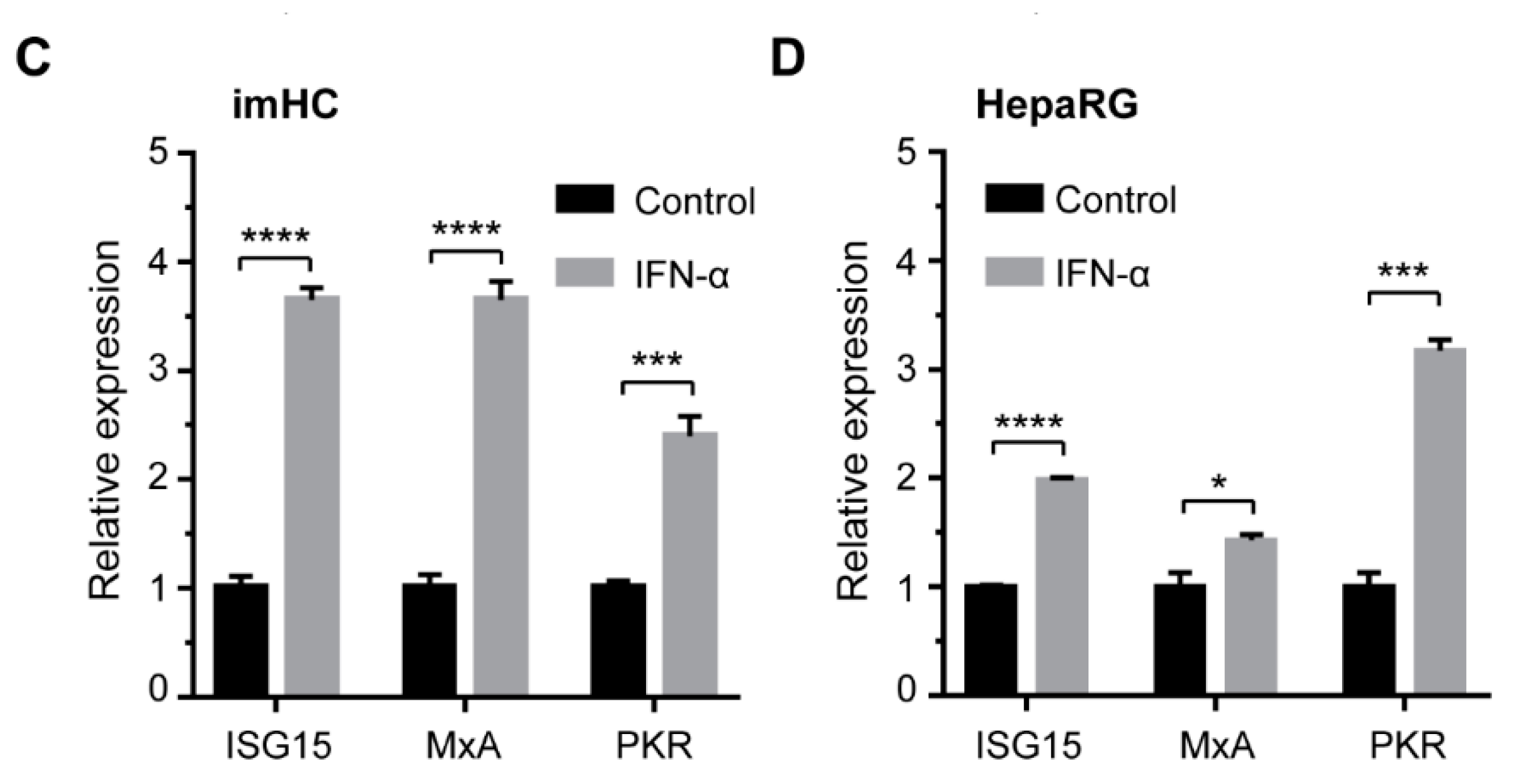
3.8. IFN-α Enhanced Antiviral Genes Expression and Innate Immune Response in Infected imHCs
3.9. HBV from Infected imHCs Spread and Secondarily Infected Naïve Hepatocytes in Co-Culture Model
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shimura, S.; Watashi, K.; Fukano, K.; Peel, M.; Sluder, A.; Kawai, F.; Iwamoto, M.; Tsukuda, S.; Takeuchi, J.S.; Miyake, T.; et al. Cyclosporin derivatives inhibit hepatitis B virus entry without interfering with NTCP transporter activity. J. Hepatol. 2017, 66, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, S.; Kakinuma, S.; Asahina, Y.; Kamiya, A.; Miyoshi, M.; Tsunoda, T.; Nitta, S.; Asano, Y.; Nagata, H.; Otani, S.; et al. Human induced pluripotent stem Cell-Derived hepatic cell lines as a new model for host interaction with hepatitis B virus. Sci. Rep. 2016, 6, 29358. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver. EASL 2017 Clinical Practice Guidelines on the management of hepatitis B virus infection. J. Hepatol. 2017, 67, 370–398. [Google Scholar] [CrossRef] [PubMed]
- Sarin, S.K.; Kumar, M.; Lau, G.K.; Abbas, Z.; Chan, H.L.Y.; Chen, C.J.; Chen, D.S.; Chen, H.L.; Chen, P.J.; Chien, R.N.; et al. Asian-Pacific clinical practice guidelines on the management of hepatitis B: A 2015 update. Hepatol. Int. 2016, 10, 1–98. [Google Scholar] [CrossRef]
- Levrero, M.; Subic, M.; Villeret, F.; Zoulim, F. Perspectives and limitations for nucleo(t)side analogs in future HBV therapies. Curr. Opin. Virol. 2018, 30, 80–89. [Google Scholar] [CrossRef]
- Liang, T.J.; Block, T.M.; McMahon, B.J.; Ghany, M.G.; Urban, S.; Guo, J.-T.; Locarnini, S.; Zoulim, F.; Chang, K.-M.; Lok, A.S. Present and future therapies of hepatitis B: From discovery to cure. Hepatology 2015, 62, 1893–1908. [Google Scholar] [CrossRef]
- Seeger, C.; Mason, W.S. Hepatitis B Virus Biology. Microbiol. Mol. Biol. Rev. 2000, 64, 51–68. [Google Scholar] [CrossRef]
- Li, W.; Urban, S. Entry of hepatitis B and hepatitis D virus into hepatocytes: Basic insights and clinical implications. J. Hepatol. 2016, 64, S32–S40. [Google Scholar] [CrossRef]
- Tu, T.; Urban, S. Virus entry and its inhibition to prevent and treat hepatitis B and hepatitis D virus infections. Curr. Opin. Virol. 2018, 30, 68–79. [Google Scholar] [CrossRef]
- Nassal, M. HBV cccDNA: Viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut 2015, 64, 1972–1984. [Google Scholar] [CrossRef]
- Lucifora, J.; Protzer, U. Attacking hepatitis B virus cccDNA—The holy grail to hepatitis B cure. J. Hepatol. 2016, 64, S41–S48. [Google Scholar] [CrossRef] [PubMed]
- Hayes, C.N.; Zhang, Y.; Makokha, G.N.; Hasan, M.Z.; Omokoko, M.D.; Chayama, K. Early events in hepatitis B virus infection: From the cell surface to the nucleus. J. Gastroenterol. Hepatol. 2016, 31, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Durantel, D.; Zoulim, F. New antiviral targets for innovative treatment concepts for hepatitis B virus and hepatitis delta virus. J. Hepatol. 2016, 64, S117–S131. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.-T.; Guo, H. Metabolism and function of hepatitis B virus cccDNA: Implications for the development of cccDNA-Targeting antiviral therapeutics. Antivir. Res. 2015, 122, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Boucle, S.; Bassit, L.; Ehteshami, M.; Schinazi, R.F. Toward Elimination of Hepatitis B Virus Using Novel Drugs, Approaches, and Combined Modalities. Clin. Liver Dis. 2016, 20, 737–749. [Google Scholar] [CrossRef][Green Version]
- Hu, J.; Lin, Y.-Y.; Chen, P.-J.; Watashi, K.; Wakita, T. Cell and Animal Models for Studying Hepatitis B Virus Infection and Drug Development. Gastroenterology 2019, 156, 338–354. [Google Scholar] [CrossRef]
- Verrier, E.R.; Colpitts, C.C.; Schuster, C.; Zeisel, M.B.; Baumert, T.F. Cell Culture Models for the Investigation of Hepatitis B and D Virus Infection. Viruses 2016, 8, 261. [Google Scholar] [CrossRef]
- Allweiss, L.; Dandri, M. Experimental in vitro and in vivo models for the study of human hepatitis B virus infection. J. Hepatol. 2016, 64, S17–S31. [Google Scholar] [CrossRef]
- Ni, Y.; Lempp, F.A.; Mehrle, S.; Nkongolo, S.; Kaufman, C.; Fälth, M.; Stindt, J.; Königer, C.; Nassal, M.; Kubitz, R.; et al. Hepatitis B and D Viruses Exploit Sodium Taurocholate Co-Transporting Polypeptide for Species-Specific Entry into Hepatocytes. Gastroenterology 2014, 146, 1070–1083. [Google Scholar] [CrossRef]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife 2012, 1, e00049. [Google Scholar] [CrossRef]
- Sun, Y.; Qi, Y.; Peng, B.; Li, W. NTCP-Reconstituted in Vitro HBV Infection System. In Hepatitis B Virus: Methods and Protocols; Guo, H., Cuconati, A., Eds.; Springer: New York, NY, USA, 2017; pp. 1–14. [Google Scholar] [CrossRef]
- Sells, M.A.; Chen, M.L.; Acs, G. Production of hepatitis B virus particles in Hep G2 cells transfected with cloned hepatitis B virus DNA. Proc. Natl. Acad. Sci. 1987, 84, 1005–1009. [Google Scholar] [CrossRef] [PubMed]
- Ladner, S.K.; Otto, M.J.; Barker, C.S.; Zaifert, K.; Wang, G.H.; Guo, J.T.; Seeger, C.; King, R.W. Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: A novel system for screening potential inhibitors of HBV replication. Antimicrob. Agents Chemother. 1997, 41, 1715–1720. [Google Scholar] [CrossRef] [PubMed]
- Shlomai, A.; Schwartz, R.E.; Ramanan, V.; Bhatta, A.; de Jong, Y.P.; Bhatia, S.N.; Rice, C.M. Modeling host interactions with hepatitis B virus using primary and induced pluripotent stem Cell-Derived hepatocellular systems. Proc. Natl. Acad. Sci. 2014, 111, 12193–12198. [Google Scholar] [CrossRef] [PubMed]
- Winer, B.Y.; Huang, T.S.; Pludwinski, E.; Heller, B.; Wojcik, F.; Lipkowitz, G.E.; Parekh, A.; Cho, C.; Shrirao, A.; Muir, T.W.; et al. Long-Term hepatitis B infection in a scalable hepatic Co-Culture system. Nat. Commun. 2017, 8, 125. [Google Scholar] [CrossRef]
- Galle, P.R.; Hagelstein, J.; Kommerell, B.; Volkmann, M.; Schranz, P.; Zentgraf, H. In vitro experimental infection of primary human hepatocytes with hepatitis B virus. Gastroenterology 1994, 106, 664–673. [Google Scholar] [CrossRef]
- Zhou, M.; Zhao, F.; Li, J.; Cheng, Z.; Tian, X.; Zhi, X.; Huang, Y.; Hu, K. Long-Term maintenance of human fetal hepatocytes and prolonged susceptibility to HBV infection by Co-Culture with Non-Parenchymal cells. J. Virol. Methods 2014, 195, 185–193. [Google Scholar] [CrossRef]
- Godoy, P.; Hewitt, N.J.; Albrecht, U.; Andersen, M.E.; Ansari, N.; Bhattacharya, S.; Bode, J.G.; Bolleyn, J.; Borner, C.; Böttger, J.; et al. Recent advances in 2D and 3D in vitro systems using primary hepatocytes, alternative hepatocyte sources and Non-Parenchymal liver cells and their use in investigating mechanisms of hepatotoxicity, cell signaling and ADME. Arch. Toxicol. 2013, 87, 1315–1530. [Google Scholar] [CrossRef]
- Khetani, S.R.; Bhatia, S.N. Microscale culture of human liver cells for drug development. Nat. Biotechnol. 2007, 26, 120. [Google Scholar] [CrossRef]
- Ploss, A.; Khetani, S.R.; Jones, C.T.; Syder, A.J.; Trehan, K.; Gaysinskaya, V.A.; Mu, K.; Ritola, K.; Rice, C.M.; Bhatia, S.N. Persistent hepatitis C virus infection in microscale primary human hepatocyte cultures. Proc. Natl. Acad. Sci. 2010, 107, 3141–3145. [Google Scholar] [CrossRef]
- March, S.; Ng, S.; Velmurugan, S.; Galstian, A.; Shan, J.; Logan, D.J.; Carpenter, A.E.; Thomas, D.; Sim, B.K.; Mota, M.M.; et al. A Microscale Human Liver Platform that Supports the Hepatic Stages of Plasmodium falciparum and vivax. Cell Host Microbe 2013, 14, 104–115. [Google Scholar] [CrossRef]
- Takayama, K.; Kawabata, K.; Nagamoto, Y.; Kishimoto, K.; Tashiro, K.; Sakurai, F.; Tachibana, M.; Kanda, K.; Hayakawa, T.; Furue, M.K.; et al. 3D spheroid culture of hESC/hiPSC-Derived Hepatocyte-Like cells for drug toxicity testing. Biomaterials 2013, 34, 1781–1789. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, Z. Opportunities and challenges in using human hepatocytes in cytochromes P450 induction assays. Expert Opin. Drug Metab. Toxicol. 2016, 12, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Sa-Ngiamsuntorn, K.; Hongeng, S.; Wongkajornsilp, A. Development of Hepatocyte-Like Cell Derived from Human Induced Pluripotent Stem cell as a Host for Clinically Isolated Hepatitis C Virus. Curr. Protoc. Stem Cell Biol. 2017, 42, 4A.13.11–14A.13.34. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, F.; Mitani, S.; Yamamoto, T.; Takayama, K.; Tachibana, M.; Watashi, K.; Wakita, T.; Iijima, S.; Tanaka, Y.; Mizuguchi, H. Human Induced-Pluripotent stem Cell-Derived Hepatocyte-Like cells as an in vitro model of human hepatitis B virus infection. Sci. Rep. 2017, 7, 45698. [Google Scholar] [CrossRef]
- Xia, Y.; Carpentier, A.; Cheng, X.; Block, P.D.; Zhao, Y.; Zhang, Z.; Protzer, U.; Liang, T.J. Human stem Cell-Derived hepatocytes as a model for hepatitis B virus infection, spreading and virus-host interactions. J. Hepatol. 2017, 66, 494–503. [Google Scholar] [CrossRef]
- Nie, Y.-Z.; Zheng, Y.-W.; Miyakawa, K.; Murata, S.; Zhang, R.-R.; Sekine, K.; Ueno, Y.; Takebe, T.; Wakita, T.; Ryo, A.; et al. Recapitulation of hepatitis B virus–host interactions in liver organoids from human induced pluripotent stem cells. EBioMedicine 2018, 35, 114–123. [Google Scholar] [CrossRef]
- Allweiss, L.; Dandri, M. The Role of cccDNA in HBV Maintenance. Viruses 2017, 9, 156. [Google Scholar] [CrossRef]
- Sa-ngiamsuntorn, K.; Wongkajornsilp, A.; Kasetsinsombat, K.; Duangsa-ard, S.; Nuntakarn, L.; Borwornpinyo, S.; Akarasereenont, P.; Limsrichamrern, S.; Hongeng, S. Upregulation of CYP 450s expression of immortalized Hepatocyte-Like cells derived from mesenchymal stem cells by enzyme inducers. BMC Biotechnol. 2011, 11, 89. [Google Scholar] [CrossRef]
- Pewkliang, Y.; Rungin, S.; Lerdpanyangam, K.; Duangmanee, A.; Kanjanasirirat, P.; Suthivanich, P.; Sa-Ngiamsuntorn, K.; Borwornpinyo, S.; Sattabongkot, J.; Patrapuvich, R.; et al. A novel immortalized Hepatocyte-Like cell line (imHC) supports in vitro liver stage development of the human malarial parasite Plasmodium vivax. Malar. J. 2018, 17, 50. [Google Scholar] [CrossRef]
- Kongmanas, K.; Punyadee, N.; Wasuworawong, K.; Songjaeng, A.; Pewkliang, Y.; Manocheewa, S.; Thiemmeca, S.; Sa-ngiamsuntorn, K.; Faull, K.F.; Hongeng, S.; et al. Immortalized Stem Cell-Derived Hepatocyte-Like Cells: An Alternative Model for Studying Dengue Pathogenesis and Therapy. Hepatology 2019. (manuscript in preparation). [Google Scholar]
- Kanebratt, K.P.; Andersson, T.B. HepaRG Cells as an in Vitro Model for Evaluation of Cytochrome P450 Induction in Humans. Drug Metab. Dispos. 2008, 36, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Zedler, U.; Langenkamp, A.; Hösel, M.; Quasdorff, M.; Esser, K.; Dienes, H.-P.; Tappertzhofen, B.; Kolanus, W.; Protzer, U. Dendritic cells take up viral antigens but do not support the early steps of hepatitis B virus infection. Hepatology 2006, 43, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Köck, J.; Theilmann, L.; Galle, P.; Schlicht, H. Hepatitis B virus nucleic acids associated with human peripheral blood mononuclear cells do not originate from replicating virus. Hepatology 1996, 23, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Wongkajornsilp, A.; Sa-Ngiamsuntorn, K.; Hongeng, S. Development of immortalized Hepatocyte-Like cells from hMSCs. Methods Mol. Biol. 2012, 826, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Gerets, H.H.J.; Tilmant, K.; Gerin, B.; Chanteux, H.; Depelchin, B.O.; Dhalluin, S.; Atienzar, F.A.J.C.B. Characterization of primary human hepatocytes, HepG2 cells, and HepaRG cells at the mRNA level and CYP activity in response to inducers and their predictivity for the detection of human hepatotoxins. Cell Biol. Toxicol. 2012, 28, 69–87. [Google Scholar] [CrossRef]
- Sureau, C.; Romet-Lemonne, J.-L.; Mullins, J.I.; Essex, M. Production of hepatitis B virus by a differentiated human hepatoma cell line after transfection with cloned circular HBV DNA. Cell 1986, 47, 37–47. [Google Scholar] [CrossRef]
- Huang, L.R.; Gäbel, Y.A.; Graf, S.; Arzberger, S.; Kurts, C.; Heikenwalder, M.; Knolle, P.A.; Protzer, U. Transfer of HBV Genomes Using Low Doses of Adenovirus Vectors Leads to Persistent Infection in Immune Competent Mice. Gastroenterology 2012, 142, 1447–1450. [Google Scholar] [CrossRef]
- Lucifora, J.; Vincent, I.E.; Berthillon, P.; Dupinay, T.; Michelet, M.; Protzer, U.; Zoulim, F.; Durantel, D.; Trepo, C.; Chemin, I. Hepatitis B virus replication in primary macaque hepatocytes: Crossing the species barrier toward a new small primate model. Hepatology 2010, 51, 1954–1960. [Google Scholar] [CrossRef]
- Hantz, O.; Parent, R.; Durantel, D.; Gripon, P.; Guguen-Guillouzo, C.; Zoulim, F. Persistence of the hepatitis B virus covalently closed circular DNA in HepaRG human hepatocyte-like cells. J. Gen. Virol. 2009, 90, 127–135. [Google Scholar] [CrossRef]
- Gripon, P.; Rumin, S.; Urban, S.; Le Seyec, J.; Glaise, D.; Cannie, I.; Guyomard, C.; Lucas, J.; Trepo, C.; Guguen-Guillouzo, C. Infection of a human hepatoma cell line by hepatitis B virus. Proc. Natl. Acad. Sci. 2002, 99, 15655–15660. [Google Scholar] [CrossRef]
- Lee, J.; Zong, L.; Krotow, A.; Qin, Y.; Jia, L.; Zhang, J.; Tong, S.; Li, J. N-Linked Glycosylation Is Not Essential for Sodium Taurocholate Cotransporting Polypeptide to Mediate Hepatitis B Virus Infection in Vitro. J. Virol. 2018, 92, e00732-18. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Prieto, A.M.; Skelton, J.K.; Wai, S.N.; Large, E.; Lussignol, M.; Vizcay-Barrena, G.; Hughes, D.; Fleck, R.A.; Thursz, M.; Catanese, M.T.; et al. 3D microfluidic liver cultures as a physiological preclinical tool for hepatitis B virus infection. Nat. Commun. 2018, 9, 682. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-C.; Chen, C.-C.; Chang, W.-C.; Tao, M.-H.; Huang, C. Entry of Hepatitis B Virus into Immortalized Human Primary Hepatocytes by Clathrin-Dependent Endocytosis. J. Virol. 2012, 86, 9443–9453. [Google Scholar] [CrossRef] [PubMed]
- Heslop, J.A.; Rowe, C.; Walsh, J.; Sison-Young, R.; Jenkins, R.; Kamalian, L.; Kia, R.; Hay, D.; Jones, R.P.; Malik, H.Z.; et al. Mechanistic evaluation of primary human hepatocyte culture using global proteomic analysis reveals a selective dedifferentiation profile. Arch. Toxicol. 2017, 91, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Kramer, K.; Sindhi, N.; Sarkar, P.; Upton, M.; Kalluri, R.J.M.; Biochemistry, C. De-differentiation of primary human hepatocytes depends on the composition of specialized liver basement membrane. Mol. Cell. Biochem. 2006, 283, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Fraczek, J.; Bolleyn, J.; Vanhaecke, T.; Rogiers, V.; Vinken, M.J.A.O.T. Primary hepatocyte cultures for Pharmaco-Toxicological studies: At the busy crossroad of various anti-dedifferentiation strategies. Arch. Toxicol. 2013, 87, 577–610. [Google Scholar] [CrossRef] [PubMed]
- Godoy, P.; Hengstler, J.G.; Ilkavets, I.; Meyer, C.; Bachmann, A.; Müller, A.; Tuschl, G.; Mueller, S.O.; Dooley, S. Extracellular matrix modulates sensitivity of hepatocytes to fibroblastoid dedifferentiation and transforming growth factor β–induced apoptosis. Hepatology 2009, 49, 2031–2043. [Google Scholar] [CrossRef]
- Michailidis, E.; Pabon, J.; Xiang, K.; Park, P.; Ramanan, V.; Hoffmann, H.-H.; Schneider, W.M.; Bhatia, S.N.; de Jong, Y.P.; Shlomai, A.; et al. A robust cell culture system supporting the complete life cycle of hepatitis B virus. Scientific reports. Sci. Rep. 2017, 7, 16616. [Google Scholar] [CrossRef]
- Watashi, K.; Sluder, A.; Daito, T.; Matsunaga, S.; Ryo, A.; Nagamori, S.; Iwamoto, M.; Nakajima, S.; Tsukuda, S.; Borroto-Esoda, K.; et al. Cyclosporin A and its analogs inhibit hepatitis B virus entry into cultured hepatocytes through targeting a membrane transporter, sodium taurocholate cotransporting polypeptide (NTCP). Hepatology 2014, 59, 1726–1737. [Google Scholar] [CrossRef]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef]
- Aravalli, R.N. Role of innate immunity in the development of hepatocellular carcinoma. World J. Gastroenterol. 2013, 19, 7500–7514. [Google Scholar] [CrossRef] [PubMed]
- Foerster, F.; Hess, M.; Gerhold-Ay, A.; Marquardt, J.U.; Becker, D.; Galle, P.R.; Schuppan, D.; Binder, H.; Bockamp, E. The immune contexture of hepatocellular carcinoma predicts clinical outcome. Sci. Rep. 2018, 8, 5351. [Google Scholar] [CrossRef] [PubMed]
- Schulze-Bergkamen, H.; Untergasser, A.; Dax, A.; Vogel, H.; Büchler, P.; Klar, E.; Lehnert, T.; Friess, H.; Büchler, M.W.; Kirschfink, M.; et al. Primary human hepatocytes—A valuable tool for investigation of apoptosis and hepatitis B virus infection. J. Hepatol. 2003, 38, 736–744. [Google Scholar] [CrossRef]
- Chai, N.; Chang, H.E.; Nicolas, E.; Gudima, S.; Chang, J.; Taylor, J. Assembly of hepatitis B virus envelope proteins onto a lentivirus pseudotype that infects primary human hepatocytes. J. Virol. 2007, 81, 10897–10904. [Google Scholar] [CrossRef]
- Schulze, A.; Mills, K.; Weiss, T.S.; Urban, S. Hepatocyte polarization is essential for the productive entry of the hepatitis B virus. Hepatology 2012, 55, 373–383. [Google Scholar] [CrossRef]
- König, A.; Döring, B.; Mohr, C.; Geipel, A.; Geyer, J.; Glebe, D. Kinetics of the bile acid transporter and hepatitis B virus receptor Na+/taurocholate cotransporting polypeptide (NTCP) in hepatocytes. J. Hepatol. 2014, 61, 867–875. [Google Scholar] [CrossRef]
- Ware, B.R.; Durham, M.J.; Monckton, C.P.; Khetani, S.R. A Cell Culture Platform to Maintain Long-Term Phenotype of Primary Human Hepatocytes and Endothelial Cells. Cell. Mol. Gastroenterol. Hepatol. 2017, 5, 187–207. [Google Scholar] [CrossRef]
- Bale, S.S.; Golberg, I.; Jindal, R.; McCarty, W.J.; Luitje, M.; Hegde, M.; Bhushan, A.; Usta, O.B.; Yarmush, M.L. Long-Term Coculture Strategies for Primary Hepatocytes and Liver Sinusoidal Endothelial Cells. Tissue Eng. Part C Methods 2015, 21, 413–422. [Google Scholar] [CrossRef]
- Gural, N.; Mancio-Silva, L.; He, J.; Bhatia, S.N. Engineered Livers for Infectious Diseases. Cell. Mol. Gastroenterol. Hepatol. 2017, 5, 131–144. [Google Scholar] [CrossRef]
- Xia, Y.; Stadler, D.; Lucifora, J.; Reisinger, F.; Webb, D.; Hösel, M.; Michler, T.; Wisskirchen, K.; Cheng, X.; Zhang, K.; et al. Interferon-γ and Tumor Necrosis Factor-α Produced by T Cells Reduce the HBV Persistence Form, cccDNA, Without Cytolysis. Gastroenterology 2016, 150, 194–205. [Google Scholar] [CrossRef]
- Qiao, Y.; Han, X.; Guan, G.; Wu, N.; Sun, J.; Pak, V.; Liang, G. TGF-β triggers HBV cccDNA degradation through AID-dependent deamination. FEBS Lett. 2016, 590, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Lucifora, J.; Xia, Y.; Reisinger, F.; Zhang, K.; Stadler, D.; Cheng, X.; Sprinzl, M.F.; Koppensteiner, H.; Makowska, Z.; Volz, T.; et al. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science 2014, 343, 1221–1228. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Tang, L.; Shu, S.; Sehgal, M.; Sheraz, M.; Liu, B.; Zhao, Q.; Cheng, J.; Zhao, X.; Zhou, T.; et al. Activation of Stimulator of Interferon Genes in Hepatocytes Suppresses the Replication of Hepatitis B Virus. Antimicrob. Agents Chemother. 2017, 61, e00771-17. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.; Koh, S.; Bertoletti, A. Immune Response in Hepatitis B Virus Infection. Cold Spring Harb. Perspect. Med. 2015, 5, a021428. [Google Scholar] [CrossRef] [PubMed]
- Maini, M.K.; Gehring, A.J. The role of innate immunity in the immunopathology and treatment of HBV infection. J. Hepatol. 2016, 64, S60–S70. [Google Scholar] [CrossRef]
- Guidotti, L.G.; Isogawa, M.; Chisari, F.V. Host-Virus interactions in hepatitis B virus infection. Curr. Opin. Immunol. 2015, 36, 61–66. [Google Scholar] [CrossRef]
- Volz, T.; Allweiss, L.; MBarek, M.B.; Warlich, M.; Lohse, A.W.; Pollok, J.M.; Alexandrov, A.; Urban, S.; Petersen, J.; Lütgehetmann, M.; et al. The entry inhibitor Myrcludex-B efficiently blocks intrahepatic virus spreading in humanized mice previously infected with hepatitis B virus. J. Hepatol. 2013, 58, 861–867. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sa-ngiamsuntorn, K.; Thongsri, P.; Pewkliang, Y.; Wongkajornsilp, A.; Kongsomboonchoke, P.; Suthivanich, P.; Borwornpinyo, S.; Hongeng, S. An Immortalized Hepatocyte-Like Cell Line (imHC) Accommodated Complete Viral Lifecycle, Viral Persistence Form, cccDNA and Eventual Spreading of a Clinically-Isolated HBV. Viruses 2019, 11, 952. https://doi.org/10.3390/v11100952
Sa-ngiamsuntorn K, Thongsri P, Pewkliang Y, Wongkajornsilp A, Kongsomboonchoke P, Suthivanich P, Borwornpinyo S, Hongeng S. An Immortalized Hepatocyte-Like Cell Line (imHC) Accommodated Complete Viral Lifecycle, Viral Persistence Form, cccDNA and Eventual Spreading of a Clinically-Isolated HBV. Viruses. 2019; 11(10):952. https://doi.org/10.3390/v11100952
Chicago/Turabian StyleSa-ngiamsuntorn, Khanit, Piyanoot Thongsri, Yongyut Pewkliang, Adisak Wongkajornsilp, Pattida Kongsomboonchoke, Phichaya Suthivanich, Suparerk Borwornpinyo, and Suradej Hongeng. 2019. "An Immortalized Hepatocyte-Like Cell Line (imHC) Accommodated Complete Viral Lifecycle, Viral Persistence Form, cccDNA and Eventual Spreading of a Clinically-Isolated HBV" Viruses 11, no. 10: 952. https://doi.org/10.3390/v11100952
APA StyleSa-ngiamsuntorn, K., Thongsri, P., Pewkliang, Y., Wongkajornsilp, A., Kongsomboonchoke, P., Suthivanich, P., Borwornpinyo, S., & Hongeng, S. (2019). An Immortalized Hepatocyte-Like Cell Line (imHC) Accommodated Complete Viral Lifecycle, Viral Persistence Form, cccDNA and Eventual Spreading of a Clinically-Isolated HBV. Viruses, 11(10), 952. https://doi.org/10.3390/v11100952