High Resolution Analysis of Respiratory Syncytial Virus Infection In Vivo

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement and Source of Nasopharyngeal Aspirates
2.2. Viruses and Cells
2.3. Label Free Mass Spectrometry and Informatics
2.4. RNA Extraction and Sequencing
3. Results
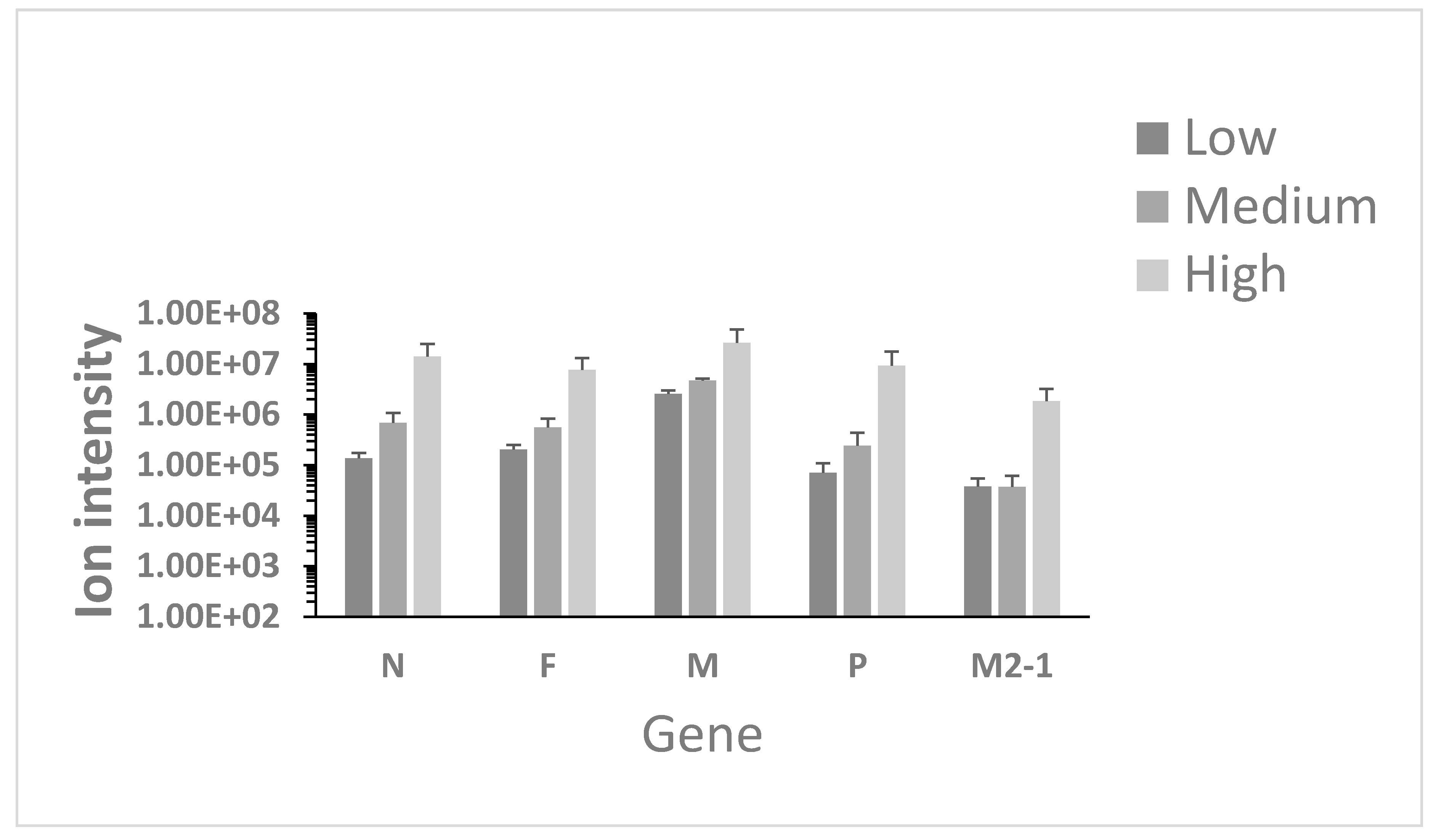
3.1. Identification of Viral Proteins in Nasopharyngeal Aspirates
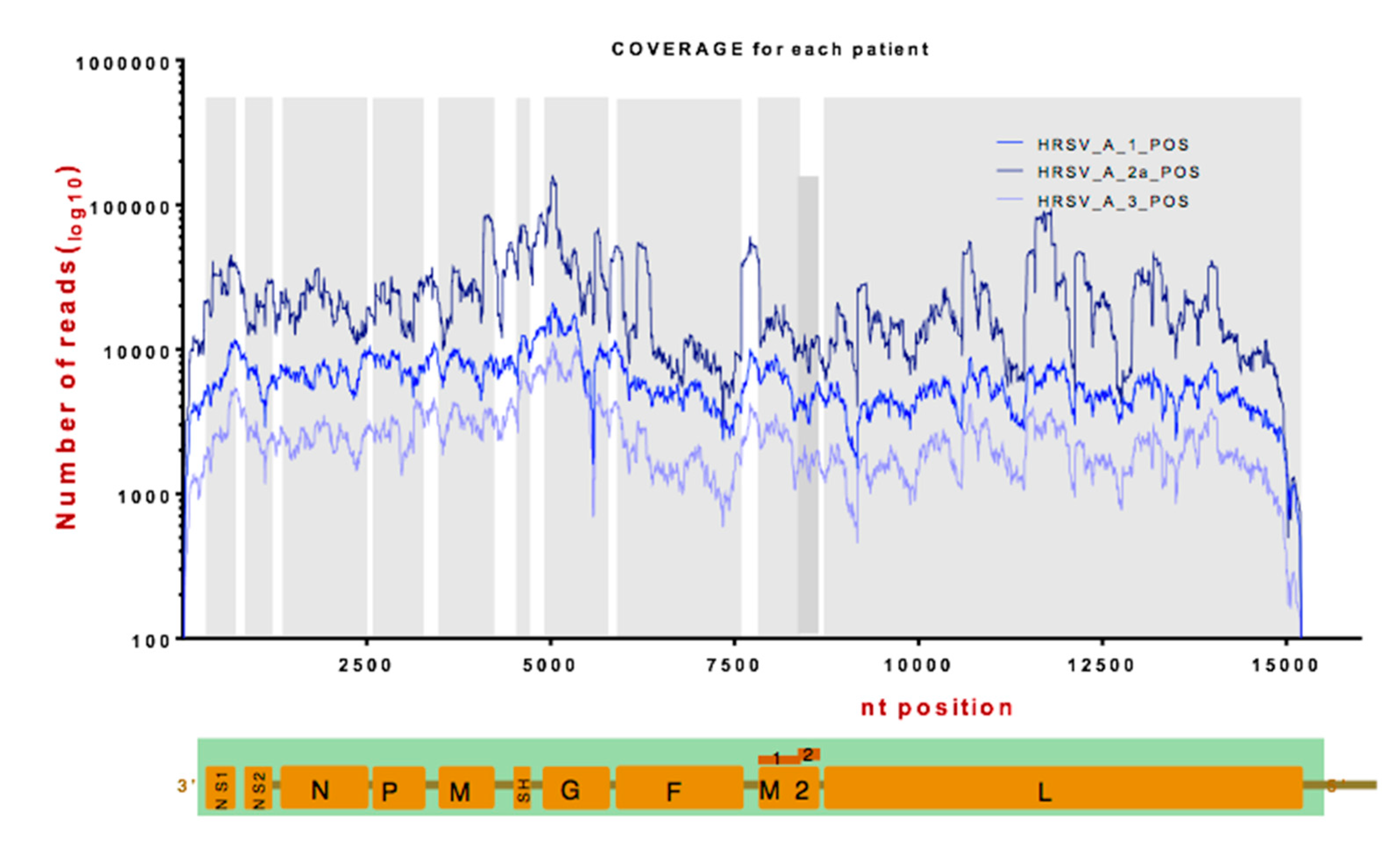
3.2. Identification of Viral RNA in Nasopharyngeal Aspirates
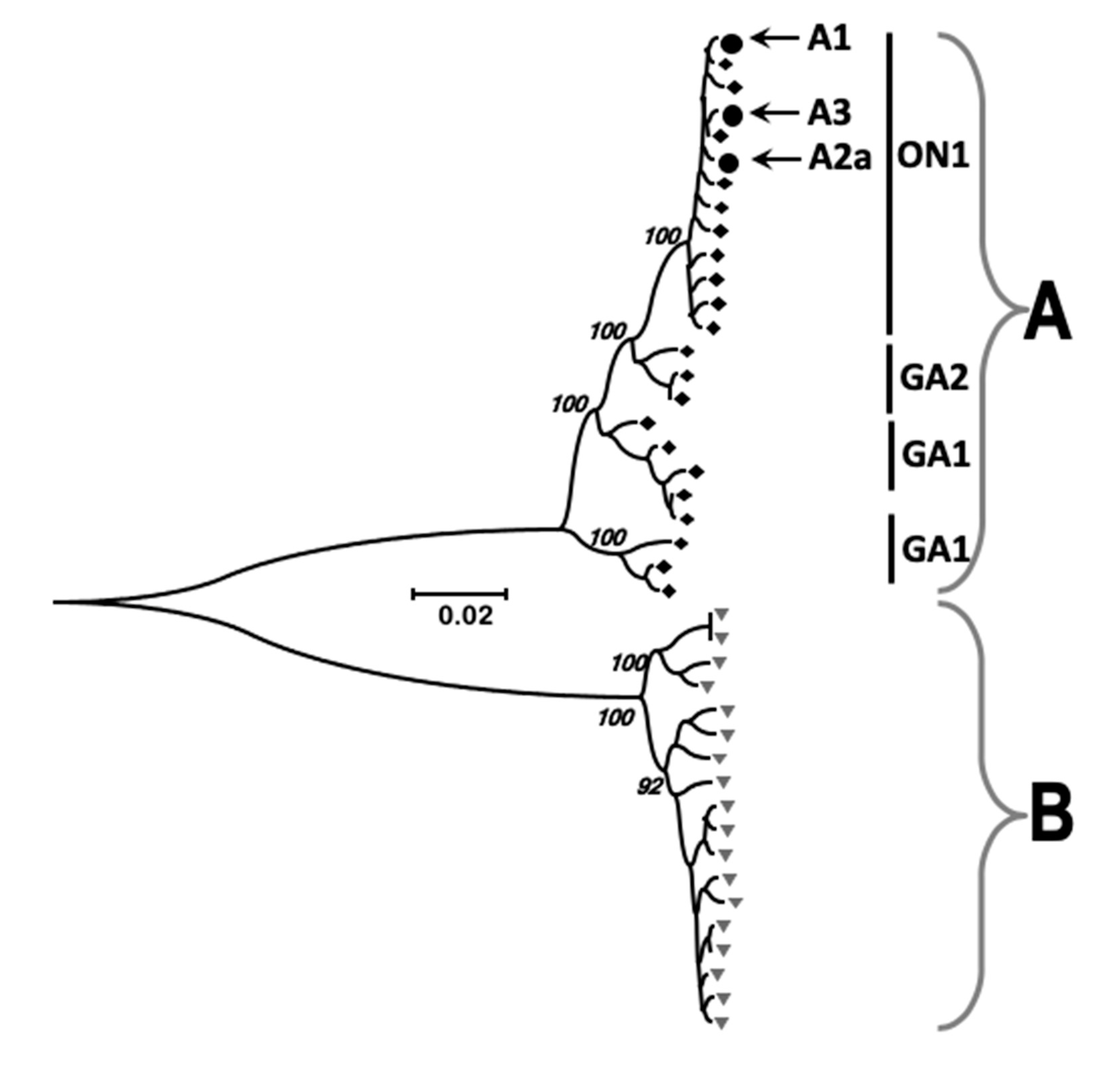
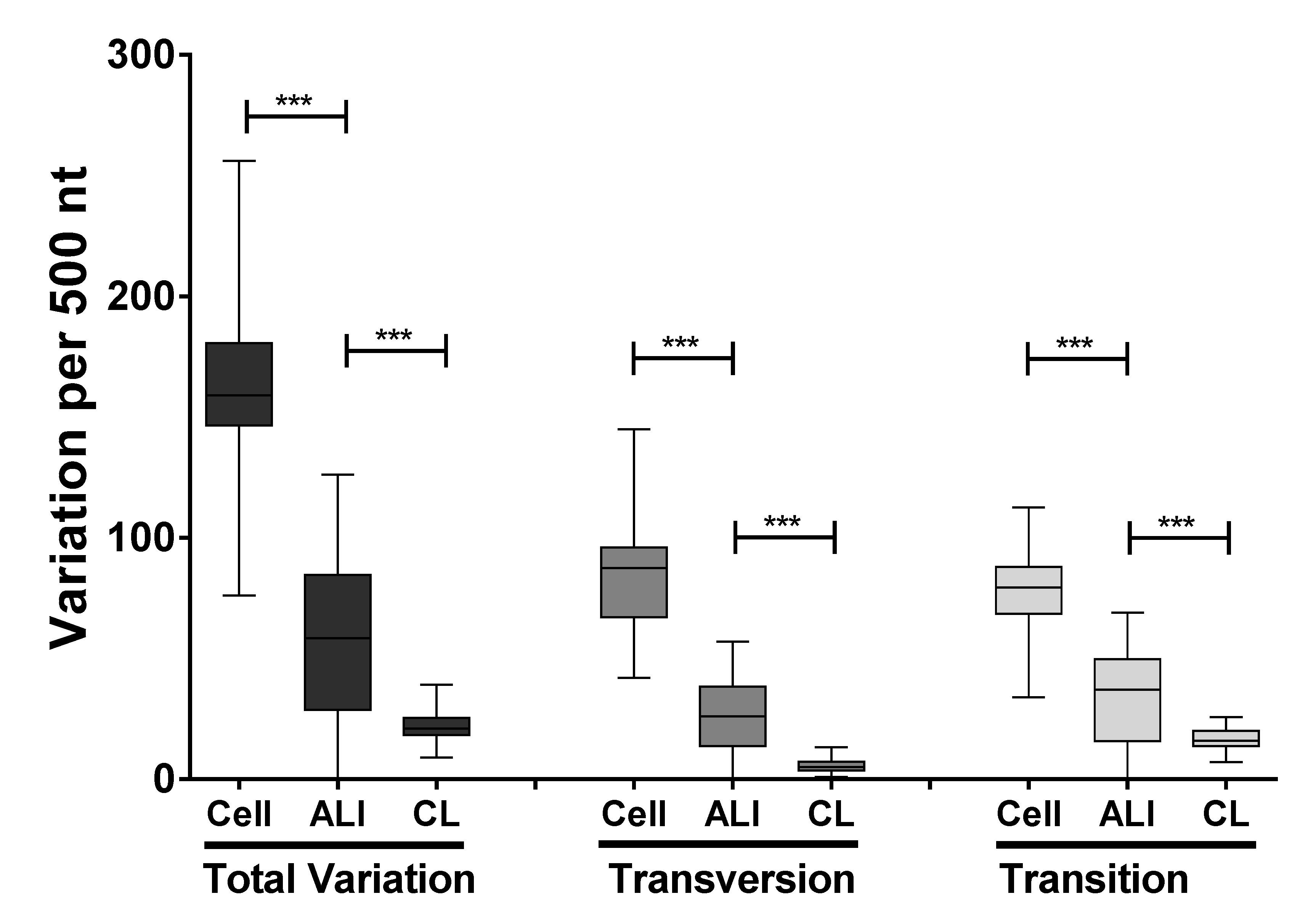
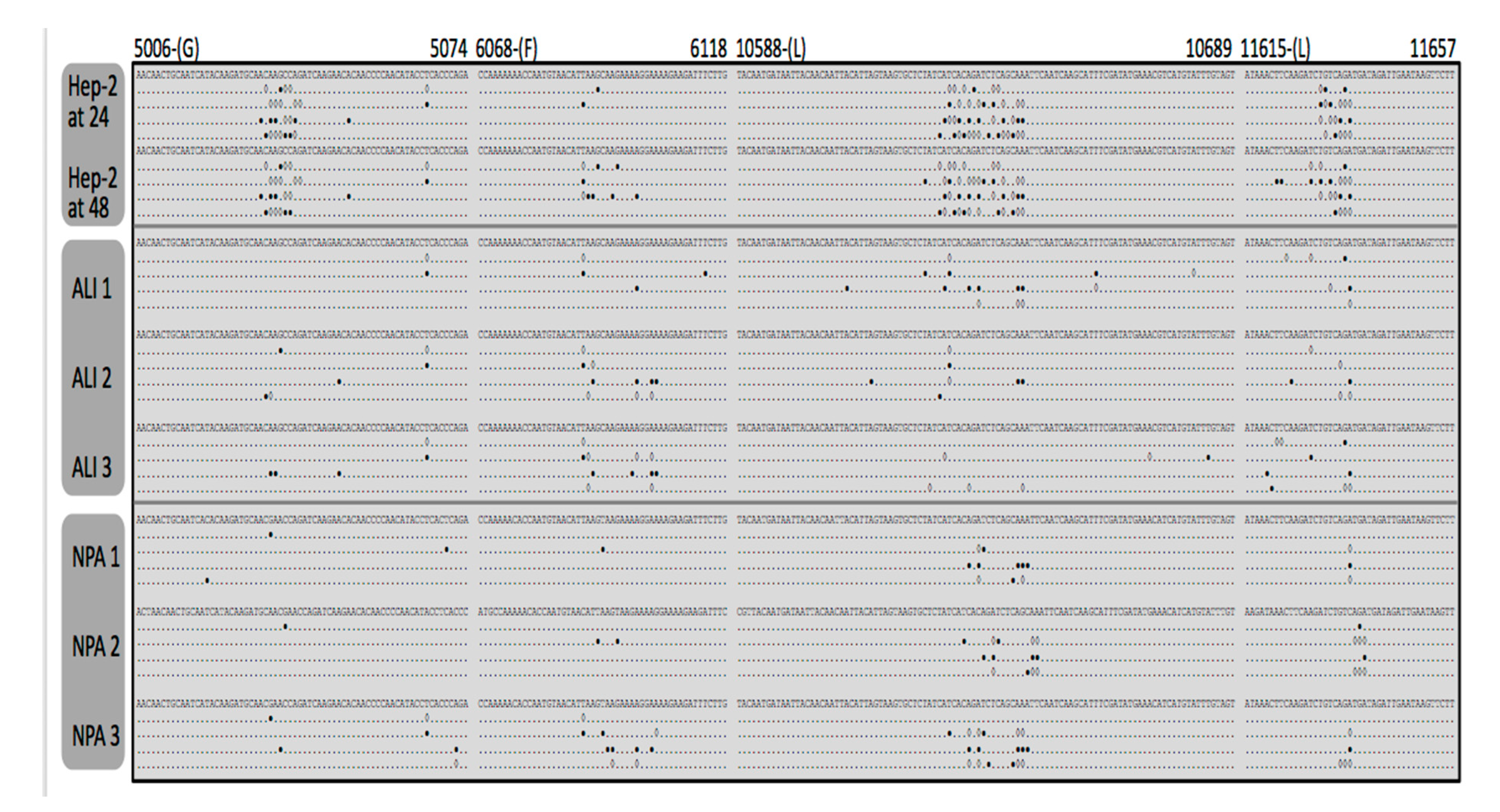
3.3. Identification and Comparison of Minor Variants in HRSV in the Clinical Samples Compared to Air Liquid Interface and Cell Culture Systems
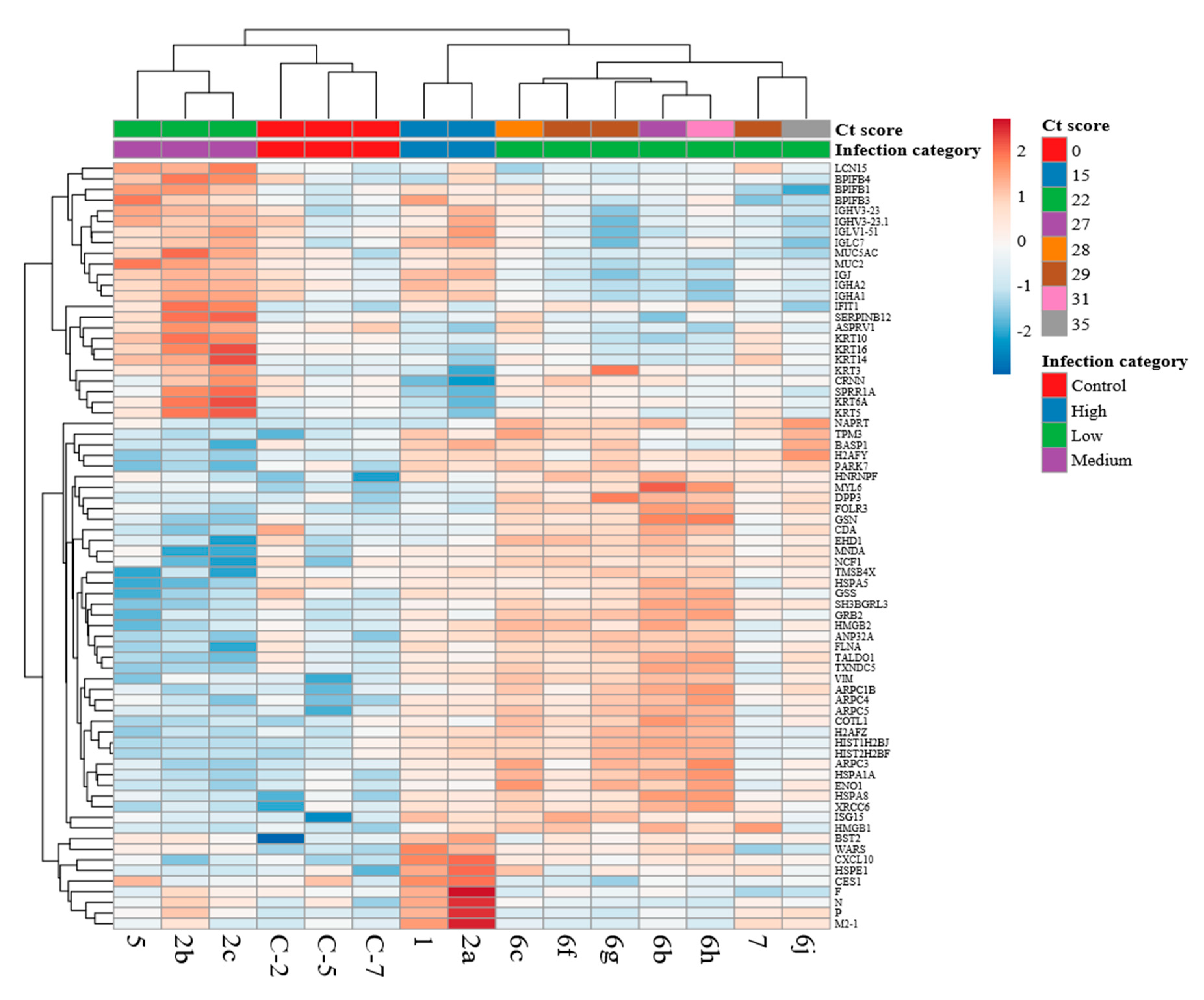
3.4. Comparison of the Host Response Between Paediatric Cases with HRSV and Control Cases
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Collins, P.L.; Graham, B.S. Viral and host factors in human respiratory syncytial virus pathogenesis. J. Virol. 2008, 82, 2040–2055. [Google Scholar] [CrossRef] [PubMed]
- Coultas, J.A.; Smyth, R.; Openshaw, P.J. Respiratory syncytial virus (RSV): A scourge from infancy to old age. Thorax 2019, 74, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Committee on Infectious Diseases. Modified recommendations for use of palivizumab for prevention of respiratory syncytial virus infections. Pediatrics 2009, 124, 1694–1701. [Google Scholar] [CrossRef] [PubMed]
- Falsey, A.R.; Hennessey, P.A.; Formica, M.A.; Cox, C.; Walsh, E.E. Respiratory Syncytial Virus Infection in Elderly and High-Risk Adults. N. Engl. J. Med. 2005, 352, 1749–1759. [Google Scholar] [CrossRef] [PubMed]
- van Drunen Littel-van den Hurk, S.; Watkiss, E.R. Pathogenesis of respiratory syncytial virus. Curr. Opin. Virol. 2012, 2, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, E.; Finelli, L.; O’Halloran, A.; Liu, P.; Karaca, Z.; Steiner, C.A.; Viboud, C.; Lipsitch, M. Hospitalizations associated with respiratory syncytial virus (RSV) and influenza in children, including children diagnosed with asthma. Epidemiology 2019, 30, 918–926. [Google Scholar] [CrossRef]
- Zhong, P.; Zhang, H.; Chen, X.; Lv, F. Clinical characteristics of the lower respiratory tract infection caused by a single infection or coinfection of the human parainfluenza virus in children. J. Med. Virol. 2019, 91, 1625–1632. [Google Scholar] [CrossRef]
- MacLellan, K.; Loney, C.; Yeo, R.P.; Bhella, D. The 24-Angstrom Structure of Respiratory Syncytial Virus Nucleocapsid Protein-RNA Decameric Rings. J. Virol. 2007, 81, 9519–9524. [Google Scholar] [CrossRef][Green Version]
- Liu, W.; Chen, D.; Tan, W.; Xu, D.; Qiu, S.; Zeng, Z.; Li, X.; Zhou, R. Epidemiology and Clinical Presentations of Respiratory Syncytial Virus Subgroups A and B Detected with Multiplex Real-Time PCR. PLoS ONE 2016, 11, e0165108. [Google Scholar] [CrossRef]
- Wu, W.; Macdonald, A.; Hiscox, J.A.; Barr, J.N. Different NF-kappaB activation characteristics of human respiratory syncytial virus subgroups A and B. Microb. Pathog. 2012, 52, 184–191. [Google Scholar] [CrossRef]
- Martinello, R.A.; Chen, M.D.; Weibel, C.; Kahn, J.S. Correlation between Respiratory Syncytial Virus Genotype and Severity of Illness. J. Infect. Dis. 2002, 186, 839–842. [Google Scholar] [CrossRef] [PubMed]
- Afonso, C.L.; Amarasinghe, G.K.; Bányai, K.; Bào, Y.; Basler, C.F.; Bavari, S.; Bejerman, N.; Blasdell, K.R.; Briand, F.-X.; Briese, T.; et al. Taxonomy of the order Mononegavirales: Update 2016. Arch. Virol. 2016, 161, 2351–2360. [Google Scholar] [CrossRef] [PubMed]
- Maes, P.; Amarasinghe, G.K.; Ayllón, M.A.; Basler, C.F.; Bavari, S.; Blasdell, K.R.; Briese, T.; Brown, P.A.; Bukreyev, A.; Balkema-Buschmann, A.; et al. Taxonomy of the order Mononegavirales: Second update 2018. Arch. Virol. 2019, 164, 1233–1244. [Google Scholar] [CrossRef] [PubMed]
- Tawar, R.G.; Duquerroy, S.; Vonrhein, C.; Varela, P.F.; Damier-Piolle, L.; Castagné, N.; MacLellan, K.; Bedouelle, H.; Bricogne, G.; Bhella, D.; et al. Crystal Structure of a Nucleocapsid-Like Nucleoprotein-RNA Complex of Respiratory Syncytial Virus. Science 2009, 326, 1279–1283. [Google Scholar] [CrossRef] [PubMed]
- Fearns, R.; Collins, P.L. Role of the M2-1 Transcription Antitermination Protein of Respiratory Syncytial Virus in Sequential Transcription. J. Virol. 1999, 73, 5852–5864. [Google Scholar] [PubMed]
- Flanagan, B.F.; Hart, C.A.; McNamara, P.S.; Smyth, R.L. Production of Chemokines in the Lungs of Infants with Severe Respiratory Syncytial Virus Bronchiolitis. J. Infect. Dis. 2005, 191, 1225–1232. [Google Scholar]
- Munday, D.C.; Emmott, E.; Surtees, R.; Lardeau, C.-H.; Wu, W.; Duprex, W.P.; Dove, B.K.; Barr, J.N.; Hiscox, J.A. Quantitative proteomic analysis of A549 cells infected with human respiratory syncytial virus. Mol. Cell. Proteom. 2010, 9, 2438–2459. [Google Scholar] [CrossRef]
- Arruvito, L.; Raiden, S.; Geffner, J. Host response to respiratory syncytial virus infection. Curr. Opin. Infect. Dis. 2015, 28, 259–266. [Google Scholar] [CrossRef]
- Russell, C.D.; Unger, S.A.; Walton, M.; Schwarze, J. The Human Immune Response to Respiratory Syncytial Virus Infection. Clin. Microbiol. Rev. 2017, 30, 481–502. [Google Scholar] [CrossRef]
- Levitz, R.; Gao, Y.; Dozmorov, I.; Song, R.; Wakeland, E.K.; Kahn, J.S. Distinct patterns of innate immune activation by clinical isolates of respiratory syncytial virus. PLoS ONE 2017, 12, e0184318. [Google Scholar] [CrossRef]
- Wu, W.; Tran, K.C.; Teng, M.N.; Heesom, K.J.; Matthews, D.A.; Barr, J.N.; Hiscox, J.A. The Interactome of the Human Respiratory Syncytial Virus NS1 Protein Highlights Multiple Effects on Host Cell Biology. J. Virol. 2012, 86, 7777–7789. [Google Scholar] [CrossRef] [PubMed]
- McCutcheon, K.M.; Jordan, R.; Mawhorter, M.E.; Noton, S.L.; Powers, J.G.; Fearns, R.; Cihlar, T.; Perron, M. The Interferon Type I/III Response to Respiratory Syncytial Virus Infection in Airway Epithelial Cells Can Be Attenuated or Amplified by Antiviral Treatment. J. Virol. 2016, 90, 1705–1717. [Google Scholar] [CrossRef] [PubMed]
- Bhoj, V.G.; Sun, Q.; Bhoj, E.J.; Somers, C.; Chen, X.; Torres, J.-P.; Mejias, A.; Gomez, A.M.; Jafri, H.; Ramilo, O.; et al. MAVS and MyD88 are essential for innate immunity but not cytotoxic T lymphocyte response against respiratory syncytial virus. Proc. Natl. Acad. Sci. USA 2008, 105, 14046–14051. [Google Scholar] [CrossRef] [PubMed]
- Bitko, V.; Shulyayeva, O.; Mazumder, B.; Musiyenko, A.; Ramaswamy, M.; Look, D.C.; Barik, S. Nonstructural proteins of respiratory syncytial virus suppress premature apoptosis by an NF-kappaB-dependent, interferon-independent mechanism and facilitate virus growth. J. Virol. 2007, 81, 1786–1795. [Google Scholar] [CrossRef] [PubMed]
- Omar, S.; Clarke, R.; Abdullah, H.; Brady, C.; Corry, J.; Winter, H.; Touzelet, O.; Power, U.F.; Lundy, F.; McGarvey, L.P.A.; et al. Respiratory virus infection up-regulates TRPV1, TRPA1 and ASICS3 receptors on airway cells. PLoS ONE 2017, 12, e0171681. [Google Scholar] [CrossRef] [PubMed]
- Leemans, A.; De Schryver, M.; Van Der Gucht, W.; Heykers, A.; Pintelon, I.; Hotard, A.L.; Moore, M.L.; Melero, J.A.; McLellan, J.S.; Graham, B.S.; et al. Antibody-Induced Internalization of the Human Respiratory Syncytial Virus Fusion Protein. J. Virol. 2017, 91, e00184-17. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Armstrong, S.D.; Xia, D.; Makepeace, B.L.; Darby, A.C.; Kadowaki, T. Draft genome of the honey bee ectoparasitic mite, Tropilaelaps mercedesae, is shaped by the parasitic life history. GigaScience 2017, 6, 1–17. [Google Scholar] [CrossRef]
- Töpfer, A.; Zagordi, O.; Prabhakaran, S.; Roth, V.; Halperin, E.; Beerenwinkel, N. Probabilistic Inference of Viral Quasispecies Subject to Recombination. J. Comput. Boil. 2013, 20, 113–123. [Google Scholar] [CrossRef]
- Carroll, M.W.; Matthews, D.A.; Hiscox, J.A.; Elmore, M.J.; Pollakis, G.; Rambaut, A.; Hewson, R.; García-Dorival, I.; Bore, J.A.; Koundouno, R.; et al. Temporal and spatial analysis of the 2014–2015 Ebola virus outbreak in West Africa. Nature 2015, 524, 97–101. [Google Scholar] [CrossRef]
- Aljabr, W.; Touzelet, O.; Pollakis, G.; Wu, W.; Munday, D.C.; Hughes, M.; Hertz-Fowler, C.; Kenny, J.; Fearns, R.; Barr, J.N.; et al. Investigating the Influence of Ribavirin on Human Respiratory Syncytial Virus RNA Synthesis by Using a High-Resolution Transcriptome Sequencing Approach. J. Virol. 2016, 90, 4876–4888. [Google Scholar] [CrossRef]
- Dowall, S.D.A.; Matthews, D.; García-Dorival, I.; Taylor, I.; Kenny, J.; Hertz-Fowler, C.; Hall, N.; Corbin-Lickfett, K.; Empig, C.; Schlunegger, K.; et al. Elucidating variations in the nucleotide sequence of Ebola virus associated with increasing pathogenicity. Genome Boil. 2014, 15, 201. [Google Scholar] [CrossRef]
- Greco, T.M.; Cristea, I.M. Proteomics Tracing the Footsteps of Infectious Disease. Mol. Cell. Proteom. 2017, 16, S5–S14. [Google Scholar] [CrossRef] [PubMed]
- Cane, P.A.; Matthews, D.A.; Pringle, C.R. Identification of variable domains of the attachment (G) protein of subgroup A respiratory syncytial viruses. J. Gen. Virol. 1991, 72, 2091–2096. [Google Scholar] [CrossRef] [PubMed]
- Cane, P.A.; Matthews, D.A.; Pringle, C.R. Frequent polymerase errors observed in a restricted area of clones derived from the attachment (G) protein gene of respiratory syncytial virus. J. Virol. 1993, 67, 1090–1093. [Google Scholar] [PubMed]
- Di Giallonardo, F.; Kok, J.; Fernandez, M.; Carter, I.; Geoghegan, J.; Dwyer, D.; Holmes, E.; Eden, J.S. Evolution of Human Respiratory Syncytial Virus (RSV) over Multiple Seasons in New South Wales, Australia. Viruses 2018, 10, 476. [Google Scholar] [CrossRef] [PubMed]
- Bose, M.E.; He, J.; Shrivastava, S.; Nelson, M.I.; Bera, J.; Halpin, R.A.; Town, C.D.; Lorenzi, H.A.; Noyola, D.E.; Falcone, V.; et al. Sequencing and Analysis of Globally Obtained Human Respiratory Syncytial Virus A and B Genomes. PLoS ONE 2015, 10, e0120098. [Google Scholar] [CrossRef] [PubMed]
- Lum, F.-M.; Lee, D.; Chua, T.-K.; Tan, J.J.L.; Lee, C.Y.P.; Liu, X.; Fang, Y.; Lee, B.; Yee, W.-X.; Rickett, N.Y.; et al. Zika Virus Infection Preferentially Counterbalances Human Peripheral Monocyte and/or NK Cell Activity. MSphere 2018, 3, e00120-18. [Google Scholar] [CrossRef]
- Villenave, R.; Shields, M.D.; Power, U.F. Respiratory syncytial virus interaction with human airway epithelium. Trends Microbiol. 2013, 21, 238–244. [Google Scholar] [CrossRef]
- Villenave, R.; Thavagnanam, S.; Sarlang, S.; Parker, J.; Douglas, I.; Skibinski, G.; Heaney, L.G.; McKaigue, J.P.; Coyle, P.V.; Shields, M.D.; et al. In vitro modeling of respiratory syncytial virus infection of pediatric bronchial epithelium, the primary target of infection in vivo. Proc. Natl. Acad. Sci. USA 2012, 109, 5040–5045. [Google Scholar] [CrossRef]
- Melero, J.A.; Pringle, C.R.; Cane, P.A. Antigenic structure, evolution and immunobiology of human respiratory syncytial virus attachment (G) protein. J. Gen. Virol. 1997, 78, 2411–2418. [Google Scholar] [CrossRef]
- Rajagopala, S.V.; Singh, H.; Patel, M.C.; Wang, W.; Tan, Y.; Shilts, M.H.; Hartert, T.V.; Boukhvalova, M.S.; Blanco, J.C.G.; Das, S.R. Cotton rat lung transcriptome reveals host immune response to Respiratory Syncytial Virus infection. Sci. Rep. 2018, 8, 11318. [Google Scholar] [CrossRef] [PubMed]
- Akram, K.M.; Moyo, N.A.; Leeming, G.H.; Bingle, L.; Jasim, S.; Hussain, S.; Schorlemmer, A.; Kipar, A.; Digard, P.; Tripp, R.A.; et al. An innate defense peptide BPIFA1/SPLUNC1 restricts influenza A virus infection. Mucosal Immunol. 2018, 11, 71. [Google Scholar] [CrossRef] [PubMed]
- Zomer-Kooijker, K.; Uiterwaal, C.S.P.M.; Van Der Gugten, A.C.; Wilbrink, B.; Bont, L.J.; Van Der Ent, C.K. Decreased lung function precedes severe respiratory syncytial virus infection and post-respiratory syncytial virus wheeze in term infants. Eur. Respir. J. 2014, 44, 666–674. [Google Scholar] [CrossRef] [PubMed]
- Openshaw, P.J.M. Antiviral Immune Responses and Lung Inflammation after Respiratory Syncytial Virus Infection. Proc. Am. Thorac. Soc. 2005, 2, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Heikkinen, T.; Marttila, J.; Salmi, A.A.; Ruuskanen, O. Nasal Swab versus Nasopharyngeal Aspirate for Isolation of Respiratory Viruses. J. Clin. Microbiol. 2002, 40, 4337–4339. [Google Scholar] [CrossRef] [PubMed]
- Chabriere, E.; Bassène, H.; Drancourt, M.; Sokhna, C. MALDI-TOF MS and point of care are disruptive diagnostic tools in Africa. New Microbes New Infect. 2018, 26, S83–S88. [Google Scholar] [CrossRef] [PubMed]
- Carroll, M.W.; Haldenby, S.; Rickett, N.Y.; Pályi, B.; Garcia-Dorival, I.; Liu, X.; Barker, G.; Bore, J.A.; Koundouno, F.R.; Williamson, E.D.; et al. Deep Sequencing of RNA from Blood and Oral Swab Samples Reveals the Presence of Nucleic Acid from a Number of Pathogens in Patients with Acute Ebola Virus Disease and Is Consistent with Bacterial Translocation across the Gut. MSphere 2017, 2, e00325-17. [Google Scholar] [CrossRef] [PubMed]
- Dave, K.A.; Norris, E.L.; Bukreyev, A.A.; Headlam, M.J.; Buchholz, U.J.; Singh, T.; Collins, P.L.; Gorman, J.J. A comprehensive proteomic view of responses of A549 type II alveolar epithelial cells to human respiratory syncytial virus infection. Mol. Cell. Proteom. 2014, 13, 3250–3269. [Google Scholar] [CrossRef] [PubMed]
- Calderaro, A.; Arcangeletti, M.C.; Rodighiero, I.; Buttrini, M.; Montecchini, S.; Simone, R.V.; Medici, M.C.; Chezzi, C.; De Conto, F. Identification of different respiratory viruses, after a cell culture step, by matrix assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF MS). Sci. Rep. 2016, 6, 36082. [Google Scholar] [CrossRef] [PubMed]
- Delorme-Axford, E.; Morosky, S.; Bomberger, J.; Stolz, D.B.; Jackson, W.T.; Coyne, C.B. BPIFB3 Regulates Autophagy and Coxsackievirus B Replication through a Noncanonical Pathway Independent of the Core Initiation Machinery. MBio 2014, 5, e02147-14. [Google Scholar] [CrossRef]
- Liu, M.; Li, H.; Xue, C.X.; Gu, L.; Qu, J.X.; Yu, X.M.; Wang, Y.M.; Liu, Y.M.; Cao, B. Differences in inflammatory marker patterns for adult community-acquired pneumonia patients induced by different pathogens. Clin. Respir. J. 2018, 12, 974–985. [Google Scholar] [CrossRef] [PubMed]
- Huong, T.N.; Tan, B.H.; Sugrue, R.J. A Proteomic-Based Workflow Using Purified Respiratory Syncytial Virus Particles to Identify Cellular Factors as Drug Targets. Adv. Struct. Saf. Stud. 2016, 1442, 175–194. [Google Scholar]
- Munday, D.C.; Wu, W.; Smith, N.; Fix, J.; Noton, S.L.; Galloux, M.; Touzelet, O.; Armstrong, S.D.; Dawson, J.M.; Aljabr, W.; et al. Interactome analysis of the human respiratory syncytial virus RNA polymerase complex identifies protein chaperones as important cofactors that promote L-protein stability and RNA synthesis. J. Virol. 2015, 89, 917–930. [Google Scholar] [CrossRef] [PubMed]
- Hägglund, S.; Blodörn, K.; Näslund, K.; Vargmar, K.; Lind, S.B.; Mi, J.; Araínga, M.; Riffault, S.; Taylor, G.; Pringle, J.; et al. Proteome analysis of bronchoalveolar lavage from calves infected with bovine respiratory syncytial virus—Insights in pathogenesis and perspectives for new treatments. PLoS ONE 2017, 12, e0186594. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aljabr, W.; Armstrong, S.; Rickett, N.Y.; Pollakis, G.; Touzelet, O.; Cloutman-Green, E.; Matthews, D.A.; Hiscox, J.A. High Resolution Analysis of Respiratory Syncytial Virus Infection In Vivo. Viruses 2019, 11, 926. https://doi.org/10.3390/v11100926
Aljabr W, Armstrong S, Rickett NY, Pollakis G, Touzelet O, Cloutman-Green E, Matthews DA, Hiscox JA. High Resolution Analysis of Respiratory Syncytial Virus Infection In Vivo. Viruses. 2019; 11(10):926. https://doi.org/10.3390/v11100926
Chicago/Turabian StyleAljabr, Waleed, Stuart Armstrong, Natasha Y. Rickett, Georgios Pollakis, Olivier Touzelet, Elaine Cloutman-Green, David A. Matthews, and Julian A. Hiscox. 2019. "High Resolution Analysis of Respiratory Syncytial Virus Infection In Vivo" Viruses 11, no. 10: 926. https://doi.org/10.3390/v11100926
APA StyleAljabr, W., Armstrong, S., Rickett, N. Y., Pollakis, G., Touzelet, O., Cloutman-Green, E., Matthews, D. A., & Hiscox, J. A. (2019). High Resolution Analysis of Respiratory Syncytial Virus Infection In Vivo. Viruses, 11(10), 926. https://doi.org/10.3390/v11100926