Overview of HCV Life Cycle with a Special Focus on Current and Possible Future Antiviral Targets




{kind=link}
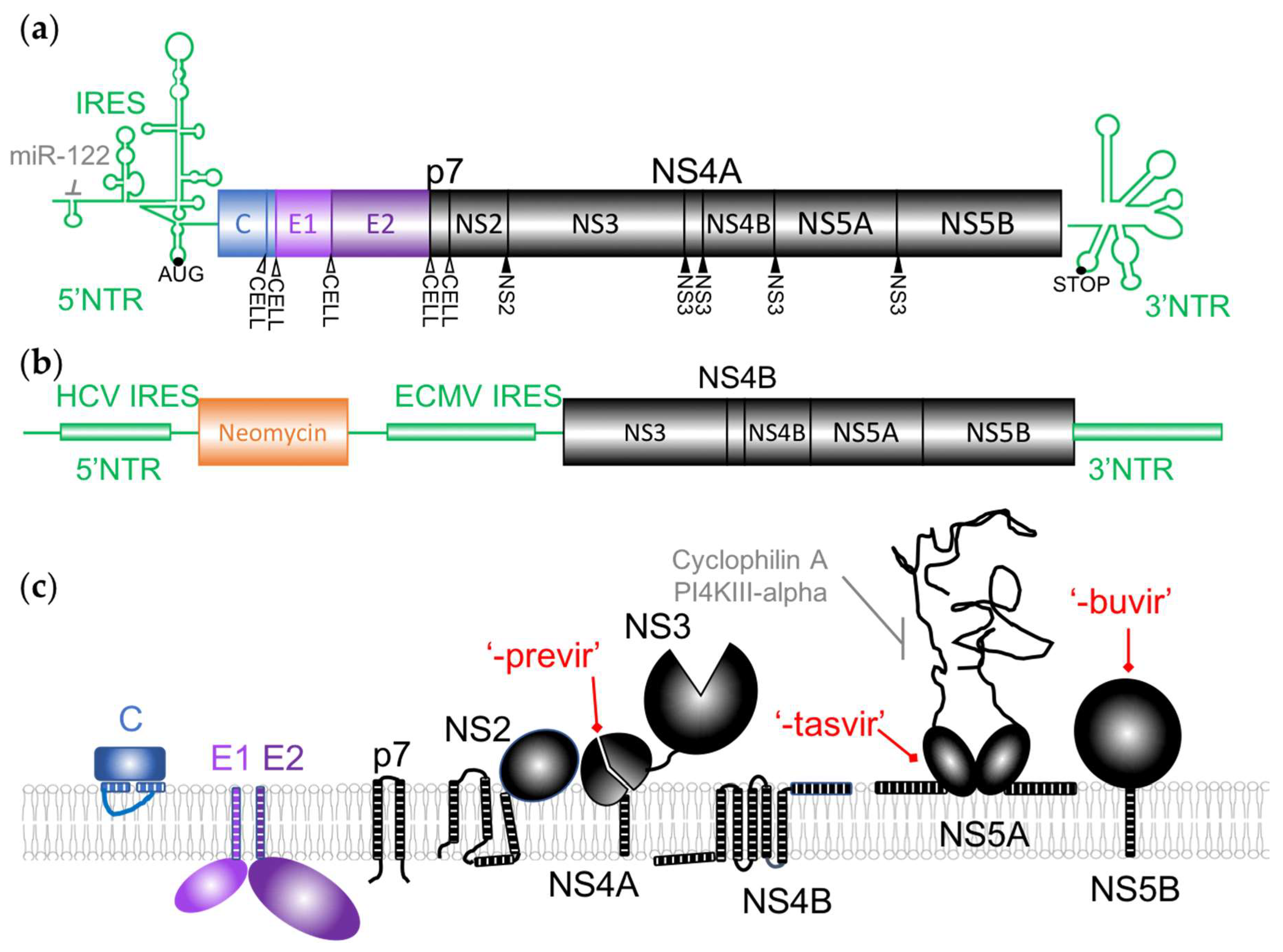{kind=link}
Abstract
1. Introduction
2. Overview of the HCV Life Cycle
2.1. Entry of HCV Particle into Hepatocytes
2.2. HCV RNA Translation and Replication
2.3. HCV Assembly, Budding and Secretion
3. Current Antiviral Targets
3.1. NS3/4A Inhibitors
3.2. NS5B Inhibitors
3.3. NS5A Inhibitors
4. Potential Future Targets
4.1. Targeting Other Viral Proteins
4.2. Host-Targeting Agents against Viral Replication and Entry
5. Concluding Remarks
Funding
Conflicts of Interest
References
- International Committee on Taxonomy of Viruses (ICTV). Available online: https://talk.ictvonline.org/ictv_wikis/flaviviridae/w/sg_flavi/56/hcv-classification (accessed on 22 July 2018).
- Choo, Q.L.; Kuo, G.; Weiner, A.J.; Overby, L.R.; Bradley, D.W.; Houghton, M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 1989, 244, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Hoofnagle, J.H.; Mullen, K.D.; Jones, D.B.; Rustgi, V.; Di Bisceglie, A.; Peters, M.; Waggoner, J.G.; Park, Y.; Jones, E.A. Treatment of chronic non-A, non-B hepatitis with recombinant human alpha interferon. A preliminary report. N. Engl. J. Med. 1986, 315, 1575–1578. [Google Scholar] [CrossRef] [PubMed]
- Poynard, T.; Marcellin, P.; Lee, S.S.; Niederau, C.; Minuk, G.S.; Ideo, G.; Bain, V.; Heathcote, J.; Zeuzem, S.; Trepo, C.; et al. Randomised trial of interferon alpha2b plus ribavirin for 48 weeks or for 24 weeks versus interferon alpha2b plus placebo for 48 weeks for treatment of chronic infection with hepatitis C virus. International Hepatitis Interventional Therapy Group (IHIT). Lancet 1998, 352, 1426–1432. [Google Scholar] [CrossRef]
- Manns, M.P.; McHutchison, J.G.; Gordon, S.C.; Rustgi, V.K.; Shiffman, M.; Reindollar, R.; Goodman, Z.D.; Koury, K.; Ling, M.; Albrecht, J.K. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: A randomised trial. Lancet 2001, 358, 958–965. [Google Scholar] [CrossRef]
- McHutchison, J.G.; Gordon, S.C.; Schiff, E.R.; Shiffman, M.L.; Lee, W.M.; Rustgi, V.K.; Goodman, Z.D.; Ling, M.H.; Cort, S.; Albrecht, J.K. Interferon alfa-2b alone or in combination with ribavirin as initial treatment for chronic hepatitis C. Hepatitis Interventional Therapy Group. N. Engl. J. Med. 1998, 339, 1485–1492. [Google Scholar] [CrossRef]
- Fried, M.W.; Shiffman, M.L.; Reddy, K.R.; Smith, C.; Marinos, G.; Gonçales, F.L.; Häussinger, D.; Diago, M.; Carosi, G.; Dhumeaux, D.; et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N. Engl. J. Med. 2002, 347, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; De Clercq, E. Current therapy for chronic hepatitis C: The role of direct-acting antivirals. Antivir. Res. 2017, 142, 83–122. [Google Scholar] [CrossRef]
- Pathak, N.; Lai, M.-L.; Chen, W.-Y.; Hsieh, B.-W.; Yu, G.-Y.; Yang, J.-M. Pharmacophore anchor models of flaviviral NS3 proteases lead to drug repurposing for DENV infection. BMC Bioinform. 2017, 18, 548. [Google Scholar] [CrossRef]
- Ogata, N.; Alter, H.J.; Miller, R.H.; Purcell, R.H. Nucleotide sequence and mutation rate of the H strain of hepatitis C virus. Proc. Natl. Acad. Sci. USA 1991, 88, 3392–3396. [Google Scholar] [CrossRef]
- Pawlotsky, J.-M. Hepatitis C Virus Resistance to Direct-Acting Antiviral Drugs in Interferon-Free Regimens. Gastroenterology 2016, 151, 70–86. [Google Scholar] [CrossRef]
- Sarrazin, C. The importance of resistance to direct antiviral drugs in HCV infection in clinical practice. J. Hepatol. 2016, 64, 486–504. [Google Scholar] [CrossRef] [PubMed]
- Halfon, P.; Sarrazin, C. Future treatment of chronic hepatitis C with direct acting antivirals: Is resistance important? Liver Int. 2012, 32, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Catanese, M.T.; Uryu, K.; Kopp, M.; Edwards, T.J.; Andrus, L.; Rice, W.J.; Silvestry, M.; Kuhn, R.J.; Rice, C.M. Ultrastructural analysis of hepatitis C virus particles. Proc. Natl. Acad. Sci. USA 2013, 110, 9505–9510. [Google Scholar] [CrossRef] [PubMed]
- Piver, E.; Boyer, A.; Gaillard, J.; Bull, A.; Beaumont, E.; Roingeard, P.; Meunier, J.-C. Ultrastructural organisation of HCV from the bloodstream of infected patients revealed by electron microscopy after specific immunocapture. Gut 2017, 66, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Denolly, S.; Granier, C.; Fontaine, N.; Pozzetto, B.; Bourlet, T.; Guérin, M.; Cosset, F.-L. A serum protein factor mediates maturation and apoB-association of HCV particles in the extracellular milieu. J. Hepatol. 2018. [Google Scholar] [CrossRef]
- Lavie, M.; Dubuisson, J. Interplay between hepatitis C virus and lipid metabolism during virus entry and assembly. Biochimie 2017, 141, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Cun, W.; Wu, X.; Shi, Q.; Tang, H.; Luo, G. Hepatitis C virus attachment mediated by apolipoprotein E binding to cell surface heparan sulfate. J. Virol. 2012, 86, 7256–7267. [Google Scholar] [CrossRef] [PubMed]
- Dao Thi, V.L.; Granier, C.; Zeisel, M.B.; Guérin, M.; Mancip, J.; Granio, O.; Penin, F.; Lavillette, D.; Bartenschlager, R.; Baumert, T.F.; et al. Characterization of hepatitis C virus particle subpopulations reveals multiple usage of the scavenger receptor BI for entry steps. J. Biol. Chem. 2012, 287, 31242–31257. [Google Scholar] [CrossRef] [PubMed]
- Bartosch, B.; Dubuisson, J.; Cosset, F.-L. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J. Exp. Med. 2003, 197, 633–642. [Google Scholar] [CrossRef]
- Hsu, M.; Zhang, J.; Flint, M.; Logvinoff, C.; Cheng-Mayer, C.; Rice, C.M.; McKeating, J.A. Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc. Natl. Acad. Sci. USA 2003, 100, 7271–7276. [Google Scholar] [CrossRef]
- Pileri, P.; Uematsu, Y.; Campagnoli, S.; Galli, G.; Falugi, F.; Petracca, R.; Weiner, A.J.; Houghton, M.; Rosa, D.; Grandi, G.; et al. Binding of hepatitis C virus to CD81. Science 1998, 282, 938–941. [Google Scholar] [CrossRef] [PubMed]
- Scarselli, E.; Ansuini, H.; Cerino, R.; Roccasecca, R.M.; Acali, S.; Filocamo, G.; Traboni, C.; Nicosia, A.; Cortese, R.; Vitelli, A. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. J. Hepatol. 2002, 21, 5017–5025. [Google Scholar] [CrossRef]
- Tong, Y.; Lavillette, D.; Li, Q.; Zhong, J. Role of Hepatitis C Virus Envelope Glycoprotein E1 in Virus Entry and Assembly. Front. Immunol. 2018, 9, 1411. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.J.; von Hahn, T.; Tscherne, D.M.; Syder, A.J.; Panis, M.; Wölk, B.; Hatziioannou, T.; McKeating, J.A.; Bieniasz, P.D.; Rice, C.M. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 2007, 446, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Ploss, A.; Evans, M.J.; Gaysinskaya, V.A.; Panis, M.; You, H.; de Jong, Y.P.; Rice, C.M. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 2009, 457, 882–886. [Google Scholar] [CrossRef] [PubMed]
- Dubuisson, J.; Cosset, F.-L. Virology and cell biology of the hepatitis C virus life cycle: An update. J. Hepatol. 2014, 61, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Lupberger, J.; Zeisel, M.B.; Xiao, F.; Thumann, C.; Fofana, I.; Zona, L.; Davis, C.; Mee, C.J.; Turek, M.; Gorke, S.; et al. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat. Med. 2011, 17, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Miao, Z.; Xie, Z.; Miao, J.; Ran, J.; Feng, Y.; Xia, X. Regulated Entry of Hepatitis C Virus into Hepatocytes. Viruses 2017, 9, 100. [Google Scholar] [CrossRef]
- Sharma, N.R.; Mateu, G.; Dreux, M.; Grakoui, A.; Cosset, F.-L.; Melikyan, G.B. Hepatitis C virus is primed by CD81 protein for low pH-dependent fusion. J. Biol. Chem. 2011, 286, 30361–30376. [Google Scholar] [CrossRef]
- Fan, H.; Qiao, L.; Kang, K.-D.; Fan, J.; Wei, W.; Luo, G. Attachment and Postattachment Receptors Important for Hepatitis C Virus Infection and Cell-to-Cell Transmission. J. Virol. 2017, 91, 280. [Google Scholar] [CrossRef]
- Ramakrishnaiah, V.; Thumann, C.; Fofana, I.; Habersetzer, F.; Pan, Q.; de Ruiter, P.E.; Willemsen, R.; Demmers, J.A.A.; Stalin Raj, V.; Jenster, G.; et al. Exosome-mediated transmission of hepatitis C virus between human hepatoma Huh7.5 cells. Proc. Natl. Acad. Sci. USA 2013, 110, 13109–13113. [Google Scholar] [CrossRef] [PubMed]
- Tsukiyama-Kohara, K.; Iizuka, N.; Kohara, M.; Nomoto, A. Internal ribosome entry site within hepatitis C virus RNA. J. Virol. 1992, 66, 1476–1483. [Google Scholar]
- Niepmann, M.; Shalamova, L.A.; Gerresheim, G.K.; Rossbach, O. Signals Involved in Regulation of Hepatitis C Virus RNA Genome Translation and Replication. Front. Microbiol. 2018, 9, 395. [Google Scholar] [CrossRef] [PubMed]
- Jopling, C.L.; Yi, M.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 2005, 309, 1577–1581. [Google Scholar] [CrossRef] [PubMed]
- Bandiera, S.; Pfeffer, S.; Baumert, T.F.; Zeisel, M.B. miR-122—A key factor and therapeutic target in liver disease. J. Hepatol. 2015, 62, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Shimakami, T.; Yamane, D.; Jangra, R.K.; Kempf, B.J.; Spaniel, C.; Barton, D.J.; Lemon, S.M. Stabilization of hepatitis C virus RNA by an Ago2-miR-122 complex. Proc. Natl. Acad. Sci. USA 2012, 109, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, V.; Körner, F.; Koch, J.; Herian, U.; Theilmann, L.; Bartenschlager, R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 1999, 285, 110–113. [Google Scholar] [CrossRef]
- Lohmann, V. Hepatitis C virus RNA replication. Curr. Top. Microbiol. Immunol. 2013, 369, 167–198. [Google Scholar]
- Romero-Brey, I.; Merz, A.; Chiramel, A.; Lee, J.-Y.; Chlanda, P.; Haselman, U.; Santarella-Mellwig, R.; Habermann, A.; Hoppe, S.; Kallis, S.; et al. Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathog. 2012, 8, e1003056. [Google Scholar] [CrossRef]
- Egger, D.; Wölk, B.; Gosert, R.; Bianchi, L.; Blum, H.E.; Moradpour, D.; Bienz, K. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J. Virol. 2002, 76, 5974–5984. [Google Scholar] [CrossRef]
- Sharma, G.; Raheja, H.; Das, S. Hepatitis C virus: Enslavement of host factors. IUBMB Life 2018, 70, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Naoumov, N.V. Cyclophilin inhibition as potential therapy for liver diseases. J. Hepatol. 2014, 61, 1166–1174. [Google Scholar] [CrossRef] [PubMed]
- Madan, V.; Paul, D.; Lohmann, V.; Bartenschlager, R. Inhibition of HCV replication by cyclophilin antagonists is linked to replication fitness and occurs by inhibition of membranous web formation. Gastroenterology 2014, 146, 1361–1372. [Google Scholar] [CrossRef] [PubMed]
- Chatterji, U.; Bobardt, M.; Tai, A.; Wood, M.; Gallay, P.A. Cyclophilin and NS5A inhibitors, but not other anti-hepatitis C virus (HCV) agents, preclude HCV-mediated formation of double-membrane-vesicle viral factories. Antimicrob. Agents Chemother. 2015, 59, 2496–2507. [Google Scholar] [CrossRef]
- Berger, K.L.; Cooper, J.D.; Heaton, N.S.; Yoon, R.; Oakland, T.E.; Jordan, T.X.; Mateu, G.; Grakoui, A.; Randall, G. Roles for endocytic trafficking and phosphatidylinositol 4-kinase III alpha in hepatitis C virus replication. Proc. Natl. Acad. Sci. USA 2009, 106, 7577–7582. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.; Evans, M.J.; Syder, A.J.; Wölk, B.; Tellinghuisen, T.L.; Liu, C.C.; Maruyama, T.; Hynes, R.O.; Burton, D.R.; McKeating, J.A.; et al. Complete replication of hepatitis C virus in cell culture. Science 2005, 309, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Wakita, T.; Pietschmann, T.; Kato, T.; Date, T.; Miyamoto, M.; Zhao, Z.; Murthy, K.; Habermann, A.; Kräusslich, H.-G.; Mizokami, M.; et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 2005, 11, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, V. Hepatitis C virus cell culture models: An encomium on basic research paving the road to therapy development. Med. Microbiol. Immunol. 2018. [Google Scholar] [CrossRef]
- Calattini, S.; Fusil, F.; Mancip, J.; Dao Thi, V.L.; Granier, C.; Gadot, N.; Scoazec, J.-Y.; Zeisel, M.B.; Baumert, T.F.; Lavillette, D.; et al. Functional and Biochemical Characterization of Hepatitis C Virus (HCV) Particles Produced in a Humanized Liver Mouse Model. J. Biol. Chem. 2015, 290, 23173–23187. [Google Scholar] [CrossRef] [PubMed]
- Miyanari, Y.; Atsuzawa, K.; Usuda, N.; Watashi, K.; Hishiki, T.; Zayas, M.; Bartenschlager, R.; Wakita, T.; Hijikata, M.; Shimotohno, K. The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell Biol. 2007, 9, 1089–1097. [Google Scholar] [CrossRef] [PubMed]
- Falcón, V.; Acosta-Rivero, N.; González, S.; Dueñas-Carrera, S.; Martinez-Donato, G.; Menéndez, I.; Garateix, R.; Silva, J.A.; Acosta, E.; Kourı, J. Ultrastructural and biochemical basis for hepatitis C virus morphogenesis. Virus Genes 2017, 53, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Zayas, M.; Long, G.; Madan, V.; Bartenschlager, R. Coordination of Hepatitis C Virus Assembly by Distinct Regulatory Regions in Nonstructural Protein 5A. PLoS Pathog. 2016, 12, e1005376. [Google Scholar] [CrossRef] [PubMed]
- Boson, B.; Denolly, S.; Turlure, F.; Chamot, C.; Dreux, M.; Cosset, F.-L. Daclatasvir Prevents Hepatitis C Virus Infectivity by Blocking Transfer of the Viral Genome to Assembly Sites. Gastroenterology 2017, 152, 895–907. [Google Scholar] [CrossRef] [PubMed]
- Popescu, C.-I.; Callens, N.; Trinel, D.; Roingeard, P.; Moradpour, D.; Descamps, V.; Duverlie, G.; Penin, F.; Héliot, L.; Rouillé, Y.; et al. NS2 protein of hepatitis C virus interacts with structural and non-structural proteins towards virus assembly. PLoS Pathog. 2011, 7, e1001278. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Anantpadma, M.; Timpe, J.M.; Shanmugam, S.; Singh, S.M.; Lemon, S.M.; Yi, M. Hepatitis C virus NS2 protein serves as a scaffold for virus assembly by interacting with both structural and nonstructural proteins. J. Virol. 2011, 85, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Jirasko, V.; Montserret, R.; Appel, N.; Janvier, A.; Eustachi, L.; Brohm, C.; Steinmann, E.; Pietschmann, T.; Penin, F.; Bartenschlager, R. Structural and functional characterization of nonstructural protein 2 for its role in hepatitis C virus assembly. J. Biol. Chem. 2008, 283, 28546–28562. [Google Scholar] [CrossRef] [PubMed]
- Denolly, S.; Mialon, C.; Bourlet, T.; Amirache, F.; Penin, F.; Lindenbach, B.; Boson, B.; Cosset, F.-L. The amino-terminus of the hepatitis C virus (HCV) p7 viroporin and its cleavage from glycoprotein E2-p7 precursor determine specific infectivity and secretion levels of HCV particle types. PLoS Pathog. 2017, 13, e1006774. [Google Scholar] [CrossRef]
- Boson, B.; Granio, O.; Bartenschlager, R.; Cosset, F.-L. A concerted action of hepatitis C virus p7 and nonstructural protein 2 regulates core localization at the endoplasmic reticulum and virus assembly. PLoS Pathog. 2011, 7, e1002144. [Google Scholar] [CrossRef]
- Stapleford, K.A.; Lindenbach, B.D. Hepatitis C virus NS2 coordinates virus particle assembly through physical interactions with the E1-E2 glycoprotein and NS3-NS4A enzyme complexes. J. Virol. 2011, 85, 1706–1717. [Google Scholar] [CrossRef]
- Gentzsch, J.; Brohm, C.; Steinmann, E.; Friesland, M.; Menzel, N.; Vieyres, G.; Perin, P.M.; Frentzen, A.; Kaderali, L.; Pietschmann, T. Hepatitis c Virus p7 is critical for capsid assembly and envelopment. PLoS Pathog. 2013, 9, e1003355. [Google Scholar] [CrossRef]
- Han, Q.; Manna, D.; Belton, K.; Cole, R.; Konan, K.V. Modulation of hepatitis C virus genome encapsidation by nonstructural protein 4B. J. Virol. 2013, 87, 7409–7422. [Google Scholar] [CrossRef] [PubMed]
- Camus, G.; Herker, E.; Modi, A.A.; Haas, J.T.; Ramage, H.R.; Farese, R.V.; Ott, M. Diacylglycerol acyltransferase-1 localizes hepatitis C virus NS5A protein to lipid droplets and enhances NS5A interaction with the viral capsid core. J. Biol. Chem. 2013, 288, 9915–9923. [Google Scholar] [CrossRef] [PubMed]
- Herker, E.; Harris, C.; Hernandez, C.; Carpentier, A.; Kaehlcke, K.; Rosenberg, A.R.; Farese, R.V.; Ott, M. Efficient hepatitis C virus particle formation requires diacylglycerol acyltransferase-1. Nat. Med. 2010, 16, 1295–1298. [Google Scholar] [CrossRef] [PubMed]
- Ariumi, Y.; Kuroki, M.; Maki, M.; Ikeda, M.; Dansako, H.; Wakita, T.; Kato, N. The ESCRT system is required for hepatitis C virus production. PLoS ONE 2011, 6, e14517. [Google Scholar] [CrossRef] [PubMed]
- Barouch-Bentov, R.; Neveu, G.; Xiao, F.; Beer, M.; Bekerman, E.; Schor, S.; Campbell, J.; Boonyaratanakornkit, J.; Lindenbach, B.; Lu, A.; et al. Hepatitis C Virus Proteins Interact with the Endosomal Sorting Complex Required for Transport (ESCRT) Machinery via Ubiquitination To Facilitate Viral Envelopment. MBio 2016, 7, e01456. [Google Scholar] [CrossRef]
- Corless, L.; Crump, C.M.; Griffin, S.D.C.; Harris, M. Vps4 and the ESCRT-III complex are required for the release of infectious hepatitis C virus particles. J. Gen. Virol. 2010, 91, 362–372. [Google Scholar] [CrossRef]
- Tamai, K.; Shiina, M.; Tanaka, N.; Nakano, T.; Yamamoto, A.; Kondo, Y.; Kakazu, E.; Inoue, J.; Fukushima, K.; Sano, K.; et al. Regulation of hepatitis C virus secretion by the Hrs-dependent exosomal pathway. Virology 2012, 422, 377–385. [Google Scholar] [CrossRef]
- Merz, A.; Long, G.; Hiet, M.-S.; Brügger, B.; Chlanda, P.; Andre, P.; Wieland, F.; Krijnse-Locker, J.; Bartenschlager, R. Biochemical and morphological properties of hepatitis C virus particles and determination of their lipidome. J. Biol. Chem. 2011, 286, 3018–3032. [Google Scholar] [CrossRef]
- Felmlee, D.J.; Coilly, A.; Chung, R.T.; Samuel, D.; Baumert, T.F. New perspectives for preventing hepatitis C virus liver graft infection. Lancet Infect. Dis. 2016, 16, 735–745. [Google Scholar] [CrossRef]
- Bartenschlager, R.; Ahlborn-Laake, L.; Mous, J.; Jacobsen, H. Nonstructural protein 3 of the hepatitis C virus encodes a serine-type proteinase required for cleavage at the NS3/4 and NS4/5 junctions. J. Virol. 1993, 67, 3835–3844. [Google Scholar]
- Eckart, M.R.; Selby, M.; Masiarz, F.; Lee, C.; Berger, K.; Crawford, K.; Kuo, C.; Kuo, G.; Houghton, M.; Choo, Q.L. The hepatitis C virus encodes a serine protease involved in processing of the putative nonstructural proteins from the viral polyprotein precursor. Biochem. Biophys. Res. Commun. 1993, 192, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Grakoui, A.; McCourt, D.W.; Wychowski, C.; Feinstone, S.M.; Rice, C.M. Characterization of the hepatitis C virus-encoded serine proteinase: Determination of proteinase-dependent polyprotein cleavage sites. J. Virol. 1993, 67, 2832–2843. [Google Scholar] [PubMed]
- Li, X.-D.; Sun, L.; Seth, R.B.; Pineda, G.; Chen, Z.J. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl. Acad. Sci. USA 2005, 102, 17717–17722. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.L.; Morgenstern, K.A.; Lin, C.; Fox, T.; Dwyer, M.D.; Landro, J.A.; Chambers, S.P.; Markland, W.; Lepre, C.A.; O’Malley, E.T.; et al. Crystal structure of the hepatitis C virus NS3 protease domain complexed with a synthetic NS4A cofactor peptide. Cell 1996, 87, 343–355. [Google Scholar] [CrossRef]
- Love, R.A.; Parge, H.E.; Wickersham, J.A.; Hostomsky, Z.; Habuka, N.; Moomaw, E.W.; Adachi, T.; Hostomska, Z. The crystal structure of hepatitis C virus NS3 proteinase reveals a trypsin-like fold and a structural zinc binding site. Cell 1996, 87, 331–342. [Google Scholar] [CrossRef]
- Brass, V.; Berke, J.M.; Montserret, R.; Blum, H.E.; Penin, F.; Moradpour, D. Structural determinants for membrane association and dynamic organization of the hepatitis C virus NS3-4A complex. Proc. Natl. Acad. Sci. USA 2008, 105, 14545–14550. [Google Scholar] [CrossRef] [PubMed]
- Pang, P.S.; Jankowsky, E.; Planet, P.J.; Pyle, A.M. The hepatitis C viral NS3 protein is a processive DNA helicase with cofactor enhanced RNA unwinding. EMBO J. 2002, 21, 1168–1176. [Google Scholar] [CrossRef]
- Tanji, Y.; Hijikata, M.; Satoh, S.; Kaneko, T.; Shimotohno, K. Hepatitis C virus-encoded nonstructural protein NS4A has versatile functions in viral protein processing. J. Virol. 1995, 69, 1575–1581. [Google Scholar]
- Kim, J.L.; Morgenstern, K.A.; Griffith, J.P.; Dwyer, M.D.; Thomson, J.A.; Murcko, M.A.; Lin, C.; Caron, P.R. Hepatitis C virus NS3 RNA helicase domain with a bound oligonucleotide: The crystal structure provides insights into the mode of unwinding. Structure 1998, 6, 89–100. [Google Scholar] [CrossRef]
- He, Y.; Weng, L.; Li, R.; Li, L.; Toyoda, T.; Zhong, J. The N-terminal helix α(0) of hepatitis C virus NS3 protein dictates the subcellular localization and stability of NS3/NS4A complex. Virology 2012, 422, 214–223. [Google Scholar] [CrossRef]
- Yan, Y.; He, Y.; Boson, B.; Wang, X.; Cosset, F.-L.; Zhong, J. A Point Mutation in the N-Terminal Amphipathic Helix α0 in NS3 Promotes Hepatitis C Virus Assembly by Altering Core Localization to the Endoplasmic Reticulum and Facilitating Virus Budding. J. Virol. 2017, 91, 02399. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Yates, J.; Liang, Y.; Lemon, S.M.; Yi, M. NS3 helicase domains involved in infectious intracellular hepatitis C virus particle assembly. J. Virol. 2008, 82, 7624–7639. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Weiner, W.S.; Schroeder, C.E.; Simpson, D.S.; Hanson, A.M.; Sweeney, N.L.; Marvin, R.K.; Ndjomou, J.; Kolli, R.; Isailovic, D.; et al. Ebselen inhibits hepatitis C virus NS3 helicase binding to nucleic acid and prevents viral replication. ACS Chem. Biol. 2014, 9, 2393–2403. [Google Scholar] [CrossRef] [PubMed]
- Beran, R.K.F.; Pyle, A.M. Hepatitis C viral NS3-4A protease activity is enhanced by the NS3 helicase. J. Biol. Chem. 2008, 283, 29929–29937. [Google Scholar] [CrossRef] [PubMed]
- Beran, R.K.F.; Serebrov, V.; Pyle, A.M. The serine protease domain of hepatitis C viral NS3 activates RNA helicase activity by promoting the binding of RNA substrate. J. Biol. Chem. 2007, 282, 34913–34920. [Google Scholar] [CrossRef] [PubMed]
- McGivern, D.R.; Masaki, T.; Lovell, W.; Hamlett, C.; Saalau-Bethell, S.; Graham, B. Protease Inhibitors Block Multiple Functions of the NS3/4A Protease-Helicase during the Hepatitis C Virus Life Cycle. J. Virol. 2015, 89, 5362–5370. [Google Scholar] [CrossRef] [PubMed]
- Ingallinella, P.; Altamura, S.; Bianchi, E.; Taliani, M.; Ingenito, R.; Cortese, R.; De Francesco, R.; Steinkühler, C.; Pessi, A. Potent peptide inhibitors of human hepatitis C virus NS3 protease are obtained by optimizing the cleavage products. Biochemistry 1998, 37, 8906–8914. [Google Scholar] [CrossRef]
- Llinàs-Brunet, M.; Bailey, M.; Déziel, R.; Fazal, G.; Gorys, V.; Goulet, S.; Halmos, T.; Maurice, R.; Poirier, M.; Poupart, M.A.; et al. Studies on the C-terminal of hexapeptide inhibitors of the hepatitis C virus serine protease. Bioorg. Med. Chem. Lett. 1998, 8, 2719–2724. [Google Scholar] [CrossRef]
- Lin, C.; Kwong, A.D.; Perni, R.B. Discovery and development of VX-950, a novel, covalent, and reversible inhibitor of hepatitis C virus NS3.4A serine protease. Infect. Disord. Drug Targets 2006, 6, 3–16. [Google Scholar] [CrossRef]
- Venkatraman, S.; Bogen, S.L.; Arasappan, A.; Bennett, F.; Chen, K.; Jao, E.; Liu, Y.-T.; Lovey, R.; Hendrata, S.; Huang, Y.; et al. Discovery of (1R,5S)-N-[3-amino-1-(cyclobutylmethyl)-2,3-dioxopropyl]-3-[2(S)-[[[(1,1-dimethylethyl)amino]carbonyl]amino]-3,3-dimethyl-1-oxobutyl]-6,6-dimethyl-3-azabicyclo[3.1.0]hexan-2(S)-carboxamide (SCH 503034), a selective, potent, orally bioavailable hepatitis C virus NS3 protease inhibitor: A potential therapeutic agent for the treatment of hepatitis C infection. J. Med. Chem. 2006, 49, 6074–6086. [Google Scholar]
- Pan, Q.; Peppelenbosch, M.P.; Janssen, H.L.; de Knegt, R.J. Telaprevir/boceprevir era: From bench to bed and back. World J. Gastroenterol. 2012, 18, 6183–6188. [Google Scholar] [CrossRef] [PubMed]
- AASLD/IDSA HCV Guidance Panel. Hepatitis C guidance: AASLD-IDSA recommendations for testing, managing, and treating adults infected with hepatitis C virus. Hepatology 2015, 62, 932–954. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver. European Association for the Study of the Liver EASL Recommendations on Treatment of Hepatitis C 2018. J. Hepatol. 2018, 69, 461–511. [Google Scholar] [CrossRef] [PubMed]
- Lontok, E.; Harrington, P.; Howe, A.; Kieffer, T.; Lennerstrand, J.; Lenz, O.; McPhee, F.; Mo, H.; Parkin, N.; Pilot-Matias, T.; et al. Hepatitis C virus drug resistance-associated substitutions: State of the art summary. Hepatology 2015, 62, 1623–1632. [Google Scholar] [CrossRef] [PubMed]
- Sorbo, M.C.; Cento, V.; Di Maio, V.C.; Howe, A.Y.M.; Garcia, F.; Perno, C.F.; Ceccherini-Silberstein, F. Hepatitis C virus drug resistance associated substitutions and their clinical relevance: Update 2018. Drug Resist. Updat. 2018, 37, 17–39. [Google Scholar] [CrossRef] [PubMed]
- Vermehren, J.; Park, J.S.; Jacobson, I.; Zeuzem, S. Challenges and perspectives of direct antivirals for the treatment of hepatitis C virus infection. J. Hepatol. 2018, 69, 1178–1187. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.H.; Purcell, R.H. Hepatitis C virus shares amino acid sequence similarity with pestiviruses and flaviviruses as well as members of two plant virus supergroups. Proc. Natl. Acad. Sci. USA 1990, 87, 2057–2061. [Google Scholar] [CrossRef]
- Behrens, S.E.; Tomei, L.; De Francesco, R. Identification and properties of the RNA-dependent RNA polymerase of hepatitis C virus. EMBO J. 1996, 15, 12–22. [Google Scholar] [CrossRef]
- Bressanelli, S.; Tomei, L.; Roussel, A.; Incitti, I.; Vitale, R.L.; Mathieu, M.; De Francesco, R.; Rey, F.A. Crystal structure of the RNA-dependent RNA polymerase of hepatitis C virus. Proc. Natl. Acad. Sci. USA 1999, 96, 13034–13039. [Google Scholar] [CrossRef]
- Lesburg, C.A.; Cable, M.B.; Ferrari, E.; Hong, Z.; Mannarino, A.F.; Weber, P.C. Crystal structure of the RNA-dependent RNA polymerase from hepatitis C virus reveals a fully encircled active site. Nat. Struct. Biol. 1999, 6, 937–943. [Google Scholar]
- Ago, H.; Adachi, T.; Yoshida, A.; Yamamoto, M.; Habuka, N.; Yatsunami, K.; Miyano, M. Crystal structure of the RNA-dependent RNA polymerase of hepatitis C virus. Structure 1999, 7, 1417–1426. [Google Scholar] [CrossRef]
- Hwang, S.B.; Park, K.J.; Kim, Y.S.; Sung, Y.C.; Lai, M.M. Hepatitis C virus NS5B protein is a membrane-associated phosphoprotein with a predominantly perinuclear localization. Virology 1997, 227, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, E.; Wright-Minogue, J.; Fang, J.W.; Baroudy, B.M.; Lau, J.Y.; Hong, Z. Characterization of soluble hepatitis C virus RNA-dependent RNA polymerase expressed in Escherichia coli. J. Virol. 1999, 73, 1649–1654. [Google Scholar] [PubMed]
- Yamashita, T.; Kaneko, S.; Shirota, Y.; Qin, W.; Nomura, T.; Kobayashi, K.; Murakami, S. RNA-dependent RNA polymerase activity of the soluble recombinant hepatitis C virus NS5B protein truncated at the C-terminal region. J. Biol. Chem. 1998, 273, 15479–15486. [Google Scholar] [CrossRef] [PubMed]
- Eltahla, A.A.; Luciani, F.; White, P.A.; Lloyd, A.R.; Bull, R.A. Inhibitors of the Hepatitis C Virus Polymerase; Mode of Action and Resistance. Viruses 2015, 7, 5206–5224. [Google Scholar] [CrossRef] [PubMed]
- Lawitz, E.; Jacobson, I.M.; Nelson, D.R.; Zeuzem, S.; Sulkowski, M.S.; Esteban, R.; Brainard, D.; McNally, J.; Symonds, W.T.; McHutchison, J.G.; et al. Development of sofosbuvir for the treatment of hepatitis C virus infection. Ann. N. Y. Acad. Sci. 2015, 1358, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Sofia, M.J. Enter Sofosbuvir: The Path to Curing HCV. Cell 2016, 167, 25–29. [Google Scholar] [CrossRef]
- Arnold, J.J.; Sharma, S.D.; Feng, J.Y.; Ray, A.S.; Smidansky, E.D.; Kireeva, M.L.; Cho, A.; Perry, J.; Vela, J.E.; Park, Y.; et al. Sensitivity of mitochondrial transcription and resistance of RNA polymerase II dependent nuclear transcription to antiviral ribonucleosides. PLoS Pathog. 2012, 8, e1003030. [Google Scholar] [CrossRef]
- Svarovskaia, E.S.; Dvory-Sobol, H.; Parkin, N.; Hebner, C.; Gontcharova, V.; Martin, R.; Ouyang, W.; Han, B.; Xu, S.; Ku, K.; et al. Infrequent development of resistance in genotype 1-6 hepatitis C virus-infected subjects treated with sofosbuvir in phase 2 and 3 clinical trials. Clin. Infect. Dis. 2014, 59, 1666–1674. [Google Scholar] [CrossRef]
- Donaldson, E.F.; Harrington, P.R.; O’Rear, J.J.; Naeger, L.K. Clinical evidence and bioinformatics characterization of potential hepatitis C virus resistance pathways for sofosbuvir. Hepatology 2015, 61, 56–65. [Google Scholar] [CrossRef]
- El Kassas, M.; Elbaz, T.; Hafez, E.; Wifi, M.N.; Esmat, G. Discovery and preclinical development of dasabuvir for the treatment of hepatitis C infection. Expert Opin. Drug Discov. 2017, 12, 635–642. [Google Scholar] [CrossRef] [PubMed]
- Hebner, C.M.; Han, B.; Brendza, K.M.; Nash, M.; Sulfab, M.; Tian, Y.; Hung, M.; Fung, W.; Vivian, R.W.; Trenkle, J.; et al. The HCV Non-Nucleoside Inhibitor Tegobuvir Utilizes a Novel Mechanism of Action to Inhibit NS5B Polymerase Function. PLoS ONE 2012, 7, e39163. [Google Scholar] [CrossRef] [PubMed]
- Tellinghuisen, T.L.; Marcotrigiano, J.; Rice, C.M. Structure of the zinc-binding domain of an essential component of the hepatitis C virus replicase. Nature 2005, 435, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Tellinghuisen, T.L.; Marcotrigiano, J.; Gorbalenya, A.E.; Rice, C.M. The NS5A protein of hepatitis C virus is a zinc metalloprotein. J. Biol. Chem. 2004, 279, 48576–48587. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Nettles, R.E.; Belema, M.; Snyder, L.B.; Nguyen, V.N.; Fridell, R.A.; Serrano-Wu, M.H.; Langley, D.R.; Sun, J.-H.; O’Boyle, D.R.; et al. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature 2010, 465, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Barakat, K.H.; Anwar-Mohamed, A.; Tuszynski, J.A.; Robins, M.J.; Tyrrell, D.L.; Houghton, M. A Refined Model of the HCV NS5A protein bound to daclatasvir explains drug-resistant mutations and activity against divergent genotypes. J. Chem. Inf. Model. 2015, 55, 362–373. [Google Scholar] [CrossRef] [PubMed]
- Berger, C.; Romero-Brey, I.; Radujkovic, D.; Terreux, R.; Zayas, M.; Paul, D.; Harak, C.; Hoppe, S.; Gao, M.; Penin, F.; et al. Daclatasvir-like inhibitors of NS5A block early biogenesis of hepatitis C virus-induced membranous replication factories, independent of RNA replication. Gastroenterology 2014, 147, 1094.e25–1105.e25. [Google Scholar] [CrossRef]
- O’Boyle, D.R.; Nower, P.T.; Sun, J.-H.; Fridell, R.; Wang, C.; Valera, L.; Gao, M. A 96-well based analysis of replicon elimination with the HCV NS5A replication complex inhibitor daclatasvir. J. Virol. Methods 2013, 193, 68–76. [Google Scholar] [CrossRef]
- Ascher, D.B.; Wielens, J.; Nero, T.L.; Doughty, L.; Morton, C.J.; Parker, M.W. Potent hepatitis C inhibitors bind directly to NS5A and reduce its affinity for RNA. Sci. Rep. 2014, 4, 4765. [Google Scholar] [CrossRef]
- Kwon, H.J.; Xing, W.; Chan, K.; Niedziela-Majka, A.; Brendza, K.M.; Kirschberg, T.; Kato, D.; Link, J.O.; Cheng, G.; Liu, X.; et al. Direct Binding of Ledipasvir to HCV NS5A: Mechanism of Resistance to an HCV Antiviral Agent. PLoS ONE 2015, 10, e0122844. [Google Scholar] [CrossRef]
- DeGoey, D.A.; Randolph, J.T.; Liu, D.; Pratt, J.; Hutchins, C.; Donner, P.; Krueger, A.C.; Matulenko, M.; Patel, S.; Motter, C.E.; et al. Discovery of ABT-267, a pan-genotypic inhibitor of HCV NS5A. J. Med. Chem. 2014, 57, 2047–2057. [Google Scholar] [CrossRef]
- Coburn, C.A.; Meinke, P.T.; Chang, W.; Fandozzi, C.M.; Graham, D.J.; Hu, B.; Huang, Q.; Kargman, S.; Kozlowski, J.; Liu, R.; et al. Discovery of MK-8742: An HCV NS5A inhibitor with broad genotype activity. ChemMedChem 2013, 8, 1930–1940. [Google Scholar] [CrossRef] [PubMed]
- Gitto, S.; Gamal, N.; Andreone, P. NS5A inhibitors for the treatment of hepatitis C infection. J. Viral Hepat. 2017, 24, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; O’Boyle, D.R.; Roberts, S. HCV NS5A replication complex inhibitors. Curr. Opin. Pharm. 2016, 30, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Ross-Thriepland, D.; Harris, M. Hepatitis C virus NS5A: Enigmatic but still promiscuous 10 years on! J. Gen. Virol. 2015, 96, 727–738. [Google Scholar] [CrossRef] [PubMed]
- McGivern, D.R.; Masaki, T.; Williford, S.; Ingravallo, P.; Feng, Z.; Lahser, F.; Asante-Appiah, E.; Neddermann, P.; De Francesco, R.; Howe, A.Y.; et al. Kinetic analyses reveal potent and early blockade of hepatitis C virus assembly by NS5A inhibitors. Gastroenterology 2014, 147, 453–462. [Google Scholar] [CrossRef]
- Guedj, J.; Dahari, H.; Rong, L.; Sansone, N.D.; Nettles, R.E.; Cotler, S.J.; Layden, T.J.; Uprichard, S.L.; Perelson, A.S. Modeling shows that the NS5A inhibitor daclatasvir has two modes of action and yields a shorter estimate of the hepatitis C virus half-life. Proc. Natl. Acad. Sci. USA 2013, 110, 3991–3996. [Google Scholar] [CrossRef]
- Sakai, A.; Claire, M.S.; Faulk, K.; Govindarajan, S.; Emerson, S.U.; Purcell, R.H.; Bukh, J. The p7 polypeptide of hepatitis C virus is critical for infectivity and contains functionally important genotype-specific sequences. Proc. Natl. Acad. Sci. USA 2003, 100, 11646–11651. [Google Scholar] [CrossRef]
- Jones, C.T.; Murray, C.L.; Eastman, D.K.; Tassello, J.; Rice, C.M. Hepatitis C virus p7 and NS2 proteins are essential for production of infectious virus. J. Virol. 2007, 81, 8374–8383. [Google Scholar] [CrossRef]
- Steinmann, E.; Penin, F.; Kallis, S.; Patel, A.H.; Bartenschlager, R.; Pietschmann, T. Hepatitis C virus p7 protein is crucial for assembly and release of infectious virions. PLoS Pathog. 2007, 3, e103. [Google Scholar] [CrossRef]
- Shanmugam, S.; Yi, M. Efficiency of E2-p7 processing modulates production of infectious hepatitis C virus. J. Virol. 2013, 87, 11255–11266. [Google Scholar] [CrossRef] [PubMed]
- Bentham, M.J.; Foster, T.L.; McCormick, C.; Griffin, S. Mutations in hepatitis C virus p7 reduce both the egress and infectivity of assembled particles via impaired proton channel function. J. Gen. Virol. 2013, 94, 2236–2248. [Google Scholar] [CrossRef] [PubMed]
- Scott, C.; Griffin, S. Viroporins: Structure, function and potential as antiviral targets. J. Gen. Virol. 2015, 96, 2000–2027. [Google Scholar] [CrossRef] [PubMed]
- Manzoor, R.; Igarashi, M.; Takada, A. Influenza a Virus M2 Protein: Roles from Ingress to Egress. Int. J. Mol. Sci. 2017, 18, 2649. [Google Scholar] [CrossRef] [PubMed]
- Griffin, S.; Stgelais, C.; Owsianka, A.M.; Patel, A.H.; Rowlands, D.; Harris, M. Genotype-dependent sensitivity of hepatitis C virus to inhibitors of the p7 ion channel. Hepatology 2008, 48, 1779–1790. [Google Scholar] [CrossRef] [PubMed]
- Van Soest, H.; van der Schaar, P.J.; Koek, G.H.; de Vries, R.A.; van Ooteghem, N.A.; van Hoek, B.; Drenth, J.P.H.; Vrolijk, J.M.; Lieverse, R.J.; Houben, P.; et al. No beneficial effects of amantadine in treatment of chronic hepatitis C patients. Dig. Liver Dis. 2010, 42, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Bichmann, L.; Wang, Y.-T.; Fischer, W.B. Docking assay of small molecule antivirals to p7 of HCV. Comput. Biol. Chem. 2014, 53, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Luscombe, C.A.; Huang, Z.; Murray, M.G.; Miller, M.; Wilkinson, J.; Ewart, G.D. A novel Hepatitis C virus p7 ion channel inhibitor, BIT225, inhibits bovine viral diarrhea virus in vitro and shows synergism with recombinant interferon-alpha-2b and nucleoside analogues. Antivir. Res. 2010, 86, 144–153. [Google Scholar] [CrossRef]
- Pavlovic, D.; Fischer, W.; Hussey, M.; Durantel, D.; Durantel, S.; Branza-Nichita, N.; Woodhouse, S.; Dwek, R.A.; Zitzmann, N. Long alkylchain iminosugars block the HCV p7 ion channel. Adv. Exp. Med. Biol. 2005, 564, 3–4. [Google Scholar]
- Wozniak, A.L.; Griffin, S.; Rowlands, D.; Harris, M.; Yi, M.; Lemon, S.M.; Weinman, S.A. Intracellular proton conductance of the hepatitis C virus p7 protein and its contribution to infectious virus production. PLoS Pathog. 2010, 6, e1001087. [Google Scholar] [CrossRef]
- Cannalire, R.; Barreca, M.L.; Manfroni, G.; Cecchetti, V. A Journey around the Medicinal Chemistry of Hepatitis C Virus Inhibitors Targeting NS4B: From Target to Preclinical Drug Candidates. J. Med. Chem. 2016, 59, 16–41. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.; Harris, M.; Fishwick, C.W.G. Identification of a lead like inhibitor of the hepatitis C virus non-structural NS2 autoprotease. Antivir. Res. 2015, 124, 54–60. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, X.; Wu, C.; Xu, H.; Liu, H. Current Drug Discovery for Anti-hepatitis C Virus Targeting NS4B. Curr. Top. Med. Chem. 2016, 16, 1362–1371. [Google Scholar] [CrossRef]
- Crouchet, E.; Wrensch, F.; Schuster, C.; Zeisel, M.B.; Baumert, T.F. Host-targeting therapies for hepatitis C virus infection: Current developments and future applications. Ther. Adv. Gastroenterol. 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, M.B.; Crouchet, E.; Baumert, T.F.; Schuster, C. Host-Targeting Agents to Prevent and Cure Hepatitis C Virus Infection. Viruses 2015, 7, 5659–5685. [Google Scholar] [CrossRef]
- Gallay, P.A.; Bobardt, M.D.; Chatterji, U.; Trepanier, D.J.; Ure, D.; Ordonez, C.; Foster, R. The Novel Cyclophilin Inhibitor CPI-431-32 Concurrently Blocks HCV and HIV-1 Infections via a Similar Mechanism of Action. PLoS ONE 2015, 10, e0134707. [Google Scholar] [CrossRef] [PubMed]
- Van der Ree, M.H.; van der Meer, A.J.; de Bruijne, J.; Maan, R.; van Vliet, A.; Welzel, T.M.; Zeuzem, S.; Lawitz, E.J.; Rodriguez-Torres, M.; Kupcova, V.; et al. Long-term safety and efficacy of microRNA-targeted therapy in chronic hepatitis C patients. Antivir. Res. 2014, 111, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Janssen, H.L.A.; Reesink, H.W.; Lawitz, E.J.; Zeuzem, S.; Rodriguez-Torres, M.; Patel, K.; van der Meer, A.J.; Patick, A.K.; Chen, A.; Zhou, Y.; et al. Treatment of HCV infection by targeting microRNA. N. Engl. J. Med. 2013, 368, 1685–1694. [Google Scholar] [CrossRef] [PubMed]
- Villareal, V.A.; Rodgers, M.A.; Costello, D.A.; Yang, P.L. Targeting host lipid synthesis and metabolism to inhibit dengue and hepatitis C viruses. Antivir. Res. 2015, 124, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Colpitts, C.C.; Chung, R.T.; Baumert, T.F. Entry Inhibitors: A Perspective for Prevention of Hepatitis C Virus Infection in Organ Transplantation. ACS Infect. Dis. 2017, 3, 620–623. [Google Scholar] [CrossRef] [PubMed]
- Douam, F.; Lavillette, D.; Cosset, F.-L. The mechanism of HCV entry into host cells. Prog. Mol. Biol. Transl. Sci. 2015, 129, 63–107. [Google Scholar] [PubMed]
- De Jong, Y.P.; Dorner, M.; Mommersteeg, M.C.; Xiao, J.W.; Balazs, A.B.; Robbins, J.B.; Winer, B.Y.; Gerges, S.; Vega, K.; Labitt, R.N.; et al. Broadly neutralizing antibodies abrogate established hepatitis C virus infection. Sci. Transl. Med. 2014, 6, 254. [Google Scholar] [CrossRef] [PubMed]
- Mailly, L.; Xiao, F.; Lupberger, J.; Wilson, G.K.; Aubert, P.; Duong, F.H.T.; Calabrese, D.; Leboeuf, C.; Fofana, I.; Thumann, C.; et al. Clearance of persistent hepatitis C virus infection using a claudin-1-targeting monoclonal antibody. Nat. Biotechnol. 2015, 33, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Rowe, I.A.; Tully, D.C.; Armstrong, M.J.; Parker, R.; Guo, K.; Barton, D.; Morse, G.D.; Venuto, C.S.; Ogilvie, C.B.; Hedegaard, D.L.; et al. Effect of scavenger receptor class B type I antagonist ITX5061 in patients with hepatitis C virus infection undergoing liver transplantation. Liver Transpl. 2016, 22, 287–297. [Google Scholar] [CrossRef]
- Bartenschlager, R.; Baumert, T.F.; Bukh, J.; Houghton, M.; Lemon, S.M.; Lindenbach, B.D.; Lohmann, V.; Moradpour, D.; Pietschmann, T.; Rice, C.M.; et al. Critical challenges and emerging opportunities in hepatitis C virus research in an era of potent antiviral therapy: Considerations for scientists and funding agencies. Virus Res. 2018, 248, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Néant, N.; Solas, C. Drug-Drug Interactions Potential of Direct Acting Antivirals for the treatment of Chronic Hepatitis C infection. Int. J. Antimicrob. Agents 2018. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Zhong, J.-Y.; Li, J.-W. Hepatitis C Virus Infection and Vaccine Development. J. Clin. Exp. Hepatol. 2018, 8, 195–204. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alazard-Dany, N.; Denolly, S.; Boson, B.; Cosset, F.-L. Overview of HCV Life Cycle with a Special Focus on Current and Possible Future Antiviral Targets. Viruses 2019, 11, 30. https://doi.org/10.3390/v11010030
Alazard-Dany N, Denolly S, Boson B, Cosset F-L. Overview of HCV Life Cycle with a Special Focus on Current and Possible Future Antiviral Targets. Viruses. 2019; 11(1):30. https://doi.org/10.3390/v11010030
Chicago/Turabian StyleAlazard-Dany, Nathalie, Solène Denolly, Bertrand Boson, and François-Loïc Cosset. 2019. "Overview of HCV Life Cycle with a Special Focus on Current and Possible Future Antiviral Targets" Viruses 11, no. 1: 30. https://doi.org/10.3390/v11010030
APA StyleAlazard-Dany, N., Denolly, S., Boson, B., & Cosset, F.-L. (2019). Overview of HCV Life Cycle with a Special Focus on Current and Possible Future Antiviral Targets. Viruses, 11(1), 30. https://doi.org/10.3390/v11010030