Mimiviridae: An Expanding Family of Highly Diverse Large dsDNA Viruses Infecting a Wide Phylogenetic Range of Aquatic Eukaryotes



Abstract
1. Introduction
2. Results
2.1. Pionnering Work on Large Aquatic Viruses
2.2. From Acanthamoeba Polyphaga Mimivirus (APMV) to the First Marine Mimiviridae
2.3. C. Roenbergensis Virus: The Prototype of a New Subfamily within the Mimiviridae
2.4. Smaller Mimiviridae Infecting Bona Fide Microalgae: Yet Another Subfamily
2.5. Recent Isolations of New Truly Giant Mimiviridae Members
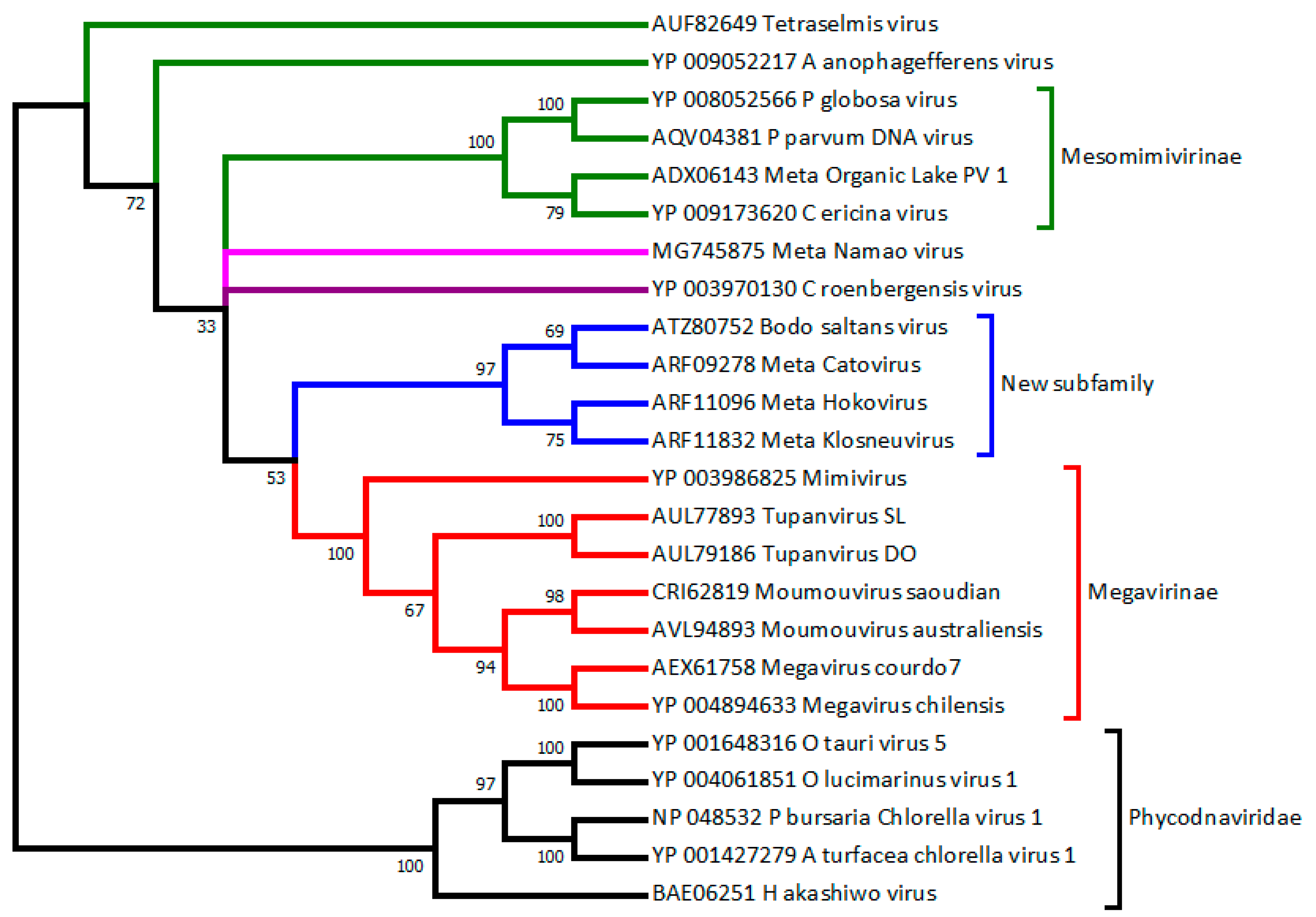
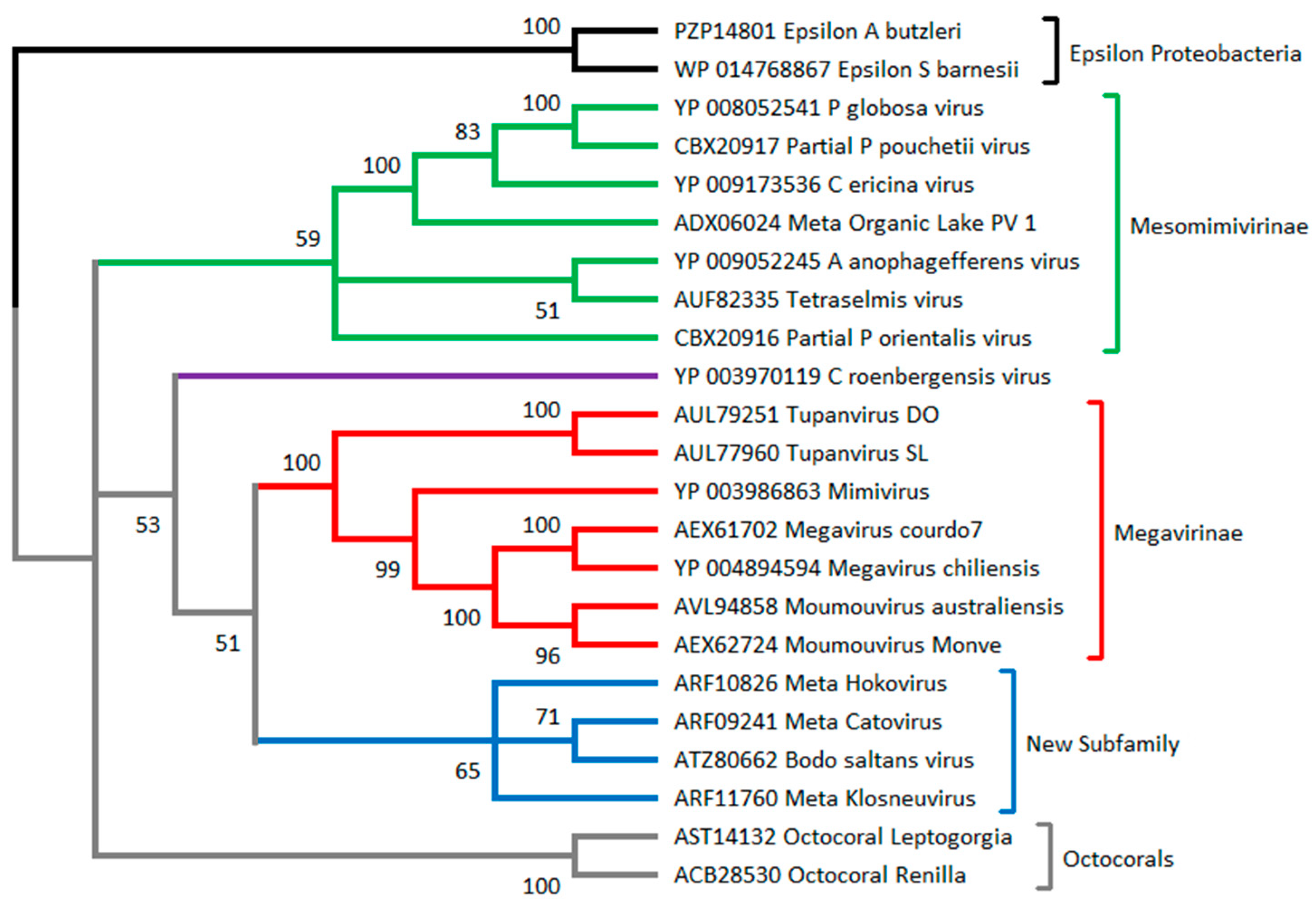
2.6. Metagenomic Contributions to the Expansion of the Mimiviridae Family
3. Discussion
3.1. Algae-Infecting Mimiviridae versus Phycodnavirididae
3.2. How to Recognize and Classify Future Members of the Mimiviridae
- A high prevalence of the strictly conserved AAAATTGA motif in the promoter regions of early transcribed genes [85].
- A high prevalence of hairpin-forming transcription termination motifs in the 3′ end of genes [86].
- The detection of a transpoviron. Transpovirons are 7-kb-long plasmid-like linear dsDNA molecules found in association with Mimivirus-relatives [88].
- The presence of a gene encoding a Mimiviridae-specific version of glutamine-dependent Asparagine synthetase (AsnS). A different, easily distinguishable homolog of this enzyme is encoded by some Prasinoviruses (e.g., OtV5) [74].
- The presence of amino-acyl tRNA synthetases (aaRS). The finding of such central components of the translational apparatus in the Mimivirus genome [2] was considered revolutionary, as the historical definition of viruses denied them the capacity to synthetize proteins [4]. Conflicting hypotheses on the evolutionary origin of these viral enzymes generated a still ongoing controversial debate. The presence of these aaRS remains a remarkable feature of the Mimiviridae, although not unique to them anymore [7]. Unfortunately, the number of virus-encoded aaRS varies greatly among the Mimiviridae members (from 0 up to 20 different ones in Tupanviruses [46]) (Table 3), which reduces the utility of their detection for classification purpose. However, the presence of aaRS in a virus genome remains a strong complementary argument for their classification within the Mimiviridae.
4. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- La Scola, B.; Audic, S.; Robert, C.; Jungang, L.; de Lamballerie, X.; Drancourt, M.; Birtles, R.; Claverie, J.M.; Raoult, D. A giant virus in amoebae. Science 2003, 299, 2033. [Google Scholar] [CrossRef] [PubMed]
- Raoult, D.; Audic, S.; Robert, C.; Abergel, C.; Renesto, P.; Ogata, H.; La Scola, B.; Suzan, M.; Claverie, J.M. The 1.2-megabase genome sequence of Mimivirus. Science 2004, 306, 1344–1350. [Google Scholar] [CrossRef] [PubMed]
- Raoult, D.; La Scola, B.; Birtles, R. The discovery and characterization of Mimivirus, the largest known virus and putative pneumonia agent. Clin. Infect. Dis. 2007, 45, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Abergel, C.; Legendre, M.; Claverie, J.M. The rapidly expanding universe of giant viruses: Mimivirus, Pandoravirus, Pithovirus and Mollivirus. FEMS Microbiol. Rev. 2015, 39, 779–796. [Google Scholar] [CrossRef] [PubMed]
- Claverie, J.M.; Abergel, C. Giant viruses: The difficult breaking of multiple epistemological barriers. Stud. Hist. Philos. Biol. Biomed. Sci. 2016, 59, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Claverie, J.M.; Ogata, H.; Audic, S.; Abergel, C.; Suhre, K.; Fournier, P.E. Mimivirus and the emerging concept of “giant” virus. Virus Res. 2006, 117, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Philippe, N.; Legendre, M.; Doutre, G.; Couté, Y.; Poirot, O.; Lescot, M.; Arslan, D.; Seltzer, V.; Bertaux, L.; Bruley, C.; et al. Pandoraviruses: Amoeba viruses with genomes up to 2.5 Mb reaching that of parasitic eukaryotes. Science 2013, 341, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, C.; Eisen, J.A.; Nene, V. New evolutionary frontiers from unusual virus genomes. Genome Biol. 2005, 6, 212. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Suzan-Monti, M.; La Scola, B.; Raoult, D. Genomic and evolutionary aspects of Mimivirus. Virus Res. 2006, 117, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Claverie, J.M. Viruses take center stage in cellular evolution. Genome Biol. 2006, 7, 110. [Google Scholar] [CrossRef] [PubMed]
- Raoult, D.; Forterre, P. Redefining viruses: Lessons from Mimivirus. Nat. Rev. Microbiol. 2008, 6, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Claverie, J.M.; Grzela, R.; Lartigue, A.; Bernadac, A.; Nitsche, S.; Vacelet, J.; Ogata, H.; Abergel, C. Mimivirus and Mimiviridae: Giant viruses with an increasing number of potential hosts, including corals and sponges. J. Invertebr. Pathol. 2009, 101, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Forterre, P. Giant viruses: Conflicts in revisiting the virus concept. Intervirology 2010, 53, 116–132. [Google Scholar] [CrossRef] [PubMed]
- Claverie, J.M.; Abergel, C. Mimivirus: The emerging paradox of quasi-autonomous viruses. Trends Genet. 2010, 26, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Claverie, J.M.; Abergel, C. Open questions about giant viruses. Adv. Virus Res. 2013, 85, 25–56. [Google Scholar] [CrossRef] [PubMed]
- Moreira, D.; Brochier-Armanet, C. Giant viruses, giant chimeras: The multiple evolutionary histories of Mimivirus genes. BMC Evol. Biol. 2008, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Moreira, D.; Lopez-Garcia, P. Ten reasons to exclude viruses from the tree of life. Nat. Rev. Microbiol. 2009, 7, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Ludmir, E.B.; Enquist, L.W. Viral genomes are part of the phylogenetic tree of life. Nat. Rev. Microbiol. 2009, 7, 615. [Google Scholar] [CrossRef] [PubMed]
- Claverie, J.M.; Ogata, H. Ten good reasons not to exclude giruses from the evolutionary picture. Nat. Rev. Microbiol. 2009, 7, 615. [Google Scholar] [CrossRef] [PubMed]
- Hegde, N.R.; Maddur, M.S.; Kaveri, S.V.; Bayry, J. Reasons to include viruses in the tree of life. Nat. Rev. Microbiol. 2009, 7, 615. [Google Scholar] [CrossRef] [PubMed]
- Navas-Castillo, J. Six comments on the ten reasons for the demotion of viruses. Nat. Rev. Microbiol. 2009, 7, 615. [Google Scholar] [CrossRef] [PubMed]
- López-García, P.; Moreira, D. Yet viruses cannot be included in the tree of life. Nat. Rev. Microbiol. 2009, 7, 615–617. [Google Scholar] [CrossRef]
- Boyer, M.; Madoui, M.A.; Gimenez, G.; La Scola, B.; Raoult, D. Phylogenetic and phyletic studies of informational genes in genomes highlight existence of a 4 domain of life including giant viruses. PLoS ONE 2010, 5, e15530. [Google Scholar] [CrossRef] [PubMed]
- Legendre, M.; Arslan, D.; Abergel, C.; Claverie, J.M. Genomics of Megavirus and the elusive fourth domain of Life. Commun. Integr. Biol. 2012, 5, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Nasir, A.; Kim, K.M.; Caetano-Anolles, G. Giant viruses coexisted with the cellular ancestors and represent a distinct supergroup along with superkingdoms Archaea, Bacteria and Eukarya. BMC Evol. Biol. 2012, 12, 156. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Senkevich, T.G.; Dolja, V.V. Compelling reasons why viruses are relevant for the origin of cells. Nat. Rev. Microbiol. 2009, 7, 615. [Google Scholar] [CrossRef] [PubMed]
- Villarreal, L.P.; Witzany, G. Viruses are essential agents within the roots and stem of the tree of life. J. Theor. Biol. 2010, 262, 698–710. [Google Scholar] [CrossRef] [PubMed]
- Nasir, A.; Sun, F.J.; Kim, K.M.; Caetano-Anollés, G. Untangling the origin of viruses and their impact on cellular evolution. Ann. N. Y. Acad. Sci. 2015, 1341, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Beijerinck, M.W. Über ein Contagium vivum fluidum als Ursache der Fleckenkrankheit der Tabaksblätter. Verhandelingen der Koninklijke akademie van Wetenschappen te Amsterdam 1898, 65, 1–22. [Google Scholar]
- Lecoq, H. Discovery of the first virus, the tobacco mosaic virus: 1892 or 1898? C. R. Acad. Sci. III 2001, 324, 929–933. [Google Scholar] [CrossRef]
- Meints, R.H.; Van Etten, J.L.; Kuczmarski, D.; Lee, K.; Ang, B. Viral infection of the symbiotic chlorella-like alga present in Hydra viridis. Virology 1981, 113, 698–703. [Google Scholar] [CrossRef]
- Van Etten, J.L.; Meints, R.H.; Burbank, D.E.; Kuczmarski, D.; Cuppels, D.A.; Lane, L.C. Isolation and characterization of a virus from the intracellular green alga symbiotic with Hydra viridis. Virology 1981, 113, 704–771. [Google Scholar] [CrossRef]
- Torrella, F.; Morita, R.Y. Evidence by electron micrographs for a high incidence of bacteriophage particles in the waters of Yaquina Bay, oregon: Ecological and taxonomical implications. Appl. Environ. Microbiol. 1979, 37, 774–778. [Google Scholar] [PubMed]
- Roux, S.; Brum, J.R.; Dutilh, B.E.; Sunagawa, S.; Duhaime, M.B.; Loy, A.; Poulos, B.T.; Solonenko, N.; Lara, E.; Poulain, J.; et al. Ecogenomics and potential biogeochemical impacts of globally abundant ocean viruses. Nature 2016, 537, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lu, Z.; Sun, L.; Ropp, S.; Kutish, G.F.; Rock, D.L.; Van Etten, J.L. Analysis of 74 kb of DNA located at the right end of the 330-kb chlorella virus PBCV-1 genome. Virology 1997, 237, 360–377. [Google Scholar] [CrossRef] [PubMed]
- Van Etten, J.L.; Meints, R.H.; Kuczmarski, D.; Burbank, D.E.; Lee, K. Viruses of symbiotic Chlorella-like algae isolated from Paramecium bursaria and Hydra viridis. Proc. Natl. Acad. Sci. USA 1982, 79, 3867–3871. [Google Scholar] [CrossRef] [PubMed]
- Van Etten, J.L. Giant Chlorella viruses. Mol. Cells (Korean Soc. Mol. Cell. Biol.) 1995, 5, 99–106. [Google Scholar]
- Van Etten, J.L.; Meints, R.H. Giant viruses infecting algae. Annu. Rev. Microbiol. 1999, 53, 447–494. [Google Scholar] [CrossRef] [PubMed]
- Venter, J.C.; Remington, K.; Heidelberg, J.F.; Halpern, A.L.; Rusch, D.; Eisen, J.A.; Wu, D.; Paulsen, I.; Nelson, K.E.; Nelson, W.; et al. Environmental genome shotgun sequencing of the Sargasso Sea. Science 2004, 304, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Claverie, J.M. Giant viruses in the oceans: The 4th Algal Virus Workshop. Virol. J. 2005, 2, 52. [Google Scholar] [CrossRef] [PubMed]
- Ghedin, E.; Claverie, J.M. Mimivirus relatives in the Sargasso Sea. Virol. J. 2005, 2, 62. [Google Scholar] [CrossRef] [PubMed]
- Monier, A.; Claverie, J.M.; Ogata, H. Taxonomic distribution of large DNA viruses in the sea. Genome Biol. 2008, 9, R106. [Google Scholar] [CrossRef] [PubMed]
- Monier, A.; Larsen, J.B.; Sandaa, R.A.; Bratbak, G.; Claverie, J.M.; Ogata, H. Marine mimivirus relatives are probably large algal viruses. Virol. J. 2008, 5, 12. [Google Scholar] [CrossRef] [PubMed]
- Ogata, H.; Ray, J.; Toyoda, K.; Sandaa, R.A.; Nagasaki, K.; Bratbak, G.; Claverie, J.M. Two new subfamilies of DNA mismatch repair proteins (MutS) specifically abundant in the marine environment. ISME J. 2011, 5, 1143–1151. [Google Scholar] [CrossRef] [PubMed]
- Arslan, D.; Legendre, M.; Seltzer, V.; Abergel, C.; Claverie, J.M. Distant Mimivirus relative with a larger genome highlights the fundamental features of Megaviridae. Proc. Natl. Acad. Sci. USA 2011, 108, 17486–17491. [Google Scholar] [CrossRef] [PubMed]
- Abrahão, J.; Silva, L.; Silva, L.S.; Khalil, J.Y.B.; Rodrigues, R.; Arantes, T.; Assis, F.; Boratto, P.; Andrade, M.; Kroon, E.G.; et al. Tailed giant Tupanvirus possesses the most complete translational apparatus of the known virosphere. Nat. Commun. 2018, 9, 749. [Google Scholar] [CrossRef] [PubMed]
- Garza, D.R.; Suttle, C.A. Large double-stranded DNA viruses which cause the lysis of a marine heterotrophic nanoflagellate (Bodo sp.) occur in natural marine viral communities. Aquat. Microb. Ecol. 1995, 9, 203–210. [Google Scholar] [CrossRef]
- Fischer, M.G.; Allen, M.J.; Wilson, W.H.; Suttle, C.A. Giant virus with a remarkable complement of genes infects marine zooplankton. Proc. Natl. Acad. Sci. USA 2010, 107, 19508–19513. [Google Scholar] [CrossRef] [PubMed]
- Richards, T.A.; Cavalier-Smith, T. Myosin domain evolution and the primary divergence of eukaryotes. Nature 2005, 436, 1113–1118. [Google Scholar] [CrossRef] [PubMed]
- Dereeper, A.; Guignon, V.; Blanc, G.; Audic, S.; Buffet, S.; Chevenet, F.; Dufayard, J.F.; Guindon, S.; Lefort, V.; Lescot, M.; et al. Phylogeny.fr: Robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008, 36, W465–W469. [Google Scholar] [CrossRef] [PubMed]
- Legendre, M.; Fabre, E.; Poirot, O.; Jeudy, S.; Lartigue, A.; Alempic, J.M.; Beucher, L.; Philippe, N.; Bertaux, L.; Christo-Foroux, E.; et al. Diversity and evolution of the emerging Pandoraviridae family. Nat. Commun. 2018, 9, 2285. [Google Scholar] [CrossRef] [PubMed]
- Sandaa, R.A.; Heldal, M.; Castberg, T.; Thyrhaug, R.; Bratbak, G. Isolation and characterization of two viruses with large genome size infecting Chrysochromulina ericina (Prymnesiophyceae) and Pyramimonas orientalis (Prasinophyceae). Virology 2001, 290, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Brussaard, C.P.; Short, S.M.; Frederickson, C.M.; Suttle, C.A. Isolation and phylogenetic analysis of novel viruses infecting the phytoplankton Phaeocystis globose (Prymnesiophyceae). Appl. Environ. Microbiol. 2004, 70, 3700–3705. [Google Scholar] [CrossRef] [PubMed]
- Baudoux, A.C.; Brussaard, C.P. Characterization of different viruses infecting the marine harmful algal bloom species Phaeocystis globosa. Virology 2005, 341, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Larsen, J.B.; Larsen, A.; Bratbak, G.; Sandaa, R.A. Phylogenetic analysis of members of the Phycodnaviridae virus family, using amplified fragments of the major capsid protein gene. Appl. Environ. Microbiol. 2008, 74, 3048–3057. [Google Scholar] [CrossRef] [PubMed]
- Santini, S.; Jeudy, S.; Bartoli, J.; Poirot, O.; Lescot, M.; Abergel, C.; Barbe, V.; Wommack, K.E.; Noordeloos, A.A.; Brussaard, C.P.; et al. Genome of Phaeocystis globosa virus PgV-16T highlights the common ancestry of the largest known DNA viruses infecting eukaryotes. Proc. Natl. Acad. Sci. USA 2013, 110, 10800–10805. [Google Scholar] [CrossRef] [PubMed]
- Moniruzzaman, M.; LeCleir, G.R.; Brown, C.M.; Gobler, C.J.; Bidle, K.D.; Wilson, W.H.; Wilhelm, S.W. Genome of brown tide virus (AaV), the little giant of the Megaviridae, elucidates NCLDV genome expansion and host-virus coevolution. Virology 2014, 466–467, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Gallot-Lavallée, L.; Blanc, G.; Claverie, J.M. Comparative Genomics of Chrysochromulina Ericina Virus and Other Microalga-Infecting Large DNA Viruses Highlights Their Intricate Evolutionary Relationship with the Established Mimiviridae Family. J. Virol. 2017, 91, JVI-00230. [Google Scholar] [CrossRef] [PubMed]
- Schvarcz, C.R.; Steward, G.F. A giant virus infecting green algae encodes key fermentation genes. Virology 2018, 518, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Sicko-Goad, L.; Walker, G. Viroplasm and large virus-like particles in the dinoflagellate Gymnodinium uberrimum. Protoplasma 1979, 99, 203–210. [Google Scholar] [CrossRef]
- Dodds, J.A.; Cole, A. Microscopy and biology of Uronema gigas, a filamentous eucaryotic green alga, and its associated tailed virus-like particle. Virology 1980, 100, 156–165. [Google Scholar] [CrossRef]
- Deeg, C.M.; Chow, C.T.; Suttle, C.A. The kinetoplastid-infecting Bodo saltans virus (BsV), a window into the most abundant giant viruses in the sea. eLife 2018, 7, e33014. [Google Scholar] [CrossRef] [PubMed]
- Williamson, S.J.; Rusch, D.B.; Yooseph, S.; Halpern, A.L.; Heidelberg, K.B.; Glass, J.I.; Andrews-Pfannkoch, C.; Fadrosh, D.; Miller, C.S.; Sutton, G.; et al. The Sorcerer II Global Ocean Sampling Expedition: Metagenomic characterization of viruses within aquatic microbial samples. PLoS ONE 2008, 3, e1456. [Google Scholar] [CrossRef] [PubMed]
- Claverie, J.M.; Abergel, C.; Ogata, H. Mimivirus. Curr. Top Microbiol. Immunol. 2009, 328, 89–121. [Google Scholar] [PubMed]
- Yau, S.; Lauro, F.M.; DeMaere, M.Z.; Brown, M.V.; Thomas, T.; Raftery, M.J.; Andrews-Pfannkoch, C.; Lewis, M.; Hoffman, J.M.; Gibson, J.A.; et al. Virophage control of antarctic algal host-virus dynamics. Proc. Natl. Acad. Sci. USA 2011, 108, 6163–6168. [Google Scholar] [CrossRef] [PubMed]
- La Scola, B.; Desnues, C.; Pagnier, I.; Robert, C.; Barrassi, L.; Fournous, G.; Merchat, M.; Suzan-Monti, M.; Forterre, P.; Koonin, E.; et al. The virophage as a unique parasite of the giant mimivirus. Nature 2008, 455, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Claverie, J.M.; Abergel, C. Mimivirus and its virophage. Annu. Rev. Genet. 2009, 43, 49–66. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhang, W.; Yan, S.; Xiao, J.; Zhang, Y.; Li, B.; Pan, Y.; Wang, Y. Diversity of virophages in metagenomic data sets. J. Virol. 2013, 87, 4225–4236. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Sun, D.; Childers, A.; McDermott, T.R.; Wang, Y.; Liles, M.R. Three novel virophage genomes discovered from Yellowstone Lake metagenomes. J. Virol. 2015, 89, 1278–1285. [Google Scholar] [CrossRef] [PubMed]
- Bekliz, M.; Colson, P.; La Scola, B. The Expanding Family of Virophages. Viruses 2016, 8, 317. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Chan, L.K.; Egan, R.; Malmstrom, R.R.; McMahon, K.D.; Sullivan, M.B. Ecogenomics of virophages and their giant virus hosts assessed through time series metagenomics. Nat. Commun. 2017, 8, 858. [Google Scholar] [CrossRef] [PubMed]
- Karsenti, E.; Acinas, S.G.; Bork, P.; Bowler, C.; de Vargas, C.; Raes, J.; Sullivan, M.; Arendt, D.; Benzoni, F.; Claverie, J.M.; et al. A holistic approach to marine eco-systems biology. PLoS Biol. 2011, 9, e1001177. [Google Scholar] [CrossRef] [PubMed]
- Hingamp, P.; Grimsley, N.; Acinas, S.G.; Clerissi, C.; Subirana, L.; Poulain, J.; Ferrera, I.; Sarmento, H.; Villar, E.; Lima-Mendez, G.; et al. Exploring nucleo-cytoplasmic large DNA viruses in Tara Oceans microbial metagenomes. ISME J. 2013, 7, 1678–1695. [Google Scholar] [CrossRef] [PubMed]
- Mozar, M.; Claverie, J.M. Expanding the Mimiviridae family using asparagine synthase as a sequence bait. Virology 2014, 466–467, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Schulz, F.; Yutin, N.; Ivanova, N.N.; Ortega, D.R.; Lee, T.K.; Vierheilig, J.; Daims, H.; Horn, M.; Wagner, M.; Jensen, G.J.; et al. Giant viruses with an expanded complement of translation system components. Science 2017, 356, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Clouthier, S.C.; Vanwalleghem, E.; Copeland, S.; Klassen, C.; Hobbs, G.; Nielsen, O.; Anderson, E.D. A new species of nucleo-cytoplasmic large DNA virus (NCLDV) associated with mortalities in Manitoba lake sturgeon Acipenser fulvescens. Dis. Aquat. Organ. 2013, 102, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Clouthier, S.C.; Anderson, E.D.; Kurath, G.; Breyta, R. Molecular systematics of sturgeon nucleocytoplasmic large DNA viruses. Mol. Phylogenet. Evol. 2018, 128, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Maumus, F.; Epert, A.; Nogué, F.; Blanc, G. Plant genomes enclose footprints of past infections by giant virus relatives. Nat. Commun. 2014, 5, 4268. [Google Scholar] [CrossRef] [PubMed]
- Maumus, F.; Blanc, G. Study of Gene Trafficking between Acanthamoeba and Giant Viruses Suggests an Undiscovered Family of Amoeba-Infecting Viruses. Genome Biol. Evol. 2016, 8, 3351–3363. [Google Scholar] [CrossRef] [PubMed]
- Brister, J.R.; Ako-Adjei, D.; Bao, Y.; Blinkova, O. NCBI viral genomes resource. Nucleic Acids Res. 2015, 43, D571–D577. [Google Scholar] [CrossRef] [PubMed]
- Pagarete, A.; Grébert, T.; Stepanova, O.; Sandaa, R.A.; Bratbak, G. Tsv-N1: A Novel DNA Algal Virus that Infects Tetraselmis striata. Viruses 2015, 7, 3937–3953. [Google Scholar] [CrossRef] [PubMed]
- Ogata, H.; Toyoda, K.; Tomaru, Y.; Nakayama, N.; Shirai, Y.; Claverie, J.M.; Nagasaki, K. Remarkable sequence similarity between the dinoflagellate-infecting marine girus and the terrestrial pathogen African swine fever virus. Virol. J. 2009, 6, 178. [Google Scholar] [CrossRef] [PubMed]
- Mackinder, L.C.; Worthy, C.A.; Biggi, G.; Hall, M.; Ryan, K.P.; Varsani, A.; Harper, G.M.; Wilson, W.H.; Brownlee, C.; Schroeder, D.C. A unicellular algal virus, Emiliania huxleyi virus 86, exploits an animal-like infection strategy. J. Gen. Virol. 2009, 90, 2306–2316. [Google Scholar] [CrossRef] [PubMed]
- Yutin, N.; Colson, P.; Raoult, D.; Koonin, E.V. Mimiviridae: Clusters of orthologous genes, reconstruction of gene repertoire evolution and proposed expansion of the giant virus family. Virol. J. 2013, 10, 106. [Google Scholar] [CrossRef] [PubMed]
- Suhre, K.; Audic, S.; Claverie, J.M. Mimivirus gene promoters exhibit an unprecedented conservation among all eukaryotes. Proc. Natl. Acad. Sci. USA 2005, 102, 14689–14693. [Google Scholar] [CrossRef] [PubMed]
- Byrne, D.; Grzela, R.; Lartigue, A.; Audic, S.; Chenivesse, S.; Encinas, S.; Claverie, J.M.; Abergel, C. The polyadenylation site of Mimivirus transcripts obeys a stringent ‘hairpin rule’. Genome Res. 2009, 19, 1233–1242. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.G.; Suttle, C.A. A virophage at the origin of large DNA transposons. Science 2011, 332, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Desnues, C.; La Scola, B.; Yutin, N.; Fournous, G.; Robert, C.; Azza, S.; Jardot, P.; Monteil, S.; Campocasso, A.; Koonin, E.V.; et al. Provirophages and transpovirons as the diverse mobilome of giant viruses. Proc. Natl. Acad. Sci. USA 2012, 109, 18078–18083. [Google Scholar] [CrossRef] [PubMed]
- Mihara, T.; Koyano, H.; Hingamp, P.; Grimsley, N.; Goto, S.; Ogata, H. Taxon Richness of “Megaviridae” Exceeds those of Bacteria and Archaea in the Ocean. Microbes Environ. 2018, 33, 162–171. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Name 1 | Accession | Genome Size (kb) | Virion Type | Virion Size (nm) | Host Phylum |
|---|---|---|---|---|---|
| Mimivirus 2 | NC_014649 | 1181 | icosahedron | 750 | Amoebozoa |
| Megavirus 2 | NC_016072 | 1259 | icosahedron | 680 | Amoebozoa |
| Moumouvirus 2 | NC_020104 | 1021 | icosahedron | 620 | Amoebozoa |
| CroV | NC_014637 | 693 | icosahedron | 300 | Heterokonta |
| PgV | NC_021312 | 460 | icosahedron | 150 | Haptophyceae |
| CeV | NC_028094 | 474 | icosahedron | 160 | Haptophyceae |
| TetV | KY322437 | 668 | icosahedron | 240 | Chlorophyta |
| BsV | MF782455 | 1386 | icosahedron | 300 | Excavata |
| AaV | NC_024697 | 371 | icosahedron | 140 | Heterokonta |
| TupanSL | KY523104 | 1439 | icosa. + tail | 450 + 550 | Amoebozoa |
| TupanDO | MF405918 | 1516 | icosa. + tail | 450 + 550 | Amoebozoa |
| Genus | Prototype | Encoded RNA pol | Genome Size (kb) | (G + C) % | Accession | Virion Ø (nm) |
|---|---|---|---|---|---|---|
| Chlorovirus | PBCV-1 | No | 330 | 40 | NC_000852 | 190 |
| Coccolithovirus 1 | EhV-86 | Yes | 407 | 40.2 | NC_007346 | 180 |
| Phaeovirus | EsV-1 | No | 336 | 51.7 | NC_002687 | 200 |
| Prasinovirus | OtV-5 | No | 187 | 45 | NC_010191 | 120 |
| Prymnesiovirus I 2 | PgV-16T | Yes | 460 | 32 | NC_021312 | 153 |
| Prymnesiovirus II 2 | PgV-01T | Unknown | ≈177 | Unknown | - | 106 |
| Raphidovirus | HaV-1 | No | 275 | 30.4 | KX008963 | 202 |
| Name | DNA Pol B | RNA 2 Pol II | aaRS 3 | MCP 4 | MutS7 | AsnS | Transpoviron | Virophage |
|---|---|---|---|---|---|---|---|---|
| Mimivirus | Yes | 8 | 4 | 4 | Yes | Yes | Yes | Yes |
| Megavirus | Yes | 8 | 7 | 4 | Yes | Yes | Yes | Yes |
| Moumouvirus | Yes | 8 | 5 | 4 | Yes | Yes | Yes | Yes |
| CroV | Yes | 8 | 1 | 4 | Yes | Yes | No | Yes |
| PgV | Yes | 8 | 0 | 2 | Yes | Yes | No | Yes |
| CeV | Yes | 8 | 0 | 3 | Yes | Yes | No | No |
| TetV | Yes | 8 | 0 | 1 | Yes | No | No | No |
| BsV | Yes | 7 | 2 | 4 | Yes | Yes | No | No |
| AaV | Yes | 8 | 0 | 2 | Yes | No | No | No |
| TupanSL | Yes | 8 | 20 | 3 | Yes | Yes | No | No |
| TupanDO | Yes | 7 | 20 | 3 | Yes | Yes | No | No |
| YlmV 1 | No | D | 0 | 1 | No | No | No | No |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Claverie, J.-M.; Abergel, C. Mimiviridae: An Expanding Family of Highly Diverse Large dsDNA Viruses Infecting a Wide Phylogenetic Range of Aquatic Eukaryotes. Viruses 2018, 10, 506. https://doi.org/10.3390/v10090506
Claverie J-M, Abergel C. Mimiviridae: An Expanding Family of Highly Diverse Large dsDNA Viruses Infecting a Wide Phylogenetic Range of Aquatic Eukaryotes. Viruses. 2018; 10(9):506. https://doi.org/10.3390/v10090506
Chicago/Turabian StyleClaverie, Jean-Michel, and Chantal Abergel. 2018. "Mimiviridae: An Expanding Family of Highly Diverse Large dsDNA Viruses Infecting a Wide Phylogenetic Range of Aquatic Eukaryotes" Viruses 10, no. 9: 506. https://doi.org/10.3390/v10090506
APA StyleClaverie, J.-M., & Abergel, C. (2018). Mimiviridae: An Expanding Family of Highly Diverse Large dsDNA Viruses Infecting a Wide Phylogenetic Range of Aquatic Eukaryotes. Viruses, 10(9), 506. https://doi.org/10.3390/v10090506