Host Shutoff in Influenza A Virus: Many Means to an End


Abstract
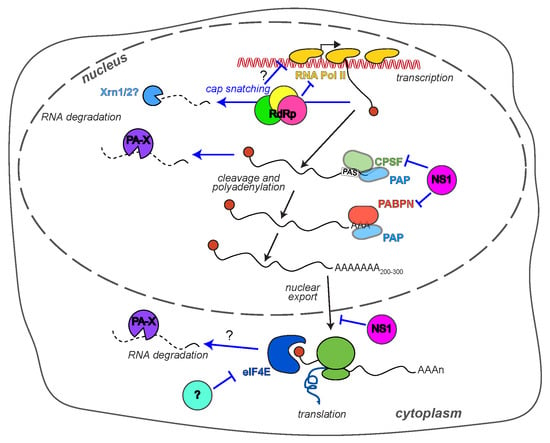
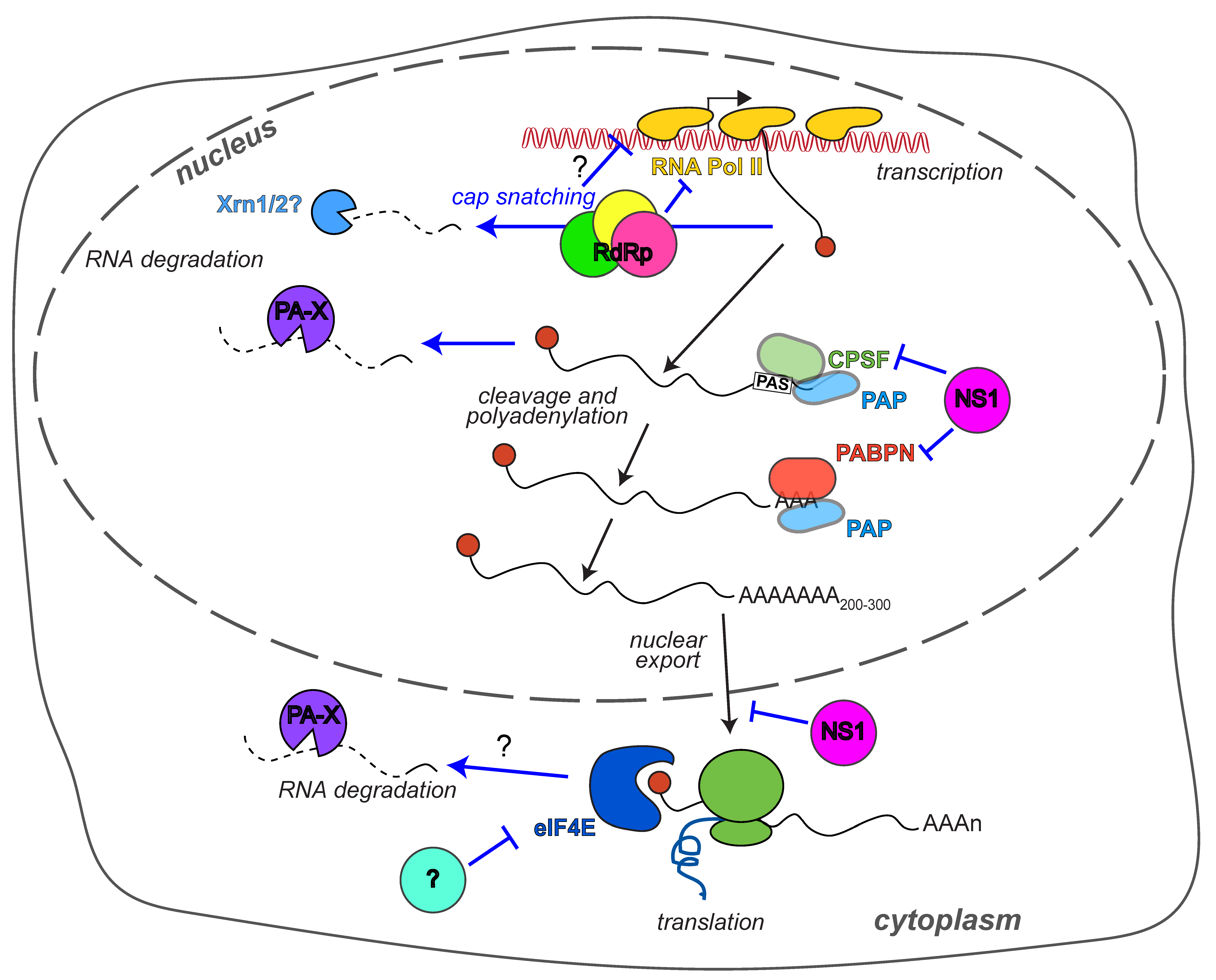
1. Introduction
2. How Does Influenza A Virus Reduce Host Gene Expression?
2.1. PA-X and RNA Degradation
2.2. The RdRp and RNA Transcription
2.3. NS1 and RNA Processing
2.4. Translation
3. Why Does Influenza A Virus Reduce Host Gene Expression?
3.1. Host Shutoff as a Mechanism of Immune Evasion
3.2. Alternative Functions of Host Shutoff
4. How Are the Multiple Host Shutoff Mechanisms Integrated?
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Skehel, J.J. Polypeptide synthesis in influenza virus-infected cells. Virology 1972, 49, 23–36. [Google Scholar] [CrossRef]
- Beloso, A.; Martínez, C.; Valcárcel, J.; Santarén, J.F.; Ortín, J. Degradation of cellular mRNA during influenza virus infection: Its possible role in protein synthesis shutoff. J. Gen. Virol. 1992, 73 Pt 3, 575–581. [Google Scholar] [CrossRef]
- Inglis, S.C. Inhibition of Host Protein Synthesis and Degradation of Cellular mRNAs during Infection by Influenza and Herpes Simplex Virus. Mol. Cell. Biol. 1982, 2, 1644–1648. [Google Scholar] [CrossRef] [PubMed]
- Katze, M.G.; Krug, R.M. Metabolism and expression of RNA polymerase II transcripts in influenza virus-infected cells. Mol. Cell. Biol. 1984, 4, 2198–2206. [Google Scholar] [CrossRef] [PubMed]
- Bercovich-Kinori, A.; Tai, J.; Gelbart, I.A.; Shitrit, A.; Ben-Moshe, S.; Drori, Y.; Itzkovitz, S.; Mandelboim, M.; Stern-Ginossar, N. A systematic view on influenza induced host shutoff. eLife 2016, 5, e18311. [Google Scholar] [CrossRef] [PubMed]
- Kwong, A.D.; Frenkel, N. Herpes simplex virus-infected cells contain a function(s) that destabilizes both host and viral mRNAs. Proc. Natl. Acad. Sci. USA 1987, 84, 1926–1930. [Google Scholar] [CrossRef] [PubMed]
- Glaunsinger, B.; Ganem, D. Lytic KSHV infection inhibits host gene expression by accelerating global mRNA turnover. Mol. Cell 2004, 13, 713–723. [Google Scholar] [CrossRef]
- Pflug, A.; Lukarska, M.; Resa-Infante, P.; Reich, S.; Cusack, S. Structural insights into RNA synthesis by the influenza virus transcription-replication machine. Virus Res. 2017, 234, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Te Velthuis, A.J.W.; Fodor, E. Influenza virus RNA polymerase: Insights into the mechanisms of viral RNA synthesis. Nat. Rev. Microbiol. 2016, 14, 479–493. [Google Scholar] [CrossRef] [PubMed]
- Dias, A.; Bouvier, D.; Crépin, T.; McCarthy, A.A.; Hart, D.J.; Baudin, F.; Cusack, S.; Ruigrok, R.W.H. The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nature 2009, 458, 914–918. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.; Bartlam, M.; Lou, Z.; Chen, S.; Zhou, J.; He, X.; Lv, Z.; Ge, R.; Li, X.; Deng, T.; et al. Crystal structure of an avian influenza polymerase PAN reveals an endonuclease active site. Nature 2009, 458, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Jagger, B.W.; Wise, H.M.; Kash, J.C.; Walters, K.-A.; Wills, N.M.; Xiao, Y.-L.; Dunfee, R.L.; Schwartzman, L.M.; Ozinsky, A.; Bell, G.L.; et al. An Overlapping Protein-Coding Region in Influenza A Virus Segment 3 Modulates the Host Response. Science 2012, 337, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Firth, A.E.; Jagger, B.W.; Wise, H.M.; Nelson, C.C.; Parsawar, K.; Wills, N.M.; Napthine, S.; Taubenberger, J.K.; Digard, P.; Atkins, J.F. Ribosomal frameshifting used in influenza A virus expression occurs within the sequence UCC_UUU_CGU and is in the +1 direction. Open Biol. 2012, 2, 120109. [Google Scholar] [CrossRef] [PubMed]
- Hara, K.; Schmidt, F.I.; Crow, M.; Brownlee, G.G. Amino Acid Residues in the N-Terminal Region of the PA Subunit of Influenza A Virus RNA Polymerase Play a Critical Role in Protein Stability, Endonuclease Activity, Cap Binding, and Virion RNA Promoter Binding. J. Virol. 2006, 80, 7789–7798. [Google Scholar] [CrossRef] [PubMed]
- Datta, K.; Wolkerstorfer, A.; Szolar, O.H.J.; Cusack, S.; Klumpp, K. Characterization of PA-N terminal domain of Influenza A polymerase reveals sequence specific RNA cleavage. Nucleic Acids Res. 2013, 41, 8289–8299. [Google Scholar] [CrossRef] [PubMed]
- Bavagnoli, L.; Cucuzza, S.; Campanini, G.; Rovida, F.; Paolucci, S.; Baldanti, F.; Maga, G. The novel influenza A virus protein PA-X and its naturally deleted variant show different enzymatic properties in comparison to the viral endonuclease PA. Nucleic Acids Res. 2015, 43, 9405–9417. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Jagger, B.W.; Wise, H.M.; Digard, P.; Holmes, E.C.; Taubenberger, J.K. Evolutionary Conservation of the PA-X Open Reading Frame in Segment 3 of Influenza A Virus. J. Virol. 2012, 86, 12411–12413. [Google Scholar] [CrossRef] [PubMed]
- Desmet, E.A.; Bussey, K.A.; Stone, R.; Takimoto, T. Identification of the N-Terminal Domain of the Influenza Virus PA Responsible for the Suppression of Host Protein Synthesis. J. Virol. 2013, 87, 3108–3118. [Google Scholar] [CrossRef] [PubMed]
- Oishi, K.; Yamayoshi, S.; Kawaoka, Y. Mapping of a Region of the PA-X Protein of Influenza A Virus That Is Important for Its Shutoff Activity. J. Virol. 2015, 89, 8661–8665. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Sun, Y.; Hu, J.; Qi, L.; Wang, J.; Xiong, X.; Wang, Y.; He, Q.; Lin, Y.; Kong, W.; et al. The contribution of PA-X to the virulence of pandemic 2009 H1N1 and highly pathogenic H5N1 avian influenza viruses. Sci. Rep. 2015, 5, 8262. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; MacDonald, L.A.; Takimoto, T. Influenza A Virus Protein PA-X Contributes to Viral Growth and Suppression of the Host Antiviral and Immune Responses. J. Virol. 2015, 89, 6442–6452. [Google Scholar] [CrossRef] [PubMed]
- Khaperskyy, D.A.; Schmaling, S.; Larkins-Ford, J.; McCormick, C.; Gaglia, M.M. Selective Degradation of Host RNA Polymerase II Transcripts by Influenza A Virus PA-X Host Shutoff Protein. PLoS Pathog. 2016, 12, e1005427. [Google Scholar] [CrossRef] [PubMed]
- Nogales, A.; Martinez-Sobrido, L.; Chiem, K.; Topham, D.J.; DeDiego, M.L. Functional Evolution of the 2009 Pandemic H1N1 Influenza NS1 And Pa in Humans. J. Virol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Chaimayo, C.; Takimoto, T.; Hayashi, T.; Chaimayo, C.; Takimoto, T. Impact of influenza PA-X on host response. Oncotarget 2015, 6, 19364–19365. [Google Scholar] [CrossRef] [PubMed]
- Gaglia, M.M.; Covarrubias, S.; Wong, W.; Glaunsinger, B.A. A Common Strategy for Host RNA Degradation by Divergent Viruses. J. Virol. 2012, 86, 9527–9530. [Google Scholar] [CrossRef] [PubMed]
- Khaperskyy, D.A.; Emara, M.M.; Johnston, B.P.; Anderson, P.; Hatchette, T.F.; McCormick, C. Influenza a virus host shutoff disables antiviral stress-induced translation arrest. PLoS Pathog. 2014, 10, e1004217. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.R.; Glaunsinger, B.A. Nuclear Import of Cytoplasmic Poly(A) Binding Protein Restricts Gene Expression via Hyperadenylation and Nuclear Retention of mRNA. Mol. Cell. Biol. 2010, 30, 4996–5008. [Google Scholar] [CrossRef] [PubMed]
- Finnen, R.L.; Hay, T.J.M.; Dauber, B.; Smiley, J.R.; Banfield, B.W. The herpes simplex virus 2 virion-associated ribonuclease vhs interferes with stress granule formation. J. Virol. 2014, 88, 12727–12739. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Chaimayo, C.; McGuinness, J.; Takimoto, T. Critical Role of the PA-X C-Terminal Domain of Influenza A Virus in Its Subcellular Localization and Shutoff Activity. J. Virol. 2016, 90, 7131–7141. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.; Everly, D.N.; Read, G.S. mRNA decay during herpesvirus infections: Interaction between a putative viral nuclease and a cellular translation factor. J. Virol. 2001, 75, 10272–10280. [Google Scholar] [CrossRef] [PubMed]
- Page, H.G.; Read, G.S. The Virion Host Shutoff Endonuclease (UL41) of Herpes Simplex Virus Interacts with the Cellular Cap-Binding Complex eIF4F. J. Virol. 2010, 84, 6886–6890. [Google Scholar] [CrossRef] [PubMed]
- Oishi, K.; Yamayoshi, S.; Kawaoka, Y. Identification of novel amino acid residues of influenza virus PA-X that are important for PA-X shutoff activity by using yeast. Virology 2018, 516, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Sun, H.; Hu, J.; Qi, L.; Wang, J.; Xiong, X.; Wang, Y.; He, Q.; Lin, Y.; Kong, W.; et al. The 20 amino acids at the C-terminus of PA-X are associated with increased influenza A virus replication and pathogenicity. J. Gen. Virol. 2015, 96, 2036–2049. [Google Scholar] [CrossRef] [PubMed]
- Feng, K.H.; Sun, M.; Iketani, S.; Holmes, E.C.; Parrish, C.R. Comparing the functions of equine and canine influenza H3N8 virus PA-X proteins: Suppression of reporter gene expression and modulation of global host gene expression. Virology 2016, 496, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Zhang, X.; Sun, Y.; Liu, Q.; Sun, H.; Xiong, X.; Jiang, M.; He, Q.; Wang, Y.; Pu, J.; et al. Truncation of C-terminal 20 amino acids in PA-X contributes to adaptation of swine influenza virus in pigs. Sci. Rep. 2016, 6, 21845. [Google Scholar] [CrossRef] [PubMed]
- Oishi, K.; Yamayoshi, S.; Kozuka-Hata, H.; Oyama, M.; Kawaoka, Y. N-Terminal Acetylation by NatB is Required for the Shutoff Activity of Influenza A Virus PA-X. Cell Rep. 2018, 24, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, O.G.; Smith, M.; Fodor, E. Association of the Influenza A Virus RNA-Dependent RNA Polymerase with Cellular RNA Polymerase II. J. Virol. 2005, 79, 5812–5818. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Gallagher, G.R.; Dai, W.; Liu, P.; Li, R.; Trombly, M.I.; Gammon, D.B.; Mello, C.C.; Wang, J.P.; Finberg, R.W. Influenza A virus preferentially snatches noncoding RNA caps. RNA 2015, 21, 2067–2075. [Google Scholar] [CrossRef] [PubMed]
- Amroun, A.; Priet, S.; de Lamballerie, X.; Quérat, G. Bunyaviridae RdRps: Structure, motifs, and RNA synthesis machinery. Crit. Rev. Microbiol. 2017, 43, 753–778. [Google Scholar] [CrossRef] [PubMed]
- Lamb, R.A.; Choppin, P.W. Synthesis of influenza virus polypeptides in cells resistant to alpha-amanitin: Evidence for the involvement of cellular RNA polymerase II in virus replication. J. Virol. 1977, 23, 816–819. [Google Scholar] [PubMed]
- Chan, A.Y.; Vreede, F.T.; Smith, M.; Engelhardt, O.G.; Fodor, E. Influenza virus inhibits RNA polymerase II elongation. Virology 2006, 351, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Bauer, D.L.V.; Tellier, M.; Martínez-Alonso, M.; Nojima, T.; Proudfoot, N.J.; Murphy, S.; Fodor, E. Influenza Virus Mounts a Two-Pronged Attack on Host RNA Polymerase II Transcription. Cell Rep. 2018, 23, 2119–2129.e3. [Google Scholar] [CrossRef] [PubMed]
- Heidemann, M.; Hintermair, C.; Voß, K.; Eick, D. Dynamic phosphorylation patterns of RNA polymerase II CTD during transcription. Biochim. Biophys. Acta BBA Gene Regul. Mech. 2013, 1829, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Lemay, J.-F.; Bachand, F. Fail-safe transcription termination: Because one is never enough. RNA Biol. 2015, 12, 927–932. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.; Pérez-González, A.; Nieto, A. Influenza Virus Infection Causes Specific Degradation of the Largest Subunit of Cellular RNA Polymerase II. J. Virol. 2007, 81, 5315–5324. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.; Pérez-González, A.; Hossain, M.J.; Chen, L.-M.; Rolling, T.; Pérez-Breña, P.; Donis, R.; Kochs, G.; Nieto, A. Attenuated Strains of Influenza A Viruses Do Not Induce Degradation of RNA Polymerase II. J. Virol. 2009, 83, 11166–11174. [Google Scholar] [CrossRef] [PubMed]
- Vreede, F.T.; Chan, A.Y.; Sharps, J.; Fodor, E. Mechanisms and functional implications of the degradation of host RNA polymerase II in influenza virus infected cells. Virology 2010, 396, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Llompart, C.M.; Nieto, A.; Rodriguez-Frandsen, A. Specific Residues of PB2 and PA Influenza Virus Polymerase Subunits Confer the Ability for RNA Polymerase II Degradation and Virus Pathogenicity in Mice. J. Virol. 2014, 88, 3455–3463. [Google Scholar] [CrossRef] [PubMed]
- García-Sastre, A.; Egorov, A.; Matassov, D.; Brandt, S.; Levy, D.E.; Durbin, J.E.; Palese, P.; Muster, T. Influenza A Virus Lacking the NS1 Gene Replicates in Interferon-Deficient Systems. Virology 1998, 252, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Hale, B.G.; Randall, R.E.; Ortín, J.; Jackson, D. The multifunctional NS1 protein of influenza A viruses. J. Gen. Virol. 2008, 89, 2359–2376. [Google Scholar] [CrossRef] [PubMed]
- Krug, R.M. Functions of the influenza A virus NS1 protein in antiviral defense. Curr. Opin. Virol. 2015, 12, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Hale, B.G.; Steel, J.; Medina, R.A.; Manicassamy, B.; Ye, J.; Hickman, D.; Hai, R.; Schmolke, M.; Lowen, A.C.; Perez, D.R.; et al. Inefficient Control of Host Gene Expression by the 2009 Pandemic H1N1 Influenza A Virus NS1 Protein. J. Virol. 2010, 84, 6909–6922. [Google Scholar] [CrossRef] [PubMed]
- Kainov, D.E.; Müller, K.H.; Theisen, L.L.; Anastasina, M.; Kaloinen, M.; Muller, C.P. Differential Effects of NS1 Proteins of Human Pandemic H1N1/2009, Avian Highly Pathogenic H5N1, and Low Pathogenic H5N2 Influenza A Viruses on Cellular Pre-mRNA Polyadenylation and mRNA Translation. J. Biol. Chem. 2011, 286, 7239–7247. [Google Scholar] [CrossRef] [PubMed]
- Kochs, G.; García-Sastre, A.; Martínez-Sobrido, L. Multiple anti-interferon actions of the influenza A virus NS1 protein. J. Virol. 2007, 81, 7011–7021. [Google Scholar] [CrossRef] [PubMed]
- Kuo, R.-L.; Zhao, C.; Malur, M.; Krug, R.M. Influenza A virus strains that circulate in humans differ in the ability of their NS1 proteins to block the activation of IRF3 and interferon-β transcription. Virology 2010, 408, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Salvatore, M.; Basler, C.F.; Parisien, J.-P.; Horvath, C.M.; Bourmakina, S.; Zheng, H.; Muster, T.; Palese, P.; García-Sastre, A. Effects of Influenza A Virus NS1 Protein on Protein Expression: The NS1 Protein Enhances Translation and is not Required for Shutoff of Host Protein Synthesis. J. Virol. 2002, 76, 1206–1212. [Google Scholar] [CrossRef] [PubMed]
- Ramos, I.; Carnero, E.; Bernal-Rubio, D.; Seibert, C.W.; Westera, L.; García-Sastre, A.; Fernandez-Sesma, A. Contribution of Double-Stranded RNA and CPSF30 Binding Domains of Influenza Virus NS1 to the Inhibition of Type I Interferon Production and Activation of Human Dendritic Cells. J. Virol. 2013, 87, 2430–2440. [Google Scholar] [CrossRef] [PubMed]
- Nemeroff, M.E.; Barabino, S.M.; Li, Y.; Keller, W.; Krug, R.M. Influenza virus NS1 protein interacts with the cellular 30 kDa subunit of CPSF and inhibits 3’end formation of cellular pre-mRNAs. Mol. Cell 1998, 1, 991–1000. [Google Scholar] [CrossRef]
- Chan, S.; Choi, E.-A.; Shi, Y. Pre-mRNA 3’-end processing complex assembly and function. Wiley Interdiscip. Rev. RNA 2011, 2, 321–335. [Google Scholar] [CrossRef] [PubMed]
- Poon, L.L.; Pritlove, D.C.; Fodor, E.; Brownlee, G.G. Direct evidence that the poly(A) tail of influenza A virus mRNA is synthesized by reiterative copying of a U track in the virion RNA template. J. Virol. 1999, 73, 3473–3476. [Google Scholar] [PubMed]
- Twu, K.Y.; Noah, D.L.; Rao, P.; Kuo, R.-L.; Krug, R.M. The CPSF30 Binding Site on the NS1A Protein of Influenza A Virus Is a Potential Antiviral Target. J. Virol. 2006, 80, 3957–3965. [Google Scholar] [CrossRef] [PubMed]
- Das, K.; Ma, L.-C.; Xiao, R.; Radvansky, B.; Aramini, J.; Zhao, L.; Marklund, J.; Kuo, R.-L.; Twu, K.Y.; Arnold, E.; et al. Structural basis for suppression of a host antiviral response by influenza A virus. Proc. Natl. Acad. Sci. USA 2008, 105, 13093–13098. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, Z.Y.; Wang, W.; Baker, C.C.; Krug, R.M. The 3’-end-processing factor CPSF is required for the splicing of single-intron pre-mRNAs in vivo. RNA 2001, 7, 920–931. [Google Scholar] [CrossRef] [PubMed]
- Ayllon, J.; Domingues, P.; Rajsbaum, R.; Miorin, L.; Schmolke, M.; Hale, B.G.; García-Sastre, A. A Single Amino Acid Substitution in the Novel H7N9 Influenza A Virus NS1 Protein Increases CPSF30 Binding and Virulence. J. Virol. 2014, 88, 12146–12151. [Google Scholar] [CrossRef] [PubMed]
- Twu, K.Y.; Kuo, R.-L.; Marklund, J.; Krug, R.M. The H5N1 Influenza Virus NS Genes Selected after 1998 Enhance Virus Replication in Mammalian Cells. J. Virol. 2007, 81, 8112–8121. [Google Scholar] [CrossRef] [PubMed]
- Nogales, A.; Chauché, C.; DeDiego, M.L.; Topham, D.J.; Parrish, C.R.; Murcia, P.R.; Martínez-Sobrido, L. The K186E Amino Acid Substitution in the Canine Influenza Virus H3N8 NS1 Protein Restores Its Ability To Inhibit Host Gene Expression. J. Virol. 2017, 91, e00877-17. [Google Scholar] [CrossRef] [PubMed]
- Noah, D.L.; Twu, K.Y.; Krug, R.M. Cellular antiviral responses against influenza A virus are countered at the posttranscriptional level by the viral NS1A protein via its binding to a cellular protein required for the 3’ end processing of cellular pre-mRNAS. Virology 2003, 307, 386–395. [Google Scholar] [CrossRef]
- Clark, A.M.; Nogales, A.; Martinez-Sobrido, L.; Topham, D.J.; DeDiego, M.L. Functional Evolution of Influenza Virus NS1 Protein in Currently Circulating Human 2009 Pandemic H1N1 Viruses. J. Virol. 2017, 91, e00721-17. [Google Scholar] [CrossRef] [PubMed]
- DeDiego, M.L.; Nogales, A.; Lambert-Emo, K.; Martinez-Sobrido, L.; Topham, D.J. NS1 Protein Mutation I64T Affects Interferon Responses and Virulence of Circulating H3N2 Human Influenza A Viruses. J. Virol. 2016, 90, 9693–9711. [Google Scholar] [CrossRef] [PubMed]
- Nogales, A.; Martinez-Sobrido, L.; Topham, D.J.; DeDiego, M.L. NS1 Protein Amino Acid Changes D189N and V194I Affect Interferon Responses, Thermosensitivity, and Virulence of Circulating H3N2 Human Influenza A Viruses. J. Virol. 2017, 91, e01930-16. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Yang, H.; Chen, W.; Cao, W.; Zhong, G.; Jiao, P.; Deng, G.; Yu, K.; Yang, C.; Bu, Z.; et al. A Naturally Occurring Deletion in Its NS Gene Contributes to the Attenuation of an H5N1 Swine Influenza Virus in Chickens. J. Virol. 2008, 82, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Iguchi, A.; Gomyou, R.; Ono, Y. Influenza Virus Inhibits Cleavage of the HSP70 Pre-mRNAs at the Polyadenylation Site. Virology 1999, 254, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Steidle, S.; Martínez-Sobrido, L.; Mordstein, M.; Lienenklaus, S.; García-Sastre, A.; Stäheli, P.; Kochs, G. Glycine 184 in Nonstructural Protein NS1 Determines the Virulence of Influenza A Virus Strain PR8 without Affecting the Host Interferon Response. J. Virol. 2010, 84, 12761–12770. [Google Scholar] [CrossRef] [PubMed]
- Forbes, N.; Selman, M.; Pelchat, M.; Jia, J.J.; Stintzi, A.; Brown, E.G. Identification of Adaptive Mutations in the Influenza A Virus Non-Structural 1 Gene That Increase Cytoplasmic Localization and Differentially Regulate Host Gene Expression. PLoS ONE 2013, 8, e84673. [Google Scholar] [CrossRef] [PubMed]
- Forbes, N.E.; Ping, J.; Dankar, S.K.; Jia, J.-J.; Selman, M.; Keleta, L.; Zhou, Y.; Brown, E.G. Multifunctional Adaptive NS1 Mutations Are Selected upon Human Influenza Virus Evolution in the Mouse. PLoS ONE 2012, 7, e31839. [Google Scholar] [CrossRef] [PubMed]
- Cheong, W.-C.; Kang, H.-R.; Yoon, H.; Kang, S.-J.; Ting, J.P.-Y.; Song, M.J. Influenza A Virus NS1 Protein Inhibits the NLRP3 Inflammasome. PLoS ONE 2015, 10, e0126456. [Google Scholar] [CrossRef] [PubMed]
- Park, H.-S.; Liu, G.; Raman, S.N.T.; Landreth, S.L.; Liu, Q.; Zhou, Y. NS1 Protein of 2009 Pandemic Influenza A Virus Inhibits Porcine NLRP3 Inflammasome-Mediated Interleukin-1 Beta Production by Suppressing ASC Ubiquitination. J. Virol. 2018, 92, e00022-18. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Li, Y.; Krug, R.M. Influenza A virus NS1 protein targetspoly(A)-binding protein II of the cellular 3′-end processing machinery. EMBO J. 1999, 18, 2273–2283. [Google Scholar] [CrossRef] [PubMed]
- Satterly, N.; Tsai, P.-L.; van Deursen, J.; Nussenzveig, D.R.; Wang, Y.; Faria, P.A.; Levay, A.; Levy, D.E.; Fontoura, B.M.A. Influenza virus targets the mRNA export machinery and the nuclear pore complex. Proc. Natl. Acad. Sci. USA 2007, 104, 1853–1858. [Google Scholar] [CrossRef] [PubMed]
- Kuss, S.K.; Mata, M.A.; Zhang, L.; Fontoura, B.M.A. Nuclear Imprisonment: Viral Strategies to Arrest Host mRNA Nuclear Export. Viruses 2013, 5, 1824–1849. [Google Scholar] [CrossRef] [PubMed]
- Panthu, B.; Terrier, O.; Carron, C.; Traversier, A.; Corbin, A.; Balvay, L.; Lina, B.; Rosa-Calatrava, M.; Ohlmann, T. The NS1 Protein from Influenza Virus Stimulates Translation Initiation by Enhancing Ribosome Recruitment to mRNAs. J. Mol. Biol. 2017, 429, 3334–3352. [Google Scholar] [CrossRef] [PubMed]
- Falcón, A.M.; Fortes, P.; Marión, R.M.; Beloso, A.; Ortín, J. Interaction of influenza virus NS1 protein and the human homologue of Staufen in vivo and in vitro. Nucleic Acids Res. 1999, 27, 2241–2247. [Google Scholar] [CrossRef] [PubMed]
- Ehrenfeld, E. Poliovirus-induced inhibition of host-cell protein synthesis. Cell 1982, 28, 435–436. [Google Scholar] [CrossRef]
- Garfinkel, M.S.; Katze, M.G. Translational control by influenza virus. Selective and cap-dependent translation of viral mRNAs in infected cells. J. Biol. Chem. 1992, 267, 9383–9390. [Google Scholar] [PubMed]
- Garfinkel, M.S.; Katze, M.G. Translational control by influenza virus. Selective translation is mediated by sequences within the viral mRNA 5’-untranslated region. J. Biol. Chem. 1993, 268, 22223–22226. [Google Scholar] [PubMed]
- Park, Y.W.; Katze, M.G. Translational control by influenza virus. Identification of cis-acting sequences and trans-acting factors which may regulate selective viral mRNA translation. J. Biol. Chem. 1995, 270, 28433–28439. [Google Scholar] [PubMed]
- Burgui, I.; Yángüez, E.; Sonenberg, N.; Nieto, A. Influenza Virus mRNA Translation Revisited: Is the eIF4E Cap-Binding Factor Required for Viral mRNA Translation? J. Virol. 2007, 81, 12427–12438. [Google Scholar] [CrossRef] [PubMed]
- Feigenblum, D.; Schneider, R.J. Modification of eukaryotic initiation factor 4F during infection by influenza virus. J. Virol. 1993, 67, 3027–3035. [Google Scholar] [PubMed]
- Yángüez, E.; Castello, A.; Welnowska, E.; Carrasco, L.; Goodfellow, I.; Nieto, A. Functional impairment of eIF4A and eIF4G factors correlates with inhibition of influenza virus mRNA translation. Virology 2011, 413, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Yángüez, E.; Rodriguez, P.; Goodfellow, I.; Nieto, A. Influenza virus polymerase confers independence of the cellular cap-binding factor eIF4E for viral mRNA translation. Virology 2012, 422, 297–307. [Google Scholar] [CrossRef] [PubMed]
- De la Luna, S.; Fortes, P.; Beloso, A.; Ortín, J. Influenza virus NS1 protein enhances the rate of translation initiation of viral mRNAs. J. Virol. 1995, 69, 2427–2433. [Google Scholar] [PubMed]
- Enami, K.; Sato, T.A.; Nakada, S.; Enami, M. Influenza virus NS1 protein stimulates translation of the M1 protein. J. Virol. 1994, 68, 1432–1437. [Google Scholar] [PubMed]
- Aragón, T.; de la Luna, S.; Novoa, I.; Carrasco, L.; Ortín, J.; Nieto, A. Eukaryotic Translation Initiation Factor 4GI is a Cellular Target for NS1 Protein, a Translational Activator of Influenza Virus. Mol. Cell. Biol. 2000, 20, 6259–6268. [Google Scholar] [CrossRef] [PubMed]
- Burgui, I.; Aragón, T.; Ortín, J.; Nieto, A. PABP1 and eIF4GI associate with influenza virus NS1 protein in viral mRNA translation initiation complexes. J. Gen. Virol. 2003, 84, 3263–3274. [Google Scholar] [CrossRef] [PubMed]
- Arias-Mireles, B.H.; de Rozieres, C.M.; Ly, K.; Joseph, S. RNA Modulates the Interaction between Influenza A Virus NS1 and Human PABP1. Biochemistry 2018, 57, 3590–3598. [Google Scholar] [CrossRef] [PubMed]
- Grimm, D.; Staeheli, P.; Hufbauer, M.; Koerner, I.; Martínez-Sobrido, L.; Solórzano, A.; García-Sastre, A.; Haller, O.; Kochs, G. Replication fitness determines high virulence of influenza A virus in mice carrying functional Mx1 resistance gene. Proc. Natl. Acad. Sci. USA 2007, 104, 6806–6811. [Google Scholar] [CrossRef] [PubMed]
- Rolling, T.; Koerner, I.; Zimmermann, P.; Holz, K.; Haller, O.; Staeheli, P.; Kochs, G. Adaptive Mutations Resulting in Enhanced Polymerase Activity Contribute to High Virulence of Influenza A Virus in Mice. J. Virol. 2009, 83, 6673–6680. [Google Scholar] [CrossRef] [PubMed]
- Talon, J.; Horvath, C.M.; Polley, R.; Basler, C.F.; Muster, T.; Palese, P.; García-Sastre, A. Activation of Interferon Regulatory Factor 3 is Inhibited by the Influenza A Virus NS1 Protein. J. Virol. 2000, 74, 7989–7996. [Google Scholar] [CrossRef] [PubMed]
- Kuo, R.-L.; Krug, R.M. Influenza A Virus Polymerase Is an Integral Component of the CPSF30-NS1A Protein Complex in Infected Cells. J. Virol. 2009, 83, 1611–1616. [Google Scholar] [CrossRef] [PubMed]
- Spesock, A.; Malur, M.; Hossain, M.J.; Chen, L.-M.; Njaa, B.L.; Davis, C.T.; Lipatov, A.S.; York, I.A.; Krug, R.M.; Donis, R.O. The Virulence of 1997 H5N1 Influenza Viruses in the Mouse Model is Increased by Correcting a Defect in Their NS1 Proteins. J. Virol. 2011, 85, 7048–7058. [Google Scholar] [CrossRef] [PubMed]
- Chauché, C.; Nogales, A.; Zhu, H.; Goldfarb, D.; Shanizza, A.I.A.; Gu, Q.; Parrish, C.R.; Martínez-Sobrido, L.; Marshall, J.F.; Murcia, P.R. Mammalian Adaptation of an Avian Influenza A Virus Involves Stepwise Changes in NS1. J. Virol. 2018, 92, e01875-17. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Mo, Y.; Wang, X.; Gu, M.; Hu, Z.; Zhong, L.; Wu, Q.; Hao, X.; Hu, S.; Liu, W.; et al. PA-X decreases the pathogenicity of highly pathogenic H5N1 influenza A virus in avian species by inhibiting virus replication and host response. J. Virol. 2015, 89, 4126–4142. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Mo, Y.; Gao, Z.; Wang, X.; Gu, M.; Liang, Y.; Cheng, X.; Hu, S.; Liu, W.; Liu, H.; et al. PA-X-associated early alleviation of the acute lung injury contributes to the attenuation of a highly pathogenic H5N1 avian influenza virus in mice. Med. Microbiol. Immunol. 2016, 205, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Xu, G.; Sun, Y.; Qi, L.; Wang, J.; Kong, W.; Sun, H.; Pu, J.; Chang, K.-C.; Liu, J. PA-X is a virulence factor in avian H9N2 influenza virus. J. Gen. Virol. 2015, 96, 2587–2594. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.-Q.; Sun, Y.-F.; Ruan, B.-Y.; Liu, X.-M.; Wang, Q.; Yang, H.-M.; Wang, S.-Y.; Zhang, P.; Wang, X.-H.; Shan, T.-L.; et al. PA-X protein decreases replication and pathogenicity of swine influenza virus in cultured cells and mouse models. Vet. Microbiol. 2017, 205, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Zhang, X.; Liu, Q.; Bing, G.; Hu, Z.; Sun, H.; Xiong, X.; Jiang, M.; He, Q.; Wang, Y.; et al. PA-X protein contributes to virulence of triple-reassortant H1N2 influenza virus by suppressing early immune responses in swine. Virology 2017, 508, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Yu, H.; Li, Y.; Ma, J.; Lang, Y.; Duff, M.; Henningson, J.; Liu, Q.; Li, Y.; Nagy, A.; et al. Impacts of different expressions of PA-X protein on 2009 pandemic H1N1 virus replication, pathogenicity and host immune responses. Virology 2017, 504, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Dauber, B.; Saffran, H.A.; Smiley, J.R. The herpes simplex virus 1 virion host shutoff protein enhances translation of viral late mRNAs by preventing mRNA overload. J. Virol. 2014, 88, 9624–9632. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.F.; Neumann, G.; Kawaoka, Y. Orthomyxoviruses. In Fields Virology, 6th ed.; David, K., Howley, M., Peter, M., Eds.; Lippincott Wiliams & Wilkins: Philadelphia, PA, USA, 2013; Volume 1, pp. 1186–1243. ISBN 978-1-4511-0563-6. [Google Scholar]
- Nogales, A.; Rodriguez, L.; DeDiego, M.L.; Topham, D.J.; Martínez-Sobrido, L. Interplay of PA-X and NS1 Proteins in Replication and Pathogenesis of a Temperature-Sensitive 2009 Pandemic H1N1 Influenza A Virus. J. Virol. 2017, 91, e00720-17. [Google Scholar] [CrossRef] [PubMed]
- Khaperskyy, D.A.; McCormick, C. Timing Is Everything: Coordinated Control of Host Shutoff by Influenza A Virus NS1 and PA-X Proteins. J. Virol. 2015, 89, 6528–6531. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Host Shutoff Protein | Residue | Strong Activity | Weak Activity | Available Information on Function | Reference |
|---|---|---|---|---|---|
| PA-X | 2 | E | na | Needs to be acetylated by NatB for full activity | [36] |
| 80 | E | na | Residues required for nuclease activity of PA/PA-X and also shown experimentally to affect shutoff activity | [10,11,12,24,32] | |
| 106 | L | na | |||
| 107 | P | na | |||
| 108 | D | na | |||
| 119 | E | na | |||
| 134 | K | na | |||
| 192–197 (aa 0-15 of X-ORF) | (varies depending on strain) | na | Required for nuclear localization and activity (WSN, Cal, PR8) | [19,22,29] | |
| 195, 198, 199, 202, 203, 206 | R/K | na | Mutation to A or E prevents nuclear import and activity (WSN, Cal, PR8) | [19,22,29] | |
| 100 | V | I, A | Changes that arose in pH1N1 and reduce PA-X activity (“weak” variants also found in WSN for 100 and 221) | [18,23] | |
| 204 | N | S | |||
| 221 | R | Q | |||
| 229 | L | S | |||
| 57 | R | Q | Amino acid differences responsible for higher shutoff activity of pH1N1 vs. WSN | [18] | |
| 62 | I | V | |||
| 65 | S | L | |||
| 4 | F | na | Important for PA-X shutoff activity, potentially by allowing nuclear import (WSN) | [32] | |
| 9 | F | na | |||
| 27 | D | na | |||
| 39 | C | na | |||
| 123 | T | na | |||
| 124 | R | na | |||
| 125 | R | na | |||
| 24 | Y | na | Important for PA-X shutoff activity presumably by structurally supporting nuclease site (WSN) | [32] | |
| 45 | C | na | |||
| 87 | A | na | |||
| 94 | I | na | |||
| 120 | I | na | |||
| 163 | L | na | |||
| 171 | I | na | |||
| 27 | D | N | Changes that increase shutoff activity of equine H3N8 PA-X in conjunction with lengthening of X-ORF isoform | [34] | |
| 231 | S | F | |||
| PA (RdRp) | 550 | L | I | Required for RdRp to direct Pol II degradation | [48] |
| PB2 (RdRp) | 504 | V | I |
| Host Shutoff Protein | Residue | Strong Activity | Weak Activity | Available Information on Function | Reference |
|---|---|---|---|---|---|
| NS1 | 103 | F | L | F103/M106 confer CPSF30 binding in many strains (not sufficient in pH1N1, canine H3N8) | [62,65] |
| 106 | M | I, V | |||
| 144 | L | na | Required for CPSF30 binding (Udorn, WSN) | [61] | |
| aa 184–188 | GLEWN | na | Required for CPSF30 binding in vitro and in cells (Udorn); K186 (instead of E186) in canine H3N8 prevents CPSF30 binding | [62,63,66,67,73] | |
| aa 223–237 | ARTARSKVRRDKMAD | na | Required for PABPN (PABII) binding (Udorn) | [63] | |
| 55 | K | E | Changes to “strong” restore strong CPSF30 binding in pH1N1 strains (with F103, M106); D189N also alters host shutoff activity in circulating H3N2 strains and 1918 H1N1; D125G appeared in mouse adaptation of H3N2 human strain | [52,68,70,74,75] | |
| 90 | I | L | |||
| 108 | K | R | |||
| 123 | V | I | |||
| 125 | D | E, G | |||
| 131 | E | K | |||
| 189 | D | G, N | |||
| 205 | S | N | |||
| 64 | I | T | Changes to “weak” in circulating H3N2 strains reduce CPSF30 binding and IFN antagonism; V194I also reduces CPSF30 binding and host shutoff activity in 1918 H1N1 NS1 | [69,70] | |
| 194 | V | I | |||
| 98 | L | S | Mutations that abolish CPSF30 binding during mouse adaptation of H3N2 strain | [74,75] | |
| 180 | V | A | |||
| 96 | E | na | Mutation causes temperature sensitive mRNA cleavage phenotype (Udorn) | [72] | |
| aa 191–195 | EALQR | deleted | Deletion in H5N1 NS1 reduces CPSF30 binding and IFN antagonism | [71] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Levene, R.E.; Gaglia, M.M. Host Shutoff in Influenza A Virus: Many Means to an End. Viruses 2018, 10, 475. https://doi.org/10.3390/v10090475
Levene RE, Gaglia MM. Host Shutoff in Influenza A Virus: Many Means to an End. Viruses. 2018; 10(9):475. https://doi.org/10.3390/v10090475
Chicago/Turabian StyleLevene, Rachel Emily, and Marta Maria Gaglia. 2018. "Host Shutoff in Influenza A Virus: Many Means to an End" Viruses 10, no. 9: 475. https://doi.org/10.3390/v10090475
APA StyleLevene, R. E., & Gaglia, M. M. (2018). Host Shutoff in Influenza A Virus: Many Means to an End. Viruses, 10(9), 475. https://doi.org/10.3390/v10090475