Molecular Determinants and the Regulation of Human Cytomegalovirus Latency and Reactivation


Abstract

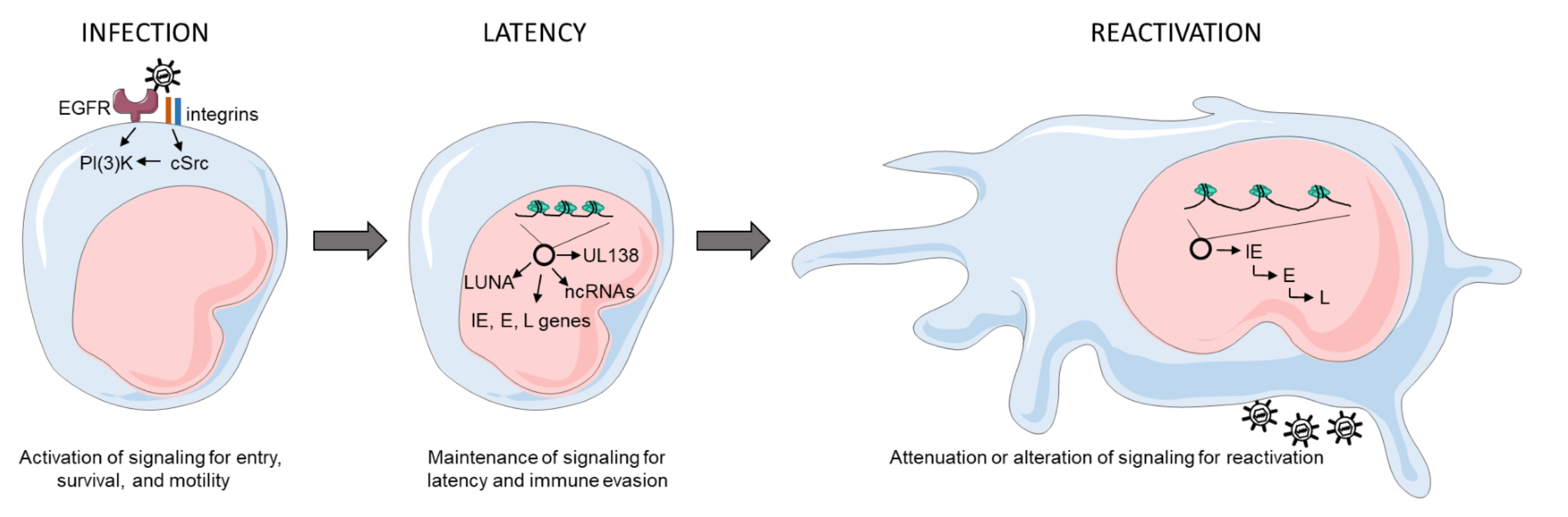{kind=link}
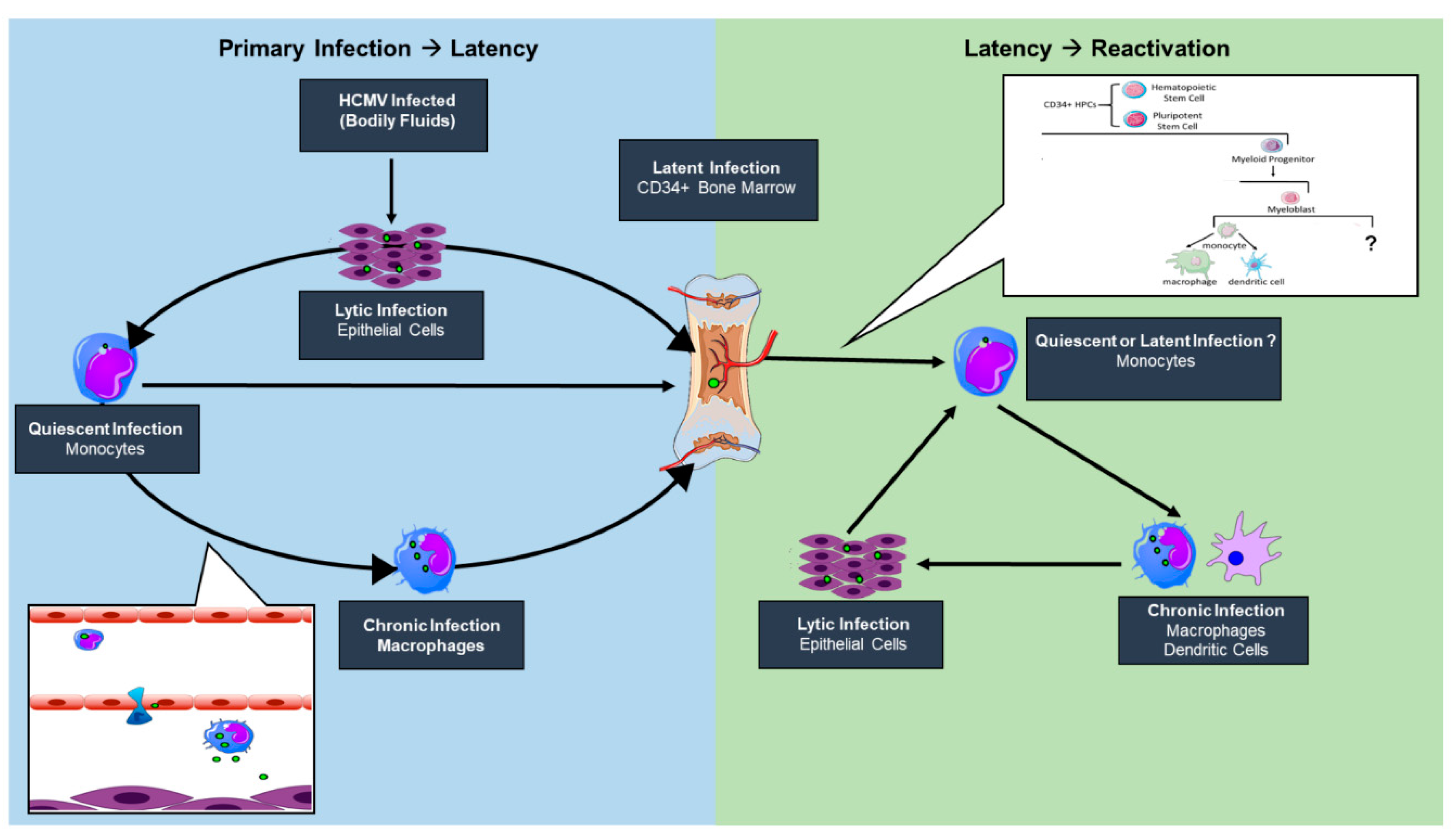{kind=link}
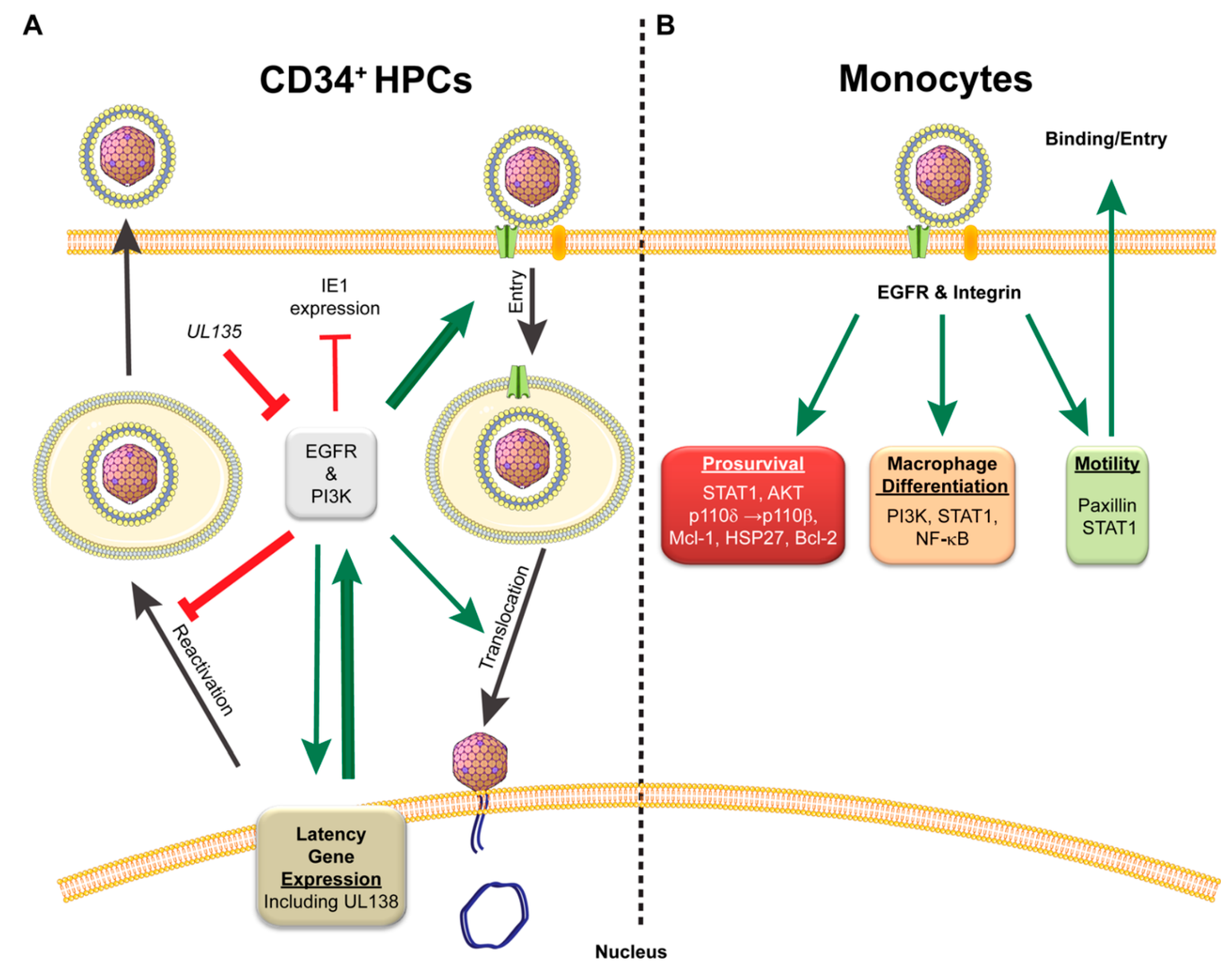{kind=link}
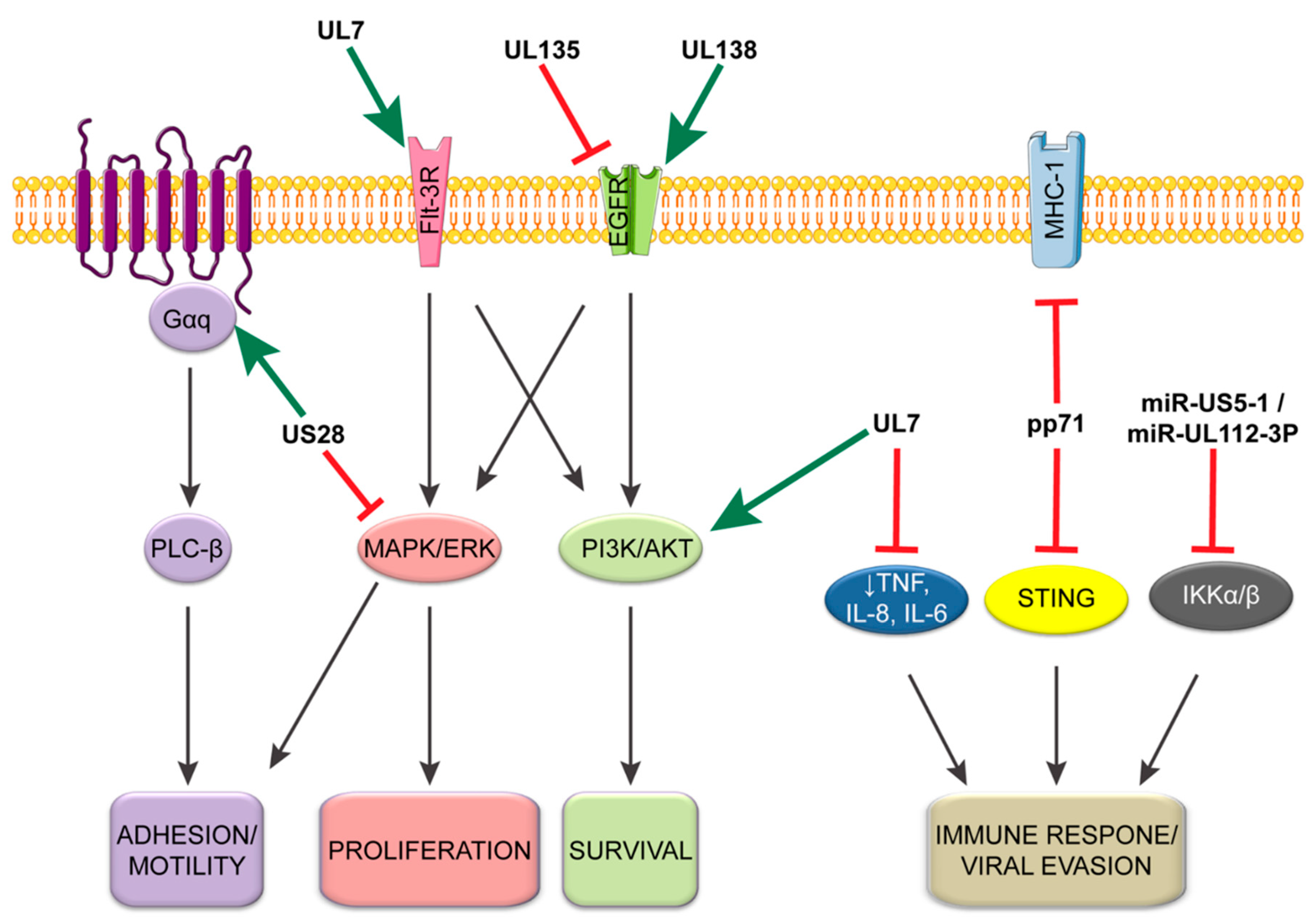{kind=link}
{kind=link}
1. Introduction
2. HCMV Latency Reservoirs
3. HCMV Latency Models
3.1. Primary Progenitor Cells
3.2. Primary Monocytes
3.3. Cell Lines
3.4. Animal Models
4. CMV-Mediated Control of Host Signaling for Latency/Reactivation
4.1. CMV Initiation of EGFR and Integrin Signaling: Starting off on the Right Receptor
4.2. CMV Manipulation of the Cellular Environment for Establishment and Maintenance of Latent Population
5. Cellular Control of the Viral Gene Expression Program for Latency/Reactivation
5.1. Cellular Transcription Factors
5.2. Chromatinization
5.3. Viral Factors and Chromatinization
5.4. Viral Gene Expression during Latency
6. Final Thoughts
Funding
Conflicts of Interest
References
- Arvin, A.M.; Fast, P.; Myers, M.; Plotkin, S.; Rabinovich, R. Vaccine development to prevent cytomegalovirus disease: Report from the national vaccine advisory committee. Clin. Infect. Dis. 2004, 39, 233–239. [Google Scholar] [PubMed]
- Cannon, M.J.; Schmid, D.S.; Hyde, T.B. Review of cytomegalovirus seroprevalence and demographic characteristics associated with infection. Rev. Med. Virol. 2010, 20, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Nogalski, M.T.; Collins-McMillen, D.; Yurochko, A.D. Overview of human cytomegalovirus pathogenesis. In Human Cytomegaloviruses: Methods and Protocols; Yurochko, A.D., Miller, W.E., Eds.; Humana Press: New York, NY, USA, 2014; pp. 15–28. [Google Scholar]
- Christensen-Quick, A.; Vanpouille, C.; Lisco, A.; Gianella, S. Cytomegalovirus and HIV persistence: Pouring gas on the fire. AIDS Res. Hum. Retroviruses 2017, 33, S23–S30. [Google Scholar] [CrossRef] [PubMed]
- Schlick, K.; Grundbichler, M.; Auberger, J.; Kern, J.M.; Hell, M.; Hohla, F.; Hopfinger, G.; Greil, R. Cytomegalovirus reactivation and its clinical impact in patients with solid tumors. Infect. Agent. Cancer 2015, 10, 45. [Google Scholar] [CrossRef] [PubMed]
- Ramanan, P.; Razonable, R.R. Cytomegalovirus infections in solid organ transplantation: A review. Infect. Chemother. 2013, 45, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Ljungman, P. The role of cytomegalovirus serostatus on outcome of hematopoietic stem cell transplantation. Curr. Opin. Hematol. 2014, 21, 466–469. [Google Scholar] [CrossRef] [PubMed]
- Bhat, V.; Joshi, A.; Sarode, R.; Chavan, P. Cytomegalovirus infection in the bone marrow transplant patient. World J. Transplant. 2015, 5, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Ljungman, P.; Griffiths, P.; Paya, C. Definitions of cytomegalovirus infection and disease in transplant recipients. Clin. Infect. Dis. 2002, 34, 1094–1097. [Google Scholar] [CrossRef] [PubMed]
- Thackeray, R.; Wright, A.; Chipman, K. Congenital cytomegalovirus reference material: A content analysis of coverage and accuracy. Matern. Child Health J. 2014, 18, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Styczynski, J. Who is the patient at risk of CMV recurrence: A review of the current scientific evidence with a focus on hematopoietic cell transplantation. Infect. Dis. Ther. 2018, 7, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Lanzieri, T.M.; Dollard, S.C.; Bialek, S.R.; Grosse, S.D. Systematic review of the birth prevalence of congenital cytomegalovirus infection in developing countries. Int. J. Infect. Dis. 2014, 22, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Adam, E.; Melnick, J.L.; DeBakey, M.E. Cytomegalovirus infection and atherosclerosis. Cent. Eur. J. Public Health 1997, 5, 99–106. [Google Scholar] [PubMed]
- Streblow, D.N.; Orloff, S.L.; Nelson, J.A. Do pathogens accelerate atherosclerosis? J. Nutr. 2001, 131, 2798S–2804S. [Google Scholar] [CrossRef] [PubMed]
- Caposio, P.; Orloff, S.L.; Streblow, D.N. The role of cytomegalovirus in angiogenesis. Virus Res. 2011, 157, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Streblow, D.N.; Dumortier, J.; Moses, A.V.; Orloff, S.L.; Nelson, J.A. Mechanisms of cytomegalovirus-accelerated vascular disease: Induction of paracrine factors that promote angiogenesis and wound healing. Curr. Top Microbiol. Immunol. 2008, 325, 397–415. [Google Scholar] [PubMed]
- Sylwester, A.W.; Mitchell, B.L.; Edgar, J.B.; Taormina, C.; Pelte, C.; Ruchti, F.; Sleath, P.R.; Grabstein, K.H.; Hosken, N.A.; Kern, F.; et al. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ t cells dominate the memory compartments of exposed subjects. J. Exp. Med. 2005, 202, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Pawelec, G. Immunosenenescence: Role of cytomegalovirus. Exp. Gerontol. 2014, 54, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Schmaltz, H.N.; Fried, L.P.; Xue, Q.L.; Walston, J.; Leng, S.X.; Semba, R.D. Chronic cytomegalovirus infection and inflammation are associated with prevalent frailty in community-dwelling older women. J. Am. Geriatr. Soc. 2005, 53, 747–754. [Google Scholar] [CrossRef] [PubMed]
- High, K.P. Chronic infection and frailty: Surrogate markers, associations, and causality. J. Am. Geriatr. Soc. 2005, 53, 906–908. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.C.; Kao, W.H.; Murakami, P.; Xue, Q.L.; Chiou, R.B.; Detrick, B.; McDyer, J.F.; Semba, R.D.; Casolaro, V.; Walston, J.D.; et al. Cytomegalovirus infection and the risk of mortality and frailty in older women: A prospective observational cohort study. Am. J. Epidemiol. 2010, 171, 1144–1152. [Google Scholar] [CrossRef] [PubMed]
- Sinzger, C.; Digel, M.; Jahn, G. Cytomegalovirus cell tropism. Curr. Top. Microbiol. Immunol. 2008, 325, 63–83. [Google Scholar] [PubMed]
- Bentz, G.L.; Jarquin-Pardo, M.; Chan, G.; Smith, M.S.; Sinzger, C.; Yurochko, A.D. Human cytomegalovirus (HCMV) infection of endothelial cells promotes naive monocyte extravasation and transfer of productive virus to enhance hematogenous dissemination of HCMV. J. Virol. 2006, 80, 11539–11555. [Google Scholar] [CrossRef] [PubMed]
- Goodrum, F.D.; Jordan, C.T.; High, K.; Shenk, T. Human cytomegalovirus gene expression during infection of primary hematopoietic progenitor cells: A model for latency. Proc. Natl. Acad. Sci. USA 2002, 99, 16255–16260. [Google Scholar] [CrossRef] [PubMed]
- Mendelson, M.; Monard, S.; Sissons, P.; Sinclair, J. Detection of endogenous human cytomegalovirus in CD34+ bone marrow progenitors. J. Gen. Virol. 1996, 77, 3099–3102. [Google Scholar] [CrossRef] [PubMed]
- Hargett, D.; Shenk, T. Experimental human cytomegalovirus latency in CD14+ monocytes. Proc. Natl. Acad. Sci. USA 2010, 107, 20039–20044. [Google Scholar] [CrossRef] [PubMed]
- Kondo, K.; Kaneshima, H.; Mocarski, E.S. Human cytomegalovirus latent infection of granulocyte-macrophage progenitors. Proc. Natl. Acad. Sci. USA 1994, 91, 11879–11883. [Google Scholar] [CrossRef] [PubMed]
- Taylor-Wiedeman, J.; Sissons, J.G.; Borysiewicz, L.K.; Sinclair, J. Monocytes are a major site of persistence of human cytomegalovirus in peripheral blood mononuclear cells. J. Gen. Virol. 1991, 72, 2059–2064. [Google Scholar] [CrossRef] [PubMed]
- Zhuravskaya, T.; Maciejewski, J.P.; Netski, D.M.; Bruening, E.; Mackintosh, F.R.; St Jeor, S. Spread of human cytomegalovirus (HCMV) after infection of human hematopoietic progenitor cells: Model of HCMV latency. Blood 1997, 90, 2482–2491. [Google Scholar] [PubMed]
- Hahn, G.; Jores, R.; Mocarski, E.S. Cytomegalovirus remains latent in a common precursor of dendritic and myeloid cells. Proc. Natl. Acad. Sci. USA 1998, 95, 3937–3942. [Google Scholar] [CrossRef] [PubMed]
- Sindre, H.; Tjoonnfjord, G.E.; Rollag, H.; Ranneberg-Nilsen, T.; Veiby, O.P.; Beck, S.; Degre, M.; Hestdal, K. Human cytomegalovirus suppression of and latency in early hematopoietic progenitor cells. Blood 1996, 88, 4526–4533. [Google Scholar] [PubMed]
- Ibanez, C.E.; Schrier, R.; Ghazal, P.; Wiley, C.; Nelson, J.A. Human cytomegalovirus productively infects primary differentiated macrophages. J. Virol. 1991, 65, 6581–6588. [Google Scholar] [PubMed]
- Reeves, M.B.; Sinclair, J.H. Circulating dendritic cells isolated from healthy seropositive donors are sites of human cytomegalovirus reactivation in vivo. J. Virol. 2013, 87, 10660–10667. [Google Scholar] [CrossRef] [PubMed]
- Soderberg-Naucler, C.; Fish, K.N.; Nelson, J.A. Reactivation of latent human cytomegalovirus by allogeneic stimulation of blood cells from healthy donors. Cell 1997, 91, 119–126. [Google Scholar] [CrossRef]
- Soderberg-Naucler, C.; Streblow, D.N.; Fish, K.N.; Allan-Yorke, J.; Smith, P.P.; Nelson, J.A. Reactivation of latent human cytomegalovirus in CD14(+) monocytes is differentiation dependent. J. Virol. 2001, 75, 7543–7554. [Google Scholar] [CrossRef] [PubMed]
- Taylor-Wiedeman, J.; Sissons, P.; Sinclair, J. Induction of endogenous human cytomegalovirus gene expression after differentiation of monocytes from healthy carriers. J. Virol. 1994, 68, 1597–1604. [Google Scholar] [PubMed]
- Mocarski, E.S.; Courcelle, C.T. Cytomegaloviruses and their replication. In Fields Virology, 4th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2001; Volume 2, pp. 2629–2673. [Google Scholar]
- Cannon, M.J.; Hyde, T.B.; Schmid, D.S. Review of cytomegalovirus shedding in bodily fluids and relevance to congenital cytomegalovirus infection. Rev. Med. Virol. 2011, 21, 240–255. [Google Scholar] [CrossRef] [PubMed]
- Halwachs-Baumann, G.; Genser, B.; Pailer, S.; Engele, H.; Rosegger, H.; Schalk, A.; Kessler, H.H.; Truschnig-Wilders, M. Human cytomegalovirus load in various body fluids of congenitally infected newborns. J. Clin. Virol. 2002, 25 (Suppl. 3), S81–S87. [Google Scholar] [CrossRef]
- Gianella, S.; Massanella, M.; Richman, D.D.; Little, S.J.; Spina, C.A.; Vargas, M.V.; Lada, S.M.; Daar, E.S.; Dube, M.P.; Haubrich, R.H.; et al. Cytomegalovirus replication in semen is associated with higher levels of proviral HIV DNA and CD4+ T cell activation during antiretroviral treatment. J. Virol. 2014, 88, 7818–7827. [Google Scholar] [CrossRef] [PubMed]
- Cojohari, O.; Peppenelli, M.A.; Chan, G.C. Human cytomegalovirus induces an atypical activation of Akt to stimulate the survival of short-lived monocytes. J. Virol. 2016, 90, 6443–6452. [Google Scholar] [CrossRef] [PubMed]
- Peppenelli, M.A.; Arend, K.C.; Cojohari, O.; Moorman, N.J.; Chan, G.C. Human cytomegalovirus stimulates the synthesis of select Akt-dependent antiapoptotic proteins during viral entry to promote survival of infected monocytes. J. Virol. 2016, 90, 3138–3147. [Google Scholar] [CrossRef] [PubMed]
- Chan, G.; Bivins-Smith, E.R.; Smith, M.S.; Smith, P.M.; Yurochko, A.D. Transcriptome analysis reveals human cytomegalovirus reprograms monocyte differentiation toward an m1 macrophage. J. Immunol. 2008, 181, 698–711. [Google Scholar] [CrossRef] [PubMed]
- Chan, G.; Bivins-Smith, E.R.; Smith, M.S.; Yurochko, A.D. NF-kappaB and phosphatidylinositol 3-kinase activity mediates the HCMV-induced atypical M1/M2 polarization of monocytes. Virus Res. 2009, 144, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Chan, G.; Nogalski, M.T.; Stevenson, E.V.; Yurochko, A.D. Human cytomegalovirus induction of a unique signalsome during viral entry into monocytes mediates distinct functional changes: A strategy for viral dissemination. J. Leukoc. Biol. 2012, 92, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Chan, G.; Nogalski, M.T.; Yurochko, A.D. Human cytomegalovirus stimulates monocyte-to-macrophage differentiation via the temporal regulation of caspase 3. J. Virol. 2012, 86, 10714–10723. [Google Scholar] [CrossRef] [PubMed]
- Yurochko, A.D. Human cytomegalovirus modulation of signal transduction. Curr. Top. Microbiol. Immunol. 2008, 325, 205–220. [Google Scholar] [PubMed]
- Smith, M.S.; Bivins-Smith, E.R.; Tilley, A.M.; Bentz, G.L.; Chan, G.; Minard, J.; Yurochko, A.D. Roles of phosphatidylinositol 3-kinase and NF-kappaB in human cytomegalovirus-mediated monocyte diapedesis and adhesion: Strategy for viral persistence. J. Virol. 2007, 81, 7683–7694. [Google Scholar] [CrossRef] [PubMed]
- Reeves, M.B.; Sinclair, J.H. Analysis of latent viral gene expression in natural and experimental latency models of human cytomegalovirus and its correlation with histone modifications at a latent promoter. J. Gen. Virol. 2010, 91, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Goodrum, F.; Caviness, K.; Zagallo, P. Human cytomegalovirus persistence. Cell. Microbiol. 2012, 14, 644–655. [Google Scholar] [CrossRef] [PubMed]
- Buehler, J.; Zeltzer, S.; Reitsma, J.; Petrucelli, A.; Umashankar, M.; Rak, M.; Zagallo, P.; Schroeder, J.; Terhune, S.; Goodrum, F. Opposing regulation of the EGF receptor: A molecular switch controlling cytomegalovirus latency and replication. PLoS Pathog. 2016, 12, e1005655. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Collins-McMillen, D.; Buehler, J.C.; Goodrum, F.D.; Yurochko, A.D. Human cytomegalovirus requires epidermal growth factor receptor signaling to enter and initiate the early steps in the establishment of latency in CD34+ human progenitor cells. J. Virol. 2017, 91, e01206. [Google Scholar] [CrossRef] [PubMed]
- Goodrum, F. Human cytomegalovirus latency: Approaching the gordian knot. Annu. Rev. Virol. 2016, 3, 333–357. [Google Scholar] [CrossRef] [PubMed]
- Poole, E.; Avdic, S.; Hodkinson, J.; Jackson, S.; Wills, M.; Slobedman, B.; Sinclair, J. Latency-associated viral interleukin-10 (IL-10) encoded by human cytomegalovirus modulates cellular IL-10 and CCL8 secretion during latent infection through changes in the cellular microRNA hsa-miR-92a. J. Virol. 2014, 88, 13947–13955. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Pan, C.; Sheng, J.; Liang, H.; Bian, Z.; Liu, Y.; Trang, P.; Wu, J. Human cytomegalovirus reprogrammes haematopoietic progenitor cells into immunosuppressive monocytes to achieve latency. Nat. Microbiol. 2018, 3, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Reeves, M.B.; Lehner, P.J.; Sissons, J.G.; Sinclair, J.H. An in vitro model for the regulation of human cytomegalovirus latency and reactivation in dendritic cells by chromatin remodelling. J. Gen. Virol. 2005, 86, 2949–2954. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.; Soderberg-Naucler, C.; Wang, F.Z.; Moller, E. Cytomegalovirus DNA can be detected in peripheral blood mononuclear cells from all seropositive and most seronegative healthy blood donors over time. Transfusion 1998, 38, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Miller, P.H.; Cheung, A.M.; Beer, P.A.; Knapp, D.J.; Dhillon, K.; Rabu, G.; Rostamirad, S.; Humphries, R.K.; Eaves, C.J. Enhanced normal short-term human myelopoiesis in mice engineered to express human-specific myeloid growth factors. Blood 2013, 121, e1-4. [Google Scholar] [CrossRef] [PubMed]
- Hogge, D.E.; Lansdorp, P.M.; Reid, D.; Gerhard, B.; Eaves, C.J. Enhanced detection, maintenance, and differentiation of primitive human hematopoietic cells in cultures containing murine fibroblasts engineered to produce human steel factor, interleukin-3, and granulocyte colony-stimulating factor. Blood 1996, 88, 3765–3773. [Google Scholar] [PubMed]
- Miller, C.L.; Eaves, C.J. Long-term culture-initiating cell assays for human and murine cells. Methods Mol. Med. 2002, 63, 123–141. [Google Scholar] [PubMed]
- Umashankar, M.; Goodrum, F. Hematopoietic long-term culture (hLTC) for human cytomegalovirus latency and reactivation. Methods Mol. Biol. 2014, 1119, 99–112. [Google Scholar] [PubMed]
- Coronel, R.; Takayama, S.; Juwono, T.; Hertel, L. Dynamics of human cytomegalovirus infection in CD34+ hematopoietic cells and derived langerhans-type dendritic cells. J. Virol. 2015, 89, 5615–5632. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, C.M.; Vanicek, J.; Murphy, E.A. Host microRNA regulation of human cytomegalovirus immediate early protein translation promotes viral latency. J. Virol. 2014, 88, 5524–5532. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, C.C.; Tarrant-Elorza, M.; Pari, G.S. Cis and trans acting factors involved in human cytomegalovirus experimental and natural latent infection of CD14 (+) monocytes and CD34 (+) cells. PLoS Pathog. 2013, 9, e1003366. [Google Scholar] [CrossRef] [PubMed]
- Saffert, R.T.; Penkert, R.R.; Kalejta, R.F. Cellular and viral control over the initial events of human cytomegalovirus experimental latency in CD34+ cells. J. Virol. 2010, 84, 5594–5604. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.S.; Bentz, G.L.; Alexander, J.S.; Yurochko, A.D. Human cytomegalovirus induces monocyte differentiation and migration as a strategy for dissemination and persistence. J. Virol. 2004, 78, 4444–4453. [Google Scholar] [CrossRef] [PubMed]
- Chan, G.; Nogalski, M.T.; Bentz, G.L.; Smith, M.S.; Parmater, A.; Yurochko, A.D. PI3K-dependent upregulation of Mcl-1 by human cytomegalovirus is mediated by epidermal growth factor receptor and inhibits apoptosis in short-lived monocytes. J. Immunol. 2010, 184, 3213–3222. [Google Scholar] [CrossRef] [PubMed]
- Chan, G.; Nogalski, M.T.; Yurochko, A.D. Activation of EGFR on monocytes is required for human cytomegalovirus entry and mediates cellular motility. Proc. Natl. Acad. Sci. USA 2009, 106, 22369–22374. [Google Scholar] [CrossRef] [PubMed]
- Nogalski, M.T.; Chan, G.; Stevenson, E.V.; Gray, S.; Yurochko, A.D. Human cytomegalovirus-regulated paxillin in monocytes links cellular pathogenic motility to the process of viral entry. J. Virol. 2011, 85, 1360–1369. [Google Scholar] [CrossRef] [PubMed]
- Nogalski, M.T.; Chan, G.C.; Stevenson, E.V.; Collins-McMillen, D.K.; Yurochko, A.D. The HCMV gH/gL/UL128-131 complex triggers the specific cellular activation required for efficient viral internalization into target monocytes. PLoS Pathog. 2013, 9, e1003463. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.S.; Bentz, G.L.; Smith, P.M.; Bivens, E.R.; Yurochko, A.D. HCMV activates PI(3)K in monocytes and promotes monocyte motility and transendothelial migration in a PI(3)K-dependent manner. J. Leukoc. Biol. 2004, 76, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Arcangeletti, M.C.; Vasile Simone, R.; Rodighiero, I.; De Conto, F.; Medici, M.C.; Maccari, C.; Chezzi, C.; Calderaro, A. Human cytomegalovirus reactivation from latency: Validation of a “switch” model in vitro. Virol. J. 2016, 13, 179. [Google Scholar] [CrossRef] [PubMed]
- Wagenknecht, N.; Reuter, N.; Scherer, M.; Reichel, A.; Muller, R.; Stamminger, T. Contribution of the major ND10 proteins PML, hDaxx and Sp100 to the regulation of human cytomegalovirus latency and lytic replication in the monocytic cell line THP-1. Viruses 2015, 7, 2884–2907. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Lee, G.C.; Chan, Y.J.; Chiou, C.J.; Ahn, J.H.; Hayward, G.S. Factors affecting human cytomegalovirus gene expression in human monocyte cell lines. Mol. Cells 1999, 9, 37–44. [Google Scholar] [PubMed]
- Beisser, P.S.; Laurent, L.; Virelizier, J.L.; Michelson, S. Human cytomegalovirus chemokine receptor gene US28 is transcribed in latently infected thp-1 monocytes. J. Virol. 2001, 75, 5949–5957. [Google Scholar] [CrossRef] [PubMed]
- Yee, L.F.; Lin, P.L.; Stinski, M.F. Ectopic expression of HCMV IE72 and IE86 proteins is sufficient to induce early gene expression but not production of infectious virus in undifferentiated promonocytic THP-1 cells. Virology 2007, 363, 174–188. [Google Scholar] [CrossRef] [PubMed]
- Saffert, R.T.; Kalejta, R.F. Human cytomegalovirus gene expression is silenced by Daxx-mediated intrinsic immune defense in model latent infections established in vitro. J. Virol. 2007, 81, 9109–9120. [Google Scholar] [CrossRef] [PubMed]
- Murayama, T.; Ohara, Y.; Obuchi, M.; Khabar, K.S.; Higashi, H.; Mukaida, N.; Matsushima, K. Human cytomegalovirus induces interleukin-8 production by a human monocytic cell line, THP-1, through acting concurrently on AP-1- and NF-kappaB-binding sites of the interleukin-8 gene. J. Virol. 1997, 71, 5692–5695. [Google Scholar] [PubMed]
- Geist, L.J.; Monick, M.M.; Stinski, M.F.; Hunninghake, G.W. The immediate early genes of human cytomegalovirus upregulate tumor necrosis factor-alpha gene expression. J. Clin. Investig. 1994, 93, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Gan, X.; Wang, H.; Yu, Y.; Yi, W.; Zhu, S.; Li, E.; Liang, Y. Epigenetically repressing human cytomegalovirus lytic infection and reactivation from latency in THP-1 model by targeting H3K9 and H3K27 histone demethylases. PLoS ONE 2017, 12, e0175390. [Google Scholar] [CrossRef] [PubMed]
- Ioudinkova, E.; Arcangeletti, M.C.; Rynditch, A.; De Conto, F.; Motta, F.; Covan, S.; Pinardi, F.; Razin, S.V.; Chezzi, C. Control of human cytomegalovirus gene expression by differential histone modifications during lytic and latent infection of a monocytic cell line. Gene 2006, 384, 120–128. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, C.M.; Murphy, E.A. A myeloid progenitor cell line capable of supporting human cytomegalovirus latency and reactivation, resulting in infectious progeny. J. Virol. 2012, 86, 9854–9865. [Google Scholar] [CrossRef] [PubMed]
- Penkert, R.R.; Kalejta, R.F. Human embryonic stem cell lines model experimental human cytomegalovirus latency. mBio 2013, 4, e00298-13. [Google Scholar] [CrossRef] [PubMed]
- Poole, E.; Lau, J.C.; Sinclair, J. Latent infection of myeloid progenitors by human cytomegalovirus protects cells from FAS-mediated apoptosis through the cellular IL-10/PEA-15 pathway. J. Gen. Virol. 2015, 96, 2355–2359. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.A.; Gil, Y.; Panet, A.; Weisblum, Y.; Oiknine-Djian, E.; Gropp, M.; Steiner, D.; Reubinoff, B.E.; Wolf, D.G. Transition toward human cytomegalovirus susceptibility in early human embryonic stem cell-derived neural precursors. J. Virol. 2015, 89, 11159–11164. [Google Scholar] [CrossRef] [PubMed]
- Andrews, P.W. Retinoic acid induces neuronal differentiation of a cloned human embryonal carcinoma cell line in vitro. Dev. Biol. 1984, 103, 285–293. [Google Scholar] [CrossRef]
- Gonczol, E.; Andrews, P.W.; Plotkin, S.A. Cytomegalovirus replicates in differentiated but not in undifferentiated human embryonal carcinoma cells. Science 1984, 224, 159–161. [Google Scholar] [CrossRef] [PubMed]
- Odeberg, J.; Wolmer, N.; Falci, S.; Westgren, M.; Seiger, A.; Soderberg-Naucler, C. Human cytomegalovirus inhibits neuronal differentiation and induces apoptosis in human neural precursor cells. J. Virol. 2006, 80, 8929–8939. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.H.; Schwartz, P.H.; Fortunato, E.A. Neonatal neural progenitor cells and their neuronal and glial cell derivatives are fully permissive for human cytomegalovirus infection. J. Virol. 2008, 82, 9994–10007. [Google Scholar] [CrossRef] [PubMed]
- Belzile, J.P.; Stark, T.J.; Yeo, G.W.; Spector, D.H. Human cytomegalovirus infection of human embryonic stem cell-derived primitive neural stem cells is restricted at several steps but leads to the persistence of viral DNA. J. Virol. 2014, 88, 4021–4039. [Google Scholar] [CrossRef] [PubMed]
- Umashankar, M.; Petrucelli, A.; Cicchini, L.; Caposio, P.; Kreklywich, C.N.; Rak, M.; Bughio, F.; Goldman, D.C.; Hamlin, K.L.; Nelson, J.A.; et al. A novel human cytomegalovirus locus modulates cell type-specific outcomes of infection. PLoS Pathog. 2011, 7, e1002444. [Google Scholar] [CrossRef] [PubMed]
- Petrucelli, A.; Rak, M.; Grainger, L.; Goodrum, F. Characterization of a novel Golgi apparatus-localized latency determinant encoded by human cytomegalovirus. J. Virol. 2009, 83, 5615–5629. [Google Scholar] [CrossRef] [PubMed]
- Petrucelli, A.; Umashankar, M.; Zagallo, P.; Rak, M.; Goodrum, F. Interactions between proteins encoded within the human cytomegalovirus UL133-UL138 locus. J. Virol. 2012, 86, 8653–8662. [Google Scholar] [CrossRef] [PubMed]
- Caviness, K.; Bughio, F.; Crawford, L.B.; Streblow, D.N.; Nelson, J.A.; Caposio, P.; Goodrum, F. Complex interplay of the UL136 isoforms balances cytomegalovirus replication and latency. mBio 2016, 7, e01986. [Google Scholar] [CrossRef] [PubMed]
- Caviness, K.; Cicchini, L.; Rak, M.; Umashankar, M.; Goodrum, F.D. Complex expression of the UL136 gene of human cytomegalovirus results in multiple proteins isoforms with unique roles in replication. J. Virol. 2014, 88, 14412–14425. [Google Scholar] [CrossRef] [PubMed]
- Rak, M.; Buehler, J.; Zeltzer, S.; Reitsma, J.; Terhune, S.; Goodrum, F. Human cytomegalovirus UL135 interacts with host adaptor proteins to regulate epidermal growth factor receptor and reactivation from latency. J. Virol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Crawford, L.B.; Streblow, D.N.; Hakki, M.; Nelson, J.A.; Caposio, P. Humanized mouse models of human cytomegalovirus infection. Curr. Opin. Virol. 2015, 13, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Crawford, L.B.; Kim, J.H.; Collins-McMillen, D.; Lee, B.J.; Landais, I.; Held, C.; Nelson, J.A.; Yurochko, A.D. Human cytomegalovirus encodes a novel FLT3 receptor ligand necessary for hematopoietic cell differentiation and viral reactivation. mBio 2018, 9, e00682-18. [Google Scholar] [CrossRef] [PubMed]
- Collins-McMillen, D.; Stevenson, E.V.; Kim, J.H.; Lee, B.J.; Cieply, S.J.; Nogalski, M.T.; Chan, G.C.; Frost, R.W., 3rd; Spohn, C.R.; Yurochko, A.D. HCMV utilizes a non-traditional stat1 activation cascade via signaling through egfr and integrins to efficiently promote the motility, differentiation, and polarization of infected monocytes. J. Virol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Kabanova, A.; Marcandalli, J.; Zhou, T.; Bianchi, S.; Baxa, U.; Tsybovsky, Y.; Lilleri, D.; Silacci-Fregni, C.; Foglierini, M. Platelet-derived growth factor-alpha receptor is the cellular receptor for human cytomegalovirus ghglgo trimer. Nat. Microbiol. 2016, 1, 16082. [Google Scholar] [CrossRef] [PubMed]
- Avraham, R.; Yarden, Y. Feedback regulation of EGFR signalling: Decision making by early and delayed loops. Nat. Rev. Mol. Cell. Biol. 2011, 12, 104–117. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, S.; Langhans, S.A. Epidermal growth factor signaling in transformed cells. Int. Rev. Cell Mol. Biol. 2015, 314, 1–41. [Google Scholar] [PubMed]
- Lupberger, J.; Duong, F.H.; Fofana, I.; Zona, L.; Xiao, F.; Thumann, C.; Durand, S.C.; Pessaux, P.; Zeisel, M.B.; Heim, M.H.; et al. Epidermal growth factor receptor signaling impairs the antiviral activity of interferon-alpha. Hepatology 2013, 58, 1225–1235. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M.; Chattopadhyay, S.; Fensterl, V.; Saikia, P.; Wetzel, J.L.; Sen, G.C. Epidermal growth factor receptor is essential for Toll-like receptor 3 signaling. Sci. Signal. 2012, 5, ra50. [Google Scholar] [CrossRef] [PubMed]
- Ortega, J.; Li, J.Y.; Lee, S.; Tong, D.; Gu, L.; Li, G.M. Phosphorylation of PCNA by EGFR inhibits mismatch repair and promotes misincorporation during DNA synthesis. Proc. Natl. Acad. Sci. USA 2015, 112, 5667–5672. [Google Scholar] [CrossRef] [PubMed]
- Luo, B.H.; Springer, T.A. Integrin structures and conformational signaling. Curr. Opin. Cell Biol. 2006, 18, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Berman, A.E.; Kozlova, N.I.; Morozevich, G.E. Integrins: Structure and signaling. Biochemistry 2003, 68, 1284–1299. [Google Scholar] [CrossRef] [PubMed]
- Caswell, P.T.; Vadrevu, S.; Norman, J.C. Integrins: Masters and slaves of endocytic transport. Nat. Rev. Mol. Cell. Biol. 2009, 10, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Cliffe, A.R.; Arbuckle, J.H.; Vogel, J.L.; Geden, M.J.; Rothbart, S.B.; Cusack, C.L.; Strahl, B.D.; Kristie, T.M.; Deshmukh, M. Neuronal stress pathway mediating a histone Methyl/Phospho switch is required for herpes simplex virus reactivation. Cell. Host Microbe 2015, 18, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Koyuncu, O.O.; MacGibeny, M.A. Compartmented neuronal cultures reveal two distinct mechanisms for alpha herpesvirus escape from genome silencing. PLoS Pathog. 2017, 13, e1006608. [Google Scholar] [CrossRef] [PubMed]
- Collins-McMillen, D.; Kim, J.H.; Nogalski, M.T.; Stevenson, E.V.; Chan, G.C.; Caskey, J.R.; Cieply, S.J.; Yurochko, A.D. Human Cytomegalovirus Promotes Survival of Infected Monocytes via a Distinct Temporal Regulation of Cellular Bcl-2 Family Proteins. J. Virol. 2015, 90, 2356–2371. [Google Scholar] [CrossRef] [PubMed]
- Maciejewski, J.P.; Bruening, E.E.; Donahue, R.E.; Sellers, S.E.; Carter, C.; Young, N.S.; St Jeor, S. Infection of mononucleated phagocytes with human cytomegalovirus. Virology 1993, 195, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Roizman, B.; Whitley, R.J. An inquiry into the molecular basis of HSV latency and reactivation. Annu. Rev. Microbiol. 2013, 67, 355–374. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Banerjee, S. The regulatory role of protein phosphorylation in human gammaherpesvirus associated cancers. Virol. Sin. 2017, 32, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Stern, J.L.; Slobedman, B. Human cytomegalovirus latent infection of myeloid cells directs monocyte migration by up-regulating monocyte chemotactic protein-1. J. Immunol. 2008, 180, 6577–6585. [Google Scholar] [CrossRef] [PubMed]
- Noriega, V.M.; Haye, K.K.; Kraus, T.A.; Kowalsky, S.R.; Ge, Y.; Moran, T.M.; Tortorella, D. Human cytomegalovirus modulates monocyte-mediated innate immune responses during short-term experimental latency in vitro. J. Virol. 2014, 88, 9391–9405. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Gao, Y.; Zhou, Q.; Zhang, Q.; Peng, Y.; Tian, K.; Wang, J.; Zheng, X. Human cytomegalovirus latent infection alters the expression of cellular and viral microRNA. Gene 2014, 536, 272–278. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.Z.; Su, S.; Gao, Y.Q.; Wang, P.P.; Huang, Z.F.; Hu, M.M.; Luo, W.W.; Li, S.; Luo, M.H.; Wang, Y.Y.; et al. Human cytomegalovirus tegument protein UL82 inhibits sting-mediated signaling to evade antiviral immunity. Cell. Host Microbe 2017, 21, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Trgovcich, J.; Cebulla, C.; Zimmerman, P.; Sedmak, D.D. Human cytomegalovirus protein pp71 disrupts major histocompatibility complex class I cell surface expression. J. Virol. 2006, 80, 951–963. [Google Scholar] [CrossRef] [PubMed]
- Cha, T.A.; Tom, E.; Kemble, G.W.; Duke, G.M.; Mocarski, E.S.; Spaete, R.R. Human cytomegalovirus clinical isolates carry at least 19 genes not found in laboratory strains. J. Virol. 1996, 70, 78–83. [Google Scholar] [PubMed]
- Murphy, E.; Yu, D.; Grimwood, J.; Schmutz, J.; Dickson, M.; Jarvis, M.A.; Hahn, G.; Nelson, J.A.; Myers, R.M.; Shenk, T.E. Coding potential of laboratory and clinical strains of human cytomegalovirus. Proc. Natl. Acad. Sci. USA 2003, 100, 14976–14981. [Google Scholar] [CrossRef] [PubMed]
- Dolan, A.; Cunningham, C.; Hector, R.D.; Hassan-Walker, A.F.; Lee, L.; Addison, C.; Dargan, D.J.; McGeoch, D.J.; Gatherer, D.; Emery, V.C.; et al. Genetic content of wild-type human cytomegalovirus. J. Gen. Virol. 2004, 85, 1301–1312. [Google Scholar] [CrossRef] [PubMed]
- Grainger, L.; Cicchini, L.; Rak, M.; Petrucelli, A.; Fitzgerald, K.D.; Semler, B.L.; Goodrum, F. Stress-inducible alternative translation initiation of human cytomegalovirus latency protein pUL138. J. Virol. 2010, 84, 9472–9486. [Google Scholar] [CrossRef] [PubMed]
- Umashankar, M.; Rak, M.; Bughio, F.; Zagallo, P.; Caviness, K.; Goodrum, F.D. Antagonistic determinants controlling replicative and latent states of human cytomegalovirus infection. J. Virol. 2014, 88, 5987–6002. [Google Scholar] [CrossRef] [PubMed]
- Jafferji, I.; Bain, M.; King, C.; Sinclair, J.H. Inhibition of epidermal growth factor receptor (EGFR) expression by human cytomegalovirus correlates with an increase in the expression and binding of Wilms’ Tumour 1 protein to the EGFR promoter. J. Gen. Virol. 2009, 90, 1569–1574. [Google Scholar] [CrossRef] [PubMed]
- Camarena, V.; Kobayashi, M.; Kim, J.Y.; Roehm, P.; Perez, R.; Gardner, J.; Wilson, A.C.; Mohr, I.; Chao, M.V. Nature and duration of growth factor signaling through receptor tyrosine kinases regulates HSV-1 latency in neurons. Cell. Host Microbe 2010, 8, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Wu, T.T.; Tchieu, J.H.; Feng, J.; Brown, H.J.; Feng, J.; Li, X.; Qi, J.; Deng, H.; Vivanco, I.; et al. Inhibition of the phosphatidylinositol 3-kinase-Akt pathway enhances gamma-2 herpesvirus lytic replication and facilitates reactivation from latency. J. Gen. Virol. 2010, 91, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Kung, C.P.; Meckes, D.G., Jr.; Raab-Traub, N. Epstein-Barr virus LMP1 activates EGFR, STAT3, and ERK through effects on PKCdelta. J. Virol. 2011, 85, 4399–4408. [Google Scholar] [CrossRef] [PubMed]
- Moepps, B.; Tulone, C.; Kern, C.; Minisini, R.; Michels, G.; Vatter, P.; Wieland, T.; Gierschik, P. Constitutive serum response factor activation by the viral chemokine receptor homologue pUS28 is differentially regulated by Galpha(q/11) and Galpha(16). Cell. Signal. 2008, 20, 1528–1537. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.E.; Zagorski, W.A.; Brenneman, J.D.; Avery, D.; Miller, J.L.; O’Connor, C.M. US28 is a potent activator of phospholipase c during hcmv infection of clinically relevant target cells. PLoS ONE 2012, 7, e50524. [Google Scholar] [CrossRef] [PubMed]
- Slinger, E.; Maussang, D.; Schreiber, A.; Siderius, M.; Rahbar, A.; Fraile-Ramos, A.; Lira, S.A.; Soderberg-Naucler, C.; Smit, M.J. HCMV-encoded chemokine receptor US28 mediates proliferative signaling through the Il-6-STAT3 axis. Sci. Signal. 2010, 3, ra58. [Google Scholar] [CrossRef] [PubMed]
- Boomker, J.M.; The, T.H.; de Leij, L.F.; Harmsen, M.C. The human cytomegalovirus-encoded receptor US28 increases the activity of the major immediate-early promoter/enhancer. Virus Res. 2006, 118, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Wen, D.Q.; Zhang, Y.Y.; Lv, L.P.; Zhou, X.P.; Yan, F.; Ma, P.; Xu, J.B. Human cytomegalovirus-encoded chemokine receptor homolog US28 stimulates the major immediate early gene promoter/enhancer via the induction of creb. J. Recept. Signal Transduct. Res. 2009, 29, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Humby, M.S.; O’Connor, C.M. Human cytomegalovirus US28 is important for latent infection of hematopoietic progenitor cells. J. Virol. 2015, 90, 2959–2970. [Google Scholar] [CrossRef] [PubMed]
- Krishna, B.A.; Poole, E.L.; Jackson, S.E. Latency-associated expression of human cytomegalovirus US28 attenuates cell signaling pathways to maintain latent infection. mBio 2017, 8, e01754-17. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.E.; Miller, W.E. The HCMV US28 vGPCR induces potent Gαq/PLC-β signaling in monocytes leading to increased adhesion to endothelial cells. Virology 2016, 497, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Todaro, M.; Zerilli, M.; Ricci-Vitiani, L.; Bini, M.; Perez Alea, M.; Maria Florena, A.; Miceli, L.; Condorelli, G.; Bonventre, S.; Di Gesu, G.; et al. Autocrine production of interleukin-4 and interleukin-10 is required for survival and growth of thyroid cancer cells. Cancer Res. 2006, 66, 1491–1499. [Google Scholar] [CrossRef] [PubMed]
- Hancock, M.H.; Hook, L.M.; Mitchell, J.; Nelson, J.A. Human Cytomegalovirus MicroRNAs miR-US5-1 and miR-UL112-3p Block Proinflammatory Cytokine Production in Response to NF-κB-Activating Factors through Direct Downregulation of IKKα and IKKβ. mBio 2017, 8, e00109-17. [Google Scholar] [CrossRef] [PubMed]
- Reeves, M.B.; Compton, T. Inhibition of inflammatory interleukin-6 activity via extracellular signal-regulated kinase-mitogen-activated protein kinase signaling antagonizes human cytomegalovirus reactivation from dendritic cells. J. Virol. 2011, 85, 12750–12758. [Google Scholar] [CrossRef] [PubMed]
- Engel, P.; Perez-Carmona, N.; Alba, M.M.; Robertson, K.; Ghazal, P.; Angulo, A. Human cytomegalovirus UL7, a homologue of the SLAM-family receptor CD229, impairs cytokine production. Immunol. Cell Biol. 2011, 89, 753–766. [Google Scholar] [CrossRef] [PubMed]
- Zwezdaryk, K.J.; Combs, J.A.; Morris, C.A.; Sullivan, D.E. Regulation of Wnt/beta-catenin signaling by herpesviruses. World J. Virol. 2016, 5, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Morrison, J.A.; Klingelhutz, A.J.; Raab-Traub, N. Epstein-Barr virus latent membrane protein 2A activates beta-catenin signaling in epithelial cells. J. Virol. 2003, 77, 12276–12284. [Google Scholar] [CrossRef] [PubMed]
- Fujimuro, M.; Hayward, S.D. The latency-associated nuclear antigen of kaposi’s sarcoma-associated herpesvirus manipulates the activity of glycogen synthase kinase-3beta. J. Virol. 2003, 77, 8019–8030. [Google Scholar] [CrossRef] [PubMed]
- Fujimuro, M.; Wu, F.Y.; ApRhys, C.; Kajumbula, H.; Young, D.B.; Hayward, G.S.; Hayward, S.D. A novel viral mechanism for dysregulation of beta-catenin in kaposi’s sarcoma-associated herpesvirus latency. Nat. Med. 2003, 9, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Shackelford, J.; Maier, C.; Pagano, J.S. Epstein-barr virus activates beta-catenin in type III latently infected B lymphocyte lines: Association with deubiquitinating enzymes. Proc. Natl. Acad. Sci. USA 2003, 100, 15572–15576. [Google Scholar] [CrossRef] [PubMed]
- Everly, D.N., Jr.; Kusano, S.; Raab-Traub, N. Accumulation of cytoplasmic beta-catenin and nuclear glycogen synthase kinase 3beta in epstein-barr virus-infected cells. J. Virol. 2004, 78, 11648–11655. [Google Scholar] [CrossRef] [PubMed]
- Jang, K.L.; Shackelford, J.; Seo, S.Y.; Pagano, J.S. Up-regulation of beta-catenin by a viral oncogene correlates with inhibition of the seven in absentia homolog 1 in b lymphoma cells. Proc. Natl. Acad. Sci. USA 2005, 102, 18431–18436. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hancock, M.; Workman, A.; Doster, A.; Jones, C. Beta-catenin, a transcription factor activated by canonical Wnt signaling, is expressed in sensory neurons of calves latently infected with bovine herpesvirus 1. J. Virol. 2016, 90, 3148–3159. [Google Scholar] [CrossRef] [PubMed]
- Workman, A.; Zhu, L.; Keel, B.N.; Smith, T.P.L.; Jones, C. The Wnt Signaling Pathway Is Differentially Expressed during the Bovine Herpesvirus 1 Latency-Reactivation Cycle: Evidence That Two Protein Kinases Associated with Neuronal Survival, Akt3 and BMPR2, Are Expressed at Higher Levels during Latency. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Workman, A.; Jones, C. Potential role for a beta-catenin coactivator (high-mobility group at-hook 1 protein) during the latency-reactivation cycle of bovine herpesvirus 1. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Angelova, M.; Zwezdaryk, K.; Ferris, M.; Shan, B.; Morris, C.A.; Sullivan, D.E. Human cytomegalovirus infection dysregulates the canonical wnt/beta-catenin signaling pathway. PLoS Pathog. 2012, 8, e1002959. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Liu, F.; Arav-Boger, R. Human Cytomegalovirus Inhibits the PARsylation Activity of Tankyrase—A Potential Strategy for Suppression of the Wnt Pathway. Viruses 2016, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; He, R.; Venkatadri, R.; Forman, M.; Arav-Boger, R. Wnt modulating agents inhibit human cytomegalovirus replication. Antimicrob. Agents Chemother. 2013, 57, 2761–2767. [Google Scholar] [CrossRef] [PubMed]
- Stinski, M.F. Sequence of protein synthesis in cells infected by human cytomegalovirus: Early and late virus-induced polypeptides. J. Virol. 1978, 26, 686–701. [Google Scholar] [PubMed]
- Wathen, M.W.; Stinski, M.F. Temporal patterns of human cytomegalovirus transcription: Mapping the viral rnas synthesized at immediate early, early, and late times after infection. J. Virol. 1982, 41, 462–477. [Google Scholar] [PubMed]
- Stinski, M.F.; Meier, J.L. Immediate-early viral gene regulation and function. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Pizzorno, M.C.; O’Hare, P.; Sha, L.; LaFemina, R.L.; Hayward, G.S. Trans-activation and autoregulation of gene expression by the immediate-early region 2 gene products of human cytomegalovirus. J. Virol. 1988, 62, 1167–1179. [Google Scholar] [PubMed]
- Malone, C.L.; Vesole, D.H.; Stinski, M.F. Transactivation of a human cytomegalovirus early promoter by gene products from the immediate-early gene IE2 and augmentation by IE1: Mutational analysis of the viral proteins. J. Virol. 1990, 64, 1498–1506. [Google Scholar] [PubMed]
- Anders, D.G.; Kerry, J.A.; Pari, G.S. DNA synthesis and late viral gene expression. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Reeves, M.B.; MacAry, P.A.; Lehner, P.J.; Sissons, J.G.; Sinclair, J.H. Latency, chromatin remodeling, and reactivation of human cytomegalovirus in the dendritic cells of healthy carriers. Proc. Natl. Acad. Sci. USA 2005, 102, 4140–4145. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, D.R.; Stenberg, R.M.; Goins, W.F.; Stinski, M.F. Promoter-regulatory region of the major immediate early gene of human cytomegalovirus. Proc. Natl. Acad. Sci. USA 1984, 81, 659–663. [Google Scholar] [CrossRef] [PubMed]
- Boshart, M.; Weber, F.; Jahn, G.; Dorsch-Hasler, K.; Fleckenstein, B.; Schaffner, W. A very strong enhancer is located upstream of an immediate early gene of human cytomegalovirus. Cell 1985, 41, 521–530. [Google Scholar] [CrossRef]
- Ghazal, P.; Lubon, H.; Fleckenstein, B.; Hennighausen, L. Binding of transcription factors and creation of a large nucleoprotein complex on the human cytomegalovirus enhancer. Proc. Natl. Acad. Sci. USA 1987, 84, 3658–3662. [Google Scholar] [CrossRef] [PubMed]
- Ghazal, P.; Lubon, H.; Hennighausen, L. Multiple sequence-specific transcription factors modulate cytomegalovirus enhancer activity in vitro. Mol. Cell. Biol. 1988, 8, 1809–1811. [Google Scholar] [CrossRef] [PubMed]
- Akrigg, A.; Wilkinson, G.W.; Oram, J.D. The structure of the major immediate early gene of human cytomegalovirus strain AD169. Virus Res. 1985, 2, 107–121. [Google Scholar] [CrossRef]
- Stinski, M.F.; Roehr, T.J. Activation of the major immediate early gene of human cytomegalovirus by cis-acting elements in the promoter-regulatory sequence and by virus-specific trans-acting components. J. Virol. 1985, 55, 431–441. [Google Scholar] [PubMed]
- Sambucetti, L.C.; Cherrington, J.M.; Wilkinson, G.W.; Mocarski, E.S. NF-kappa B activation of the cytomegalovirus enhancer is mediated by a viral transactivator and by T cell stimulation. EMBO J. 1989, 8, 4251–4258. [Google Scholar] [PubMed]
- Hunninghake, G.W.; Monick, M.M.; Liu, B.; Stinski, M.F. The promoter-regulatory region of the major immediate-early gene of human cytomegalovirus responds to T-lymphocyte stimulation and contains functional cyclic amp-response elements. J. Virol. 1989, 63, 3026–3033. [Google Scholar] [PubMed]
- Lang, D.; Fickenscher, H.; Stamminger, T. Analysis of proteins binding to the proximal promoter region of the human cytomegalovirus IE-1/2 enhancer/promoter reveals both consensus and aberrant recognition sequences for transcription factors Sp1 and CREB. Nucleic Acids Res. 1992, 20, 3287–3295. [Google Scholar] [CrossRef] [PubMed]
- Wade, E.J.; Klucher, K.M.; Spector, D.H. An AP-1 binding site is the predominant cis-acting regulatory element in the 1.2-kilobase early RNA promoter of human cytomegalovirus. J. Virol. 1992, 66, 2407–2417. [Google Scholar] [PubMed]
- LaFemina, R.; Hayward, G.S. Constitutive and retinoic acid-inducible expression of cytomegalovirus immediate-early genes in human teratocarcinoma cells. J. Virol. 1986, 58, 434–440. [Google Scholar] [PubMed]
- Angulo, A.; Suto, C.; Heyman, R.A.; Ghazal, P. Characterization of the sequences of the human cytomegalovirus enhancer that mediate differential regulation by natural and synthetic retinoids. Mol. Endocrinol. 1996, 10, 781–793. [Google Scholar] [PubMed]
- Ghazal, P.; DeMattei, C.; Giulietti, E.; Kliewer, S.A.; Umesono, K.; Evans, R.M. Retinoic acid receptors initiate induction of the cytomegalovirus enhancer in embryonal cells. Proc. Natl. Acad. Sci. USA 1992, 89, 7630–7634. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.J.; Chiou, C.J.; Huang, Q.; Hayward, G.S. Synergistic interactions between overlapping binding sites for the serum response factor and ELK-1 proteins mediate both basal enhancement and phorbol ester responsiveness of primate cytomegalovirus major immediate-early promoters in monocyte and T-lymphocyte cell types. J. Virol. 1996, 70, 8590–8605. [Google Scholar] [PubMed]
- Netterwald, J.; Yang, S.; Wang, W.; Ghanny, S.; Cody, M.; Soteropoulos, P.; Tian, B.; Dunn, W.; Liu, F.; Zhu, H. Two gamma interferon-activated site-like elements in the human cytomegalovirus major immediate-early promoter/enhancer are important for viral replication. J. Virol. 2005, 79, 5035–5046. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Y.; Ni, Y.S.; Saifudeen, Z.; Asiedu, C.K.; Supakar, P.C.; Ehrlich, M. Increasing binding of a transcription factor immediately downstream of the cap site of a cytomegalovirus gene represses expression. Nucleic Acids Res. 1995, 23, 3026–3033. [Google Scholar] [CrossRef] [PubMed]
- Zweidler-Mckay, P.A.; Grimes, H.L.; Flubacher, M.M.; Tsichlis, P.N. Gfi-1 encodes a nuclear zinc finger protein that binds DNA and functions as a transcriptional repressor. Mol. Cell. Biol. 1996, 16, 4024–4034. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Baillie, J.; Sissons, J.G.; Sinclair, J.H. The transcription factor YY1 binds to negative regulatory elements in the human cytomegalovirus major immediate early enhancer/promoter and mediates repression in non-permissive cells. Nucleic Acids Res. 1994, 22, 2453–2459. [Google Scholar] [CrossRef] [PubMed]
- Bain, M.; Mendelson, M.; Sinclair, J. Ets-2 repressor factor (ERF) mediates repression of the human cytomegalovirus major immediate-early promoter in undifferentiated non-permissive cells. J. Gen. Virol. 2003, 84, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Kothari, S.; Baillie, J.; Sissons, J.G.; Sinclair, J.H. The 21bp repeat element of the human cytomegalovirus major immediate early enhancer is a negative regulator of gene expression in undifferentiated cells. Nucleic Acids Res. 1991, 19, 1767–1771. [Google Scholar] [CrossRef] [PubMed]
- Hennighausen, L.; Fleckenstein, B. Nuclear factor 1 interacts with five DNA elements in the promoter region of the human cytomegalovirus major immediate early gene. EMBO J. 1986, 5, 1367–1371. [Google Scholar] [PubMed]
- Jeang, K.T.; Rawlins, D.R.; Rosenfeld, P.J.; Shero, J.H.; Kelly, T.J.; Hayward, G.S. Multiple tandemly repeated binding sites for cellular nuclear factor 1 that surround the major immediate-early promoters of simian and human cytomegalovirus. J. Virol. 1987, 61, 1559–1570. [Google Scholar] [PubMed]
- Shelbourn, S.L.; Kothari, S.K.; Sissons, J.G.; Sinclair, J.H. Repression of human cytomegalovirus gene expression associated with a novel immediate early regulatory region binding factor. Nucleic Acids Res. 1989, 17, 9165–9171. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.H.; Oka, T.; Asai, T.; Okada, T.; Merrills, B.W.; Gertson, P.N.; Whitson, R.H.; Itakura, K. Repression by a differentiation-specific factor of the human cytomegalovirus enhancer. Nucleic Acids Res. 1996, 24, 1695–1701. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.A.; Reynolds-Kohler, C.; Smith, B.A. Negative and positive regulation by a short segment in the 5′-flanking region of the human cytomegalovirus major immediate-early gene. Mol. Cell. Biol. 1987, 7, 4125–4129. [Google Scholar] [CrossRef] [PubMed]
- Lubon, H.; Ghazal, P.; Hennighausen, L.; Reynolds-Kohler, C.; Lockshin, C.; Nelson, J. Cell-specific activity of the modulator region in the human cytomegalovirus major immediate-early gene. Mol. Cell. Biol. 1989, 9, 1342–1345. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.A.; Groudine, M. Transcriptional regulation of the human cytomegalovirus major immediate-early gene is associated with induction of DNase I-hypersensitive sites. Mol. Cell. Biol. 1986, 6, 452–461. [Google Scholar] [CrossRef] [PubMed]
- Keller, M.J.; Wu, A.W.; Andrews, J.I.; McGonagill, P.W.; Tibesar, E.E.; Meier, J.L. Reversal of human cytomegalovirus major immediate-early enhancer/promoter silencing in quiescently infected cells via the cyclic amp signaling pathway. J. Virol. 2007, 81, 6669–6681. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Liu, X.; Wu, A.W.; McGonagill, P.W.; Keller, M.J.; Galle, C.S.; Meier, J.L. Breaking human cytomegalovirus major immediate-early gene silence by vasoactive intestinal peptide stimulation of the protein kinase A-CREB-TORC2 signaling cascade in human pluripotent embryonal NTera2 cells. J. Virol. 2009, 83, 6391–6403. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yuan, J.; Wu, A.W.; McGonagill, P.W.; Galle, C.S.; Meier, J.L. Phorbol ester-induced human cytomegalovirus major immediate-early (MIE) enhancer activation through PKC-delta, CREB, and NF-kappaB desilences MIE gene expression in quiescently infected human pluripotent NTera2 cells. J. Virol. 2010, 84, 8495–8508. [Google Scholar] [CrossRef] [PubMed]
- Prosch, S.; Wuttke, R.; Kruger, D.H.; Volk, H.D. NF-kappaB—A potential therapeutic target for inhibition of human cytomegalovirus (re)activation? Biol. Chem. 2002, 383, 1601–1609. [Google Scholar] [CrossRef] [PubMed]
- Meier, J.L. Reactivation of the human cytomegalovirus major immediate-early regulatory region and viral replication in embryonal ntera2 cells: Role of trichostatin a, retinoic acid, and deletion of the 21-base-pair repeats and modulator. J. Virol. 2001, 75, 1581–1593. [Google Scholar] [CrossRef] [PubMed]
- Wright, E.; Bain, M.; Teague, L.; Murphy, J.; Sinclair, J. Ets-2 repressor factor recruits histone deacetylase to silence human cytomegalovirus immediate-early gene expression in non-permissive cells. J. Gen. Virol. 2005, 86, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.M.; Inouye, C.; Zeng, Y.; Bearss, D.; Seto, E. Transcriptional repression by YY1 is mediated by interaction with a mammalian homolog of the yeast global regulator RPD3. Proc. Natl. Acad. Sci. USA 1996, 93, 12845–12850. [Google Scholar] [CrossRef] [PubMed]
- Kew, V.G.; Yuan, J.; Meier, J.; Reeves, M.B. Mitogen and stress activated kinases act co-operatively with creb during the induction of human cytomegalovirus immediate-early gene expression from latency. PLoS Pathog. 2014, 10, e1004195. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, P.M. Epigenetics and genetics of viral latency. Cell. Host Microbe 2016, 19, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Hummel, M.; Yan, S.; Li, Z.; Varghese, T.K.; Abecassis, M. Transcriptional reactivation of murine cytomegalovirus ie gene expression by 5-aza-2′-deoxycytidine and trichostatin A in latently infected cells despite lack of methylation of the major immediate-early promoter. J. Gen. Virol. 2007, 88, 1097–1102. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.C.; Fischle, W.; Verdin, E.; Sinclair, J.H. Control of cytomegalovirus lytic gene expression by histone acetylation. EMBO J. 2002, 21, 1112–1120. [Google Scholar] [CrossRef] [PubMed]
- Varnum, S.M.; Streblow, D.N.; Monroe, M.E.; Smith, P.; Auberry, K.J.; Pasa-Tolic, L.; Wang, D.; Camp, D.G., 2nd; Rodland, K.; Wiley, S.; et al. Identification of proteins in human cytomegalovirus (HCMV) particles: The HCMV proteome. J. Virol. 2004, 78, 10960–10966. [Google Scholar] [CrossRef] [PubMed]
- Groves, I.J.; Reeves, M.B.; Sinclair, J.H. Lytic infection of permissive cells with human cytomegalovirus is regulated by an intrinsic ‘pre-immediate-early’ repression of viral gene expression mediated by histone post-translational modification. J. Gen. Virol. 2009, 90, 2364–2374. [Google Scholar] [CrossRef] [PubMed]
- Nitzsche, A.; Paulus, C.; Nevels, M. Temporal dynamics of cytomegalovirus chromatin assembly in productively infected human cells. J. Virol. 2008, 82, 11167–11180. [Google Scholar] [CrossRef] [PubMed]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Cuevas-Bennett, C.; Shenk, T. Dynamic histone H3 acetylation and methylation at human cytomegalovirus promoters during replication in fibroblasts. J. Virol. 2008, 82, 9525–9536. [Google Scholar] [CrossRef] [PubMed]
- Abraham, C.G.; Kulesza, C.A. Polycomb repressive complex 2 silences human cytomegalovirus transcription in quiescent infection models. J. Virol. 2013, 87, 13193–13205. [Google Scholar] [CrossRef] [PubMed]
- Tavalai, N.; Papior, P.; Rechter, S.; Leis, M.; Stamminger, T. Evidence for a role of the cellular ND10 protein PML in mediating intrinsic immunity against human cytomegalovirus infections. J. Virol. 2006, 80, 8006–8018. [Google Scholar] [CrossRef] [PubMed]
- Tavalai, N.; Papior, P.; Rechter, S.; Stamminger, T. Nuclear domain 10 components promyelocytic leukemia protein and hDaxx independently contribute to an intrinsic antiviral defense against human cytomegalovirus infection. J. Virol. 2008, 82, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Woodhall, D.L.; Groves, I.J.; Reeves, M.B.; Wilkinson, G.; Sinclair, J.H. Human Daxx-mediated repression of human cytomegalovirus gene expression correlates with a repressive chromatin structure around the major immediate early promoter. J. Biol. Chem. 2006, 281, 37652–37660. [Google Scholar] [CrossRef] [PubMed]
- Lukashchuk, V.; McFarlane, S.; Everett, R.D.; Preston, C.M. Human cytomegalovirus protein PP71 displaces the chromatin-associated factor ATRX from nuclear domain 10 at early stages of infection. J. Virol. 2008, 82, 12543–12554. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Kalejta, R.F. Human cytomegalovirus protein PP71 induces Daxx sumoylation. J. Virol. 2009, 83, 6591–6598. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Kalejta, R.F. Proteasome-dependent, ubiquitin-independent degradation of Daxx by the viral PP71 protein in human cytomegalovirus-infected cells. Virology 2007, 367, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Stinski, M.F. Human cytomegalovirus contains a tegument protein that enhances transcription from promoters with upstream ATF and AP-1 cis-acting elements. J. Virol. 1992, 66, 4434–4444. [Google Scholar] [PubMed]
- Cantrell, S.R.; Bresnahan, W.A. Interaction between the human cytomegalovirus UL82 gene product (pp71) and hdaxx regulates immediate-early gene expression and viral replication. J. Virol. 2005, 79, 7792–7802. [Google Scholar] [CrossRef] [PubMed]
- Cantrell, S.R.; Bresnahan, W.A. Human cytomegalovirus (HCMV) UL82 gene product (pp71) relieves hDaxx-mediated repression of HCMV replication. J. Virol. 2006, 80, 6188–6191. [Google Scholar] [CrossRef] [PubMed]
- Preston, C.M.; Nicholl, M.J. Role of the cellular protein hdaxx in human cytomegalovirus immediate-early gene expression. J. Gen. Virol. 2006, 87, 1113–1121. [Google Scholar] [CrossRef] [PubMed]
- Ishov, A.M.; Vladimirova, O.V.; Maul, G.G. Daxx-mediated accumulation of human cytomegalovirus tegument protein pp71 at ND10 facilitates initiation of viral infection at these nuclear domains. J. Virol. 2002, 76, 7705–7712. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, H.; Sindre, H.; Stamminger, T. Functional interaction between the pp71 protein of human cytomegalovirus and the PML-interacting protein human Daxx. J. Virol. 2002, 76, 5769–5783. [Google Scholar] [CrossRef] [PubMed]
- Nevels, M.; Paulus, C.; Shenk, T. Human cytomegalovirus immediate-early 1 protein facilitates viral replication by antagonizing histone deacetylation. Proc. Natl. Acad. Sci. USA 2004, 101, 17234–17239. [Google Scholar] [CrossRef] [PubMed]
- Park, J.J.; Kim, Y.E.; Pham, H.T.; Kim, E.T.; Chung, Y.H.; Ahn, J.H. Functional interaction of the human cytomegalovirus IE2 protein with histone deacetylase 2 in infected human fibroblasts. J. Gen. Virol. 2007, 88, 3214–3223. [Google Scholar] [CrossRef] [PubMed]
- Ishov, A.M.; Stenberg, R.M.; Maul, G.G. Human cytomegalovirus immediate early interaction with host nuclear structures: Definition of an immediate transcript environment. J. Cell Biol. 1997, 138, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Albright, E.R.; Lee, J.H.; Jacobs, D. Cellular defense against latent colonization foiled by human cytomegalovirus UL138 protein. Sci. Adv. 2015, 1, e1501164. [Google Scholar] [CrossRef] [PubMed]
- Goodrum, F.; Reeves, M.; Sinclair, J.; High, K.; Shenk, T. Human cytomegalovirus sequences expressed in latently infected individuals promote a latent infection in vitro. Blood 2007, 110, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.E.; Olsen, L. Structural basis of histone demethylase KDM6B histone 3 lysine 27 specificity. Biochemistry 2018, 57, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Minton, E.J.; Tysoe, C.; Sinclair, J.H.; Sissons, J.G. Human cytomegalovirus infection of the monocyte/macrophage lineage in bone marrow. J. Virol. 1994, 68, 4017–4021. [Google Scholar] [PubMed]
- Sinclair, J.H.; Baillie, J.; Bryant, L.A.; Taylor-Wiedeman, J.A.; Sissons, J.G. Repression of human cytomegalovirus major immediate early gene expression in a monocytic cell line. J. Gen. Virol. 1992, 73 Pt 2, 433–435. [Google Scholar] [CrossRef]
- Stinski, M.F. Cytomegalovirus promoter for expression in mammalian cells. In Gene Expression Systems: Using Nature for the Art of Expression, 1st ed.; Fernandez, J.M., Hoeffler, J.P., Eds.; Elsevier-Academic Press: Cambridge, MA, USA, 1999; pp. 211–233. [Google Scholar]
- Goodrum, F.; Jordan, C.T.; Terhune, S.S.; High, K.; Shenk, T. Differential outcomes of human cytomegalovirus infection in primitive hematopoietic cell subpopulations. Blood 2004, 104, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Cheung, A.K.; Abendroth, A.; Cunningham, A.L.; Slobedman, B. Viral gene expression during the establishment of human cytomegalovirus latent infection in myeloid progenitor cells. Blood 2006, 108, 3691–3699. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Caviness, K.; Buehler, J.; Smithey, M.; Nikolich-Zugich, J.; Goodrum, F. Transcriptome-wide characterization of human cytomegalovirus in natural infection and experimental latency. Proc. Natl. Acad. Sci. USA 2017, 114. [Google Scholar] [CrossRef] [PubMed]
- Tarrant-Elorza, M.; Rossetto, C.C.; Pari, G.S. Maintenance and replication of the human cytomegalovirus genome during latency. Cell. Host Microbe 2014, 16, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Kondo, K.; Xu, J.; Mocarski, E.S. Human cytomegalovirus latent gene expression in granulocyte-macrophage progenitors in culture and in seropositive individuals. Proc. Natl. Acad. Sci. USA 1996, 93, 11137–11142. [Google Scholar] [CrossRef] [PubMed]
- Maciejewski, J.P.; St Jeor, S.C. Human cytomegalovirus infection of human hematopoietic progenitor cells. Leuk. Lymphoma. 1999, 33, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Price, A.M.; Luftig, M.A. To be or not IIb: A multi-step process for epstein-barr virus latency establishment and consequences for b cell tumorigenesis. PLoS Pathog. 2015, 11, e1004656. [Google Scholar] [CrossRef] [PubMed]
- Cliffe, A.R.; Wilson, A.C. Restarting lytic gene transcription at the onset of herpes simplex virus reactivation. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Arend, K.C.; Ziehr, B.; Vincent, H.A.; Moorman, N.J. Multiple transcripts encode full-length human cytomegalovirus IE1 and IE2 proteins during lytic infection. J. Virol. 2016, 90, 8855–8865. [Google Scholar] [CrossRef] [PubMed]
- Shnayder, M.; Nachshon, A.; Krishna, B.; Poole, E.; Boshkov, A.; Binyamin, A.; Maza, I.; Sinclair, J.; Schwartz, M.; Stern-Ginossar, N. Defining the transcriptional landscape during cytomegalovirus latency with single-cell RNA sequencing. mBio 2018, 9. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Collins-McMillen, D.; Buehler, J.; Peppenelli, M.; Goodrum, F. Molecular Determinants and the Regulation of Human Cytomegalovirus Latency and Reactivation. Viruses 2018, 10, 444. https://doi.org/10.3390/v10080444
Collins-McMillen D, Buehler J, Peppenelli M, Goodrum F. Molecular Determinants and the Regulation of Human Cytomegalovirus Latency and Reactivation. Viruses. 2018; 10(8):444. https://doi.org/10.3390/v10080444
Chicago/Turabian StyleCollins-McMillen, Donna, Jason Buehler, Megan Peppenelli, and Felicia Goodrum. 2018. "Molecular Determinants and the Regulation of Human Cytomegalovirus Latency and Reactivation" Viruses 10, no. 8: 444. https://doi.org/10.3390/v10080444
APA StyleCollins-McMillen, D., Buehler, J., Peppenelli, M., & Goodrum, F. (2018). Molecular Determinants and the Regulation of Human Cytomegalovirus Latency and Reactivation. Viruses, 10(8), 444. https://doi.org/10.3390/v10080444