Structure and Analysis of R1 and R2 Pyocin Receptor-Binding Fibers


Abstract

1. Introduction
2. Materials and Methods
2.1. Engineering and Choice of the Construct for Expression
2.2. Protein Expression and Purification
2.3. Crystallization, Data Collection, and Structure Determination
2.4. Molecular Graphics, Analysis of Surface Properties, and Bioinformatics
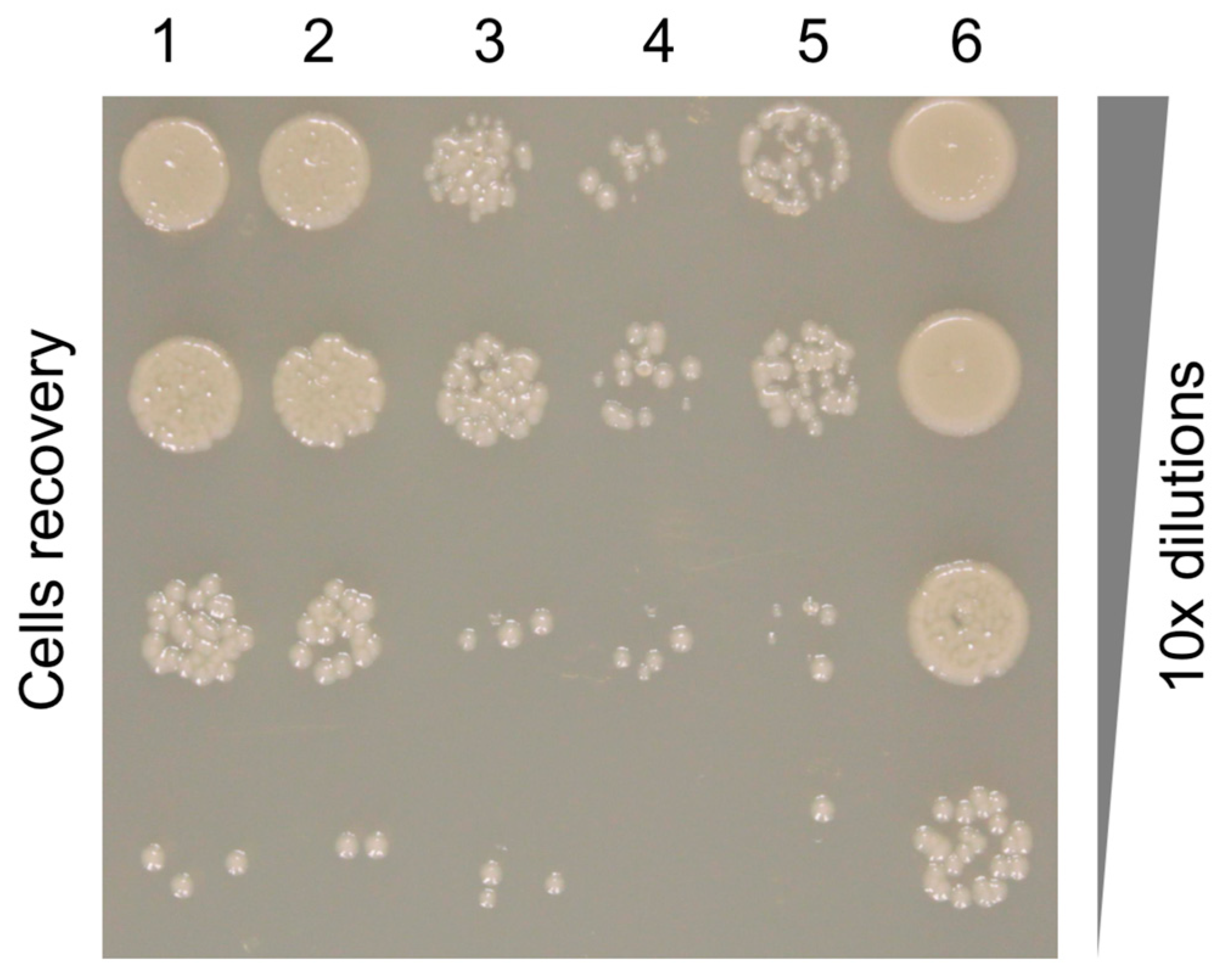
2.5. Pyocin Fiber Competition Assay
3. Results
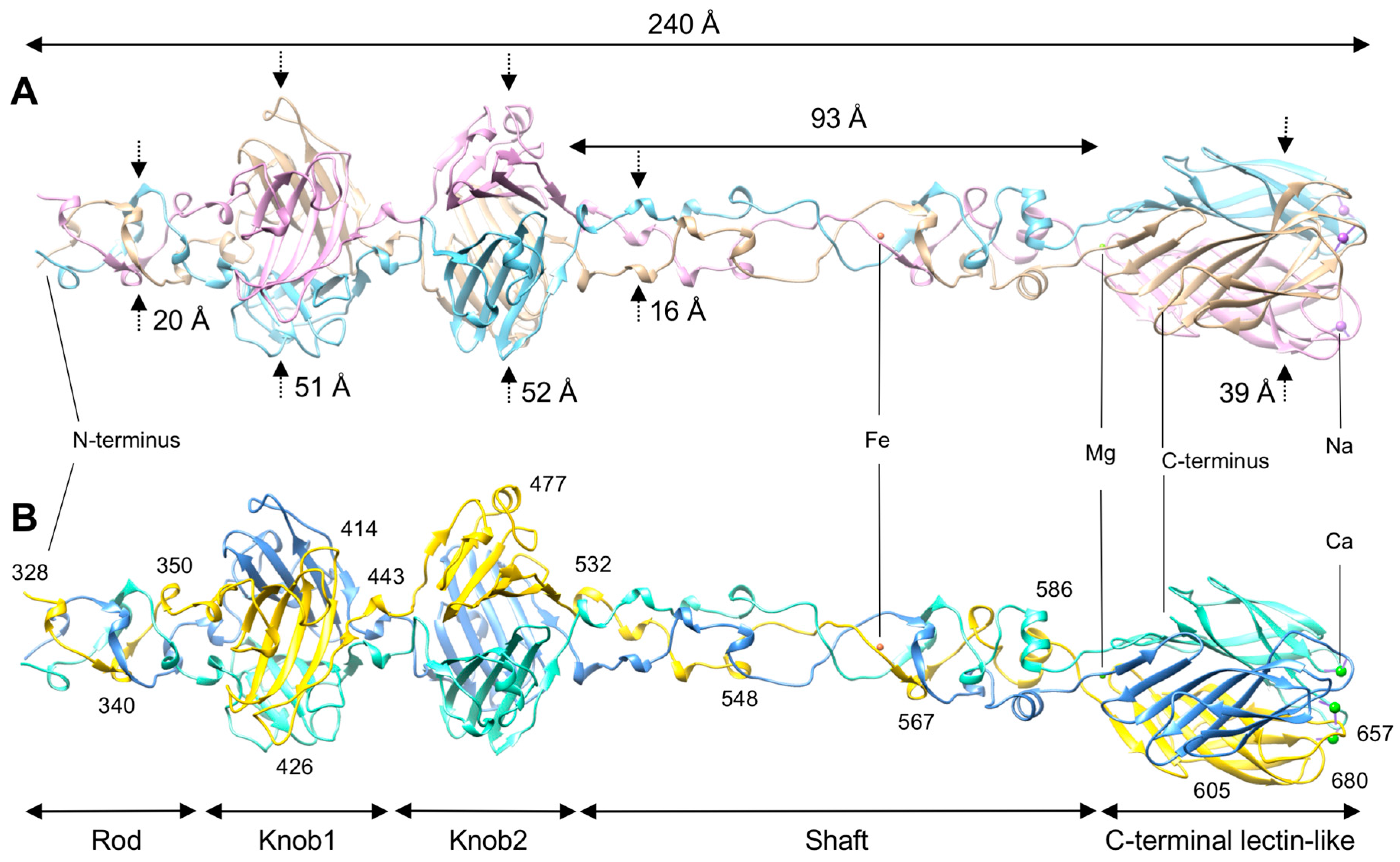
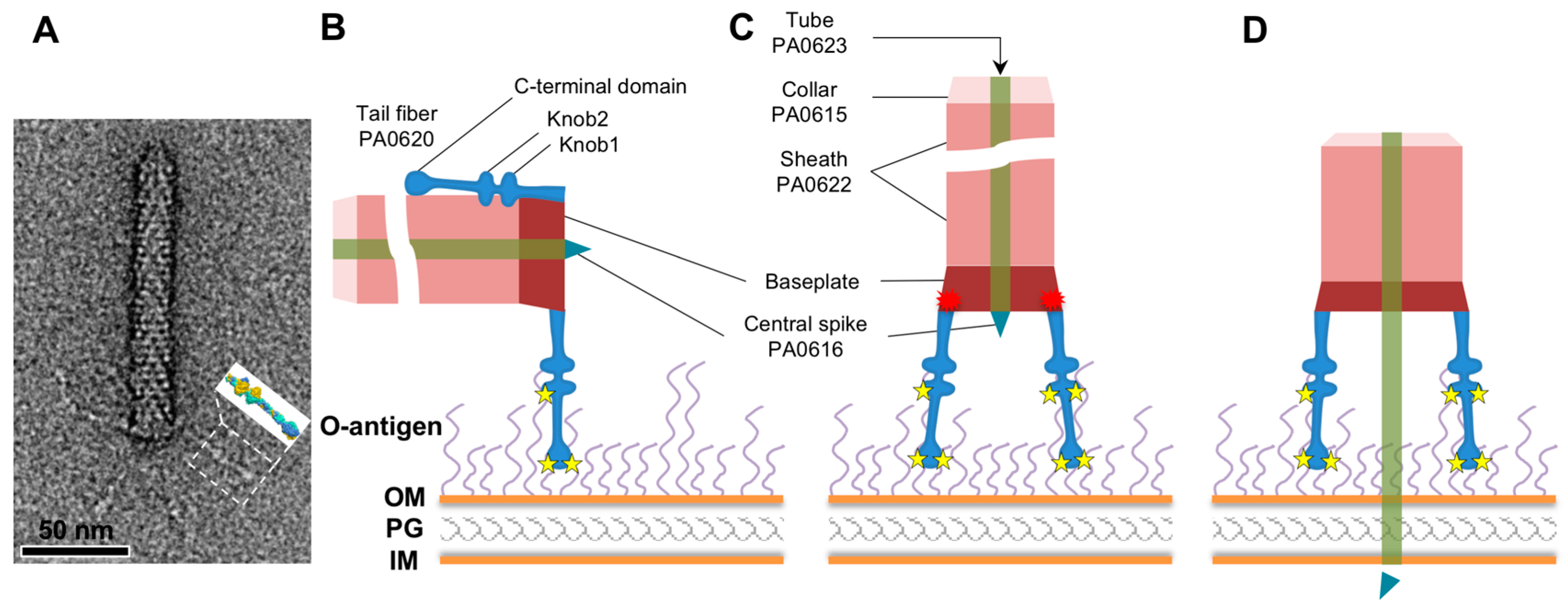
3.1. R1 and R2 Fibers Are Structurally Similar
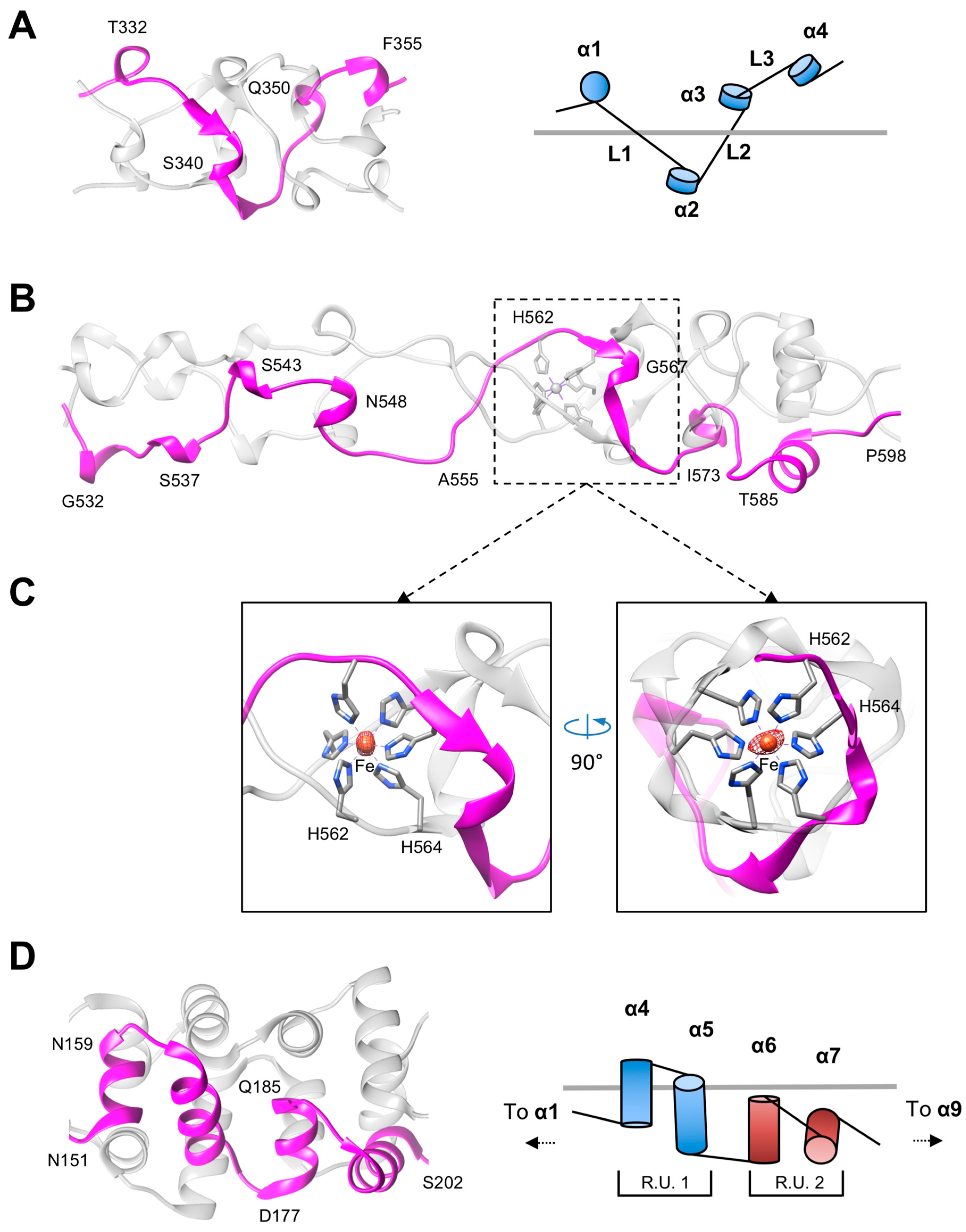
3.2. The Rod and Shaft Domains Are Built Using a Helix-Plus-Turn Motif
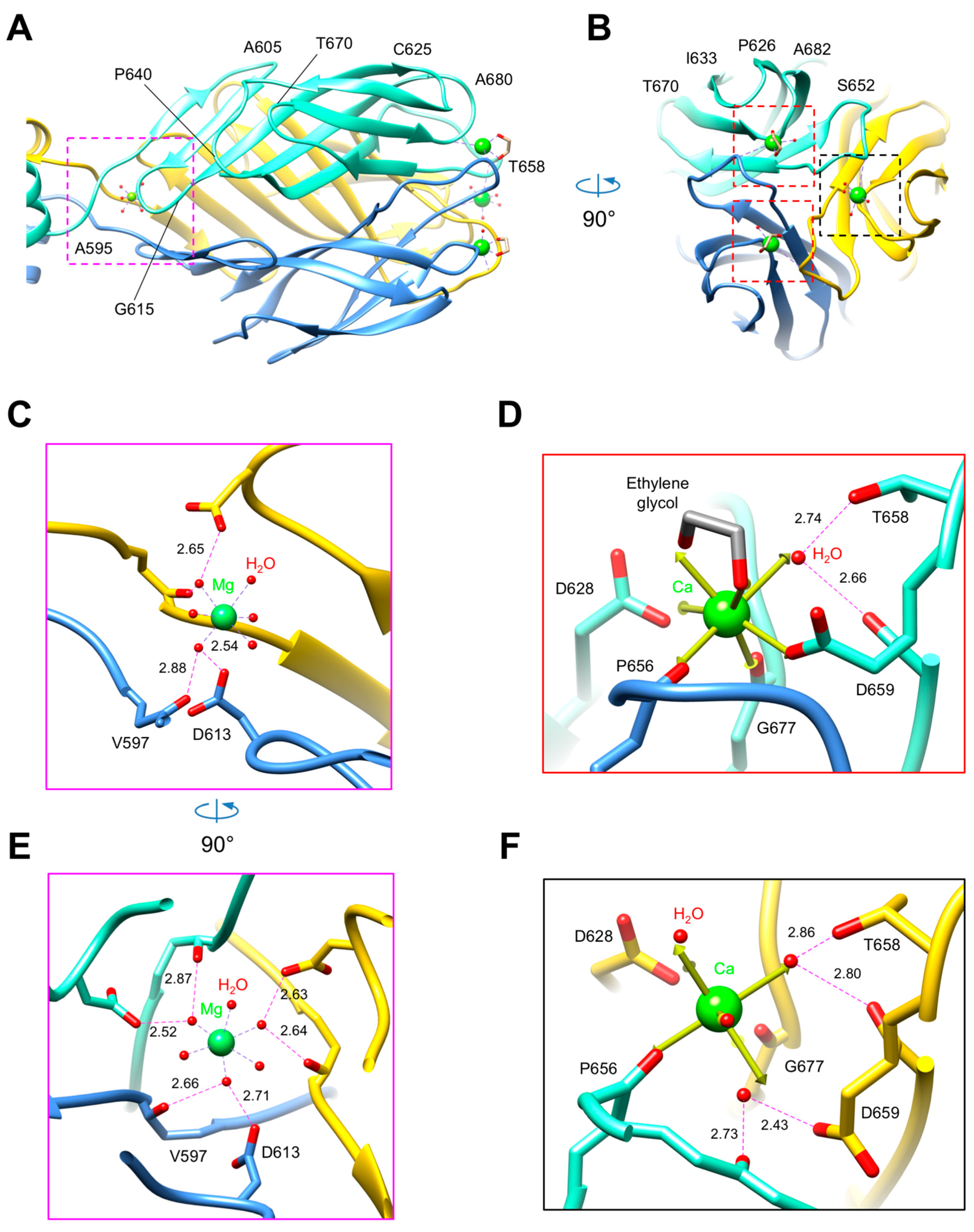
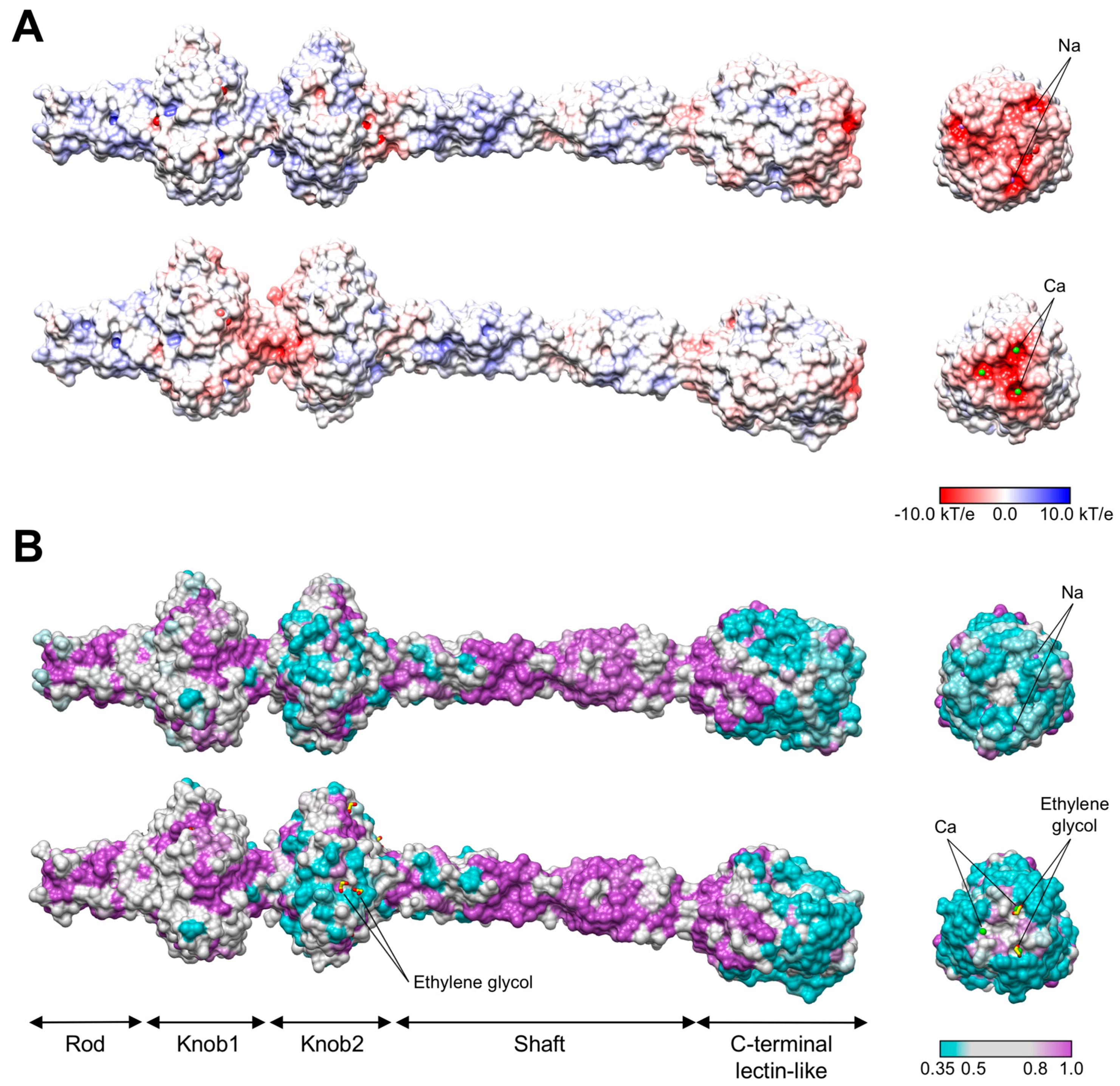
3.3. R1 and R2 Fibers Contain Buried and Solvent Exposed Metal Ions
3.4. A Small Compound is Buried in the Hydrophobic Interior of the Knob2 and Shaft Domains
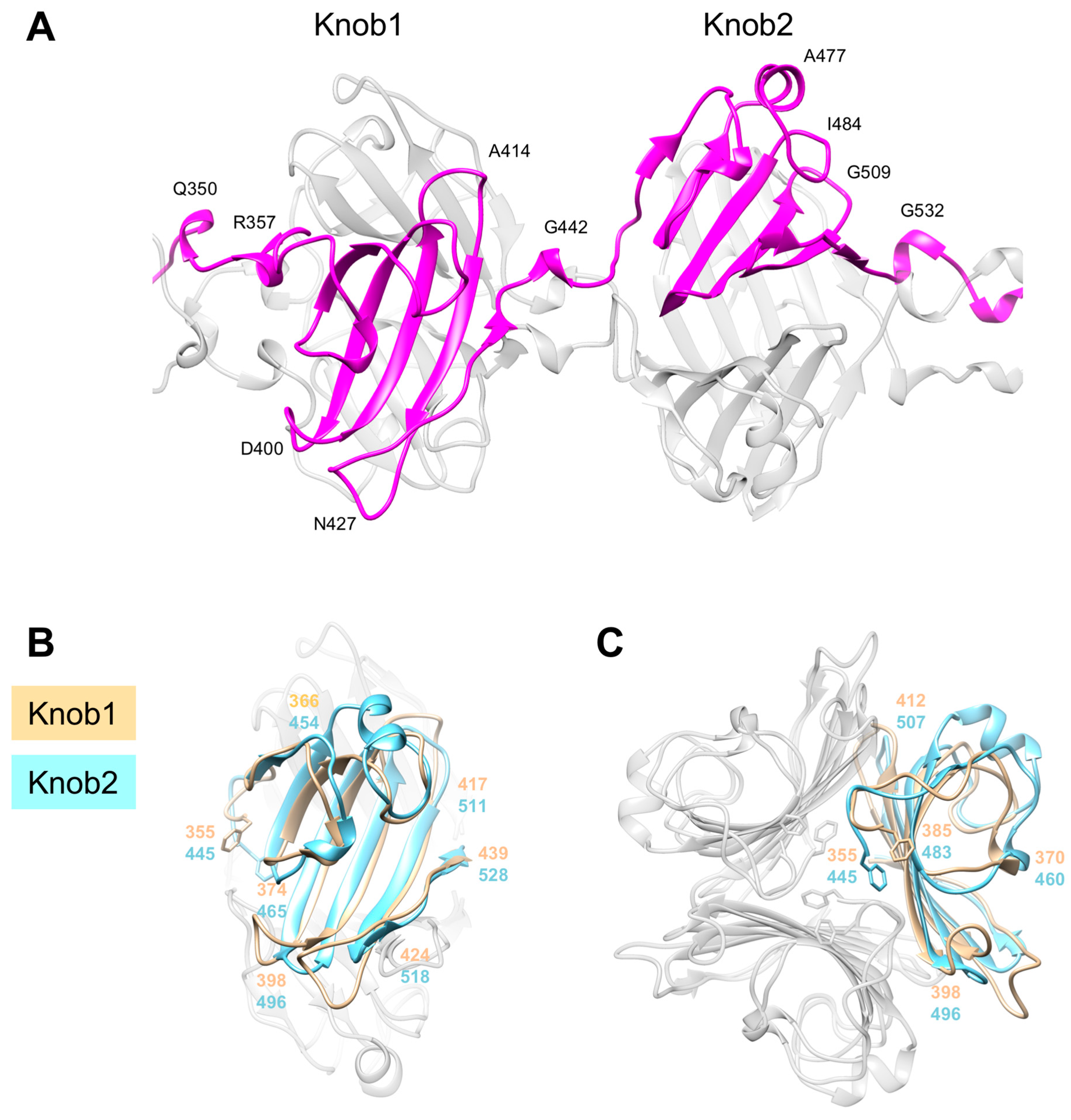
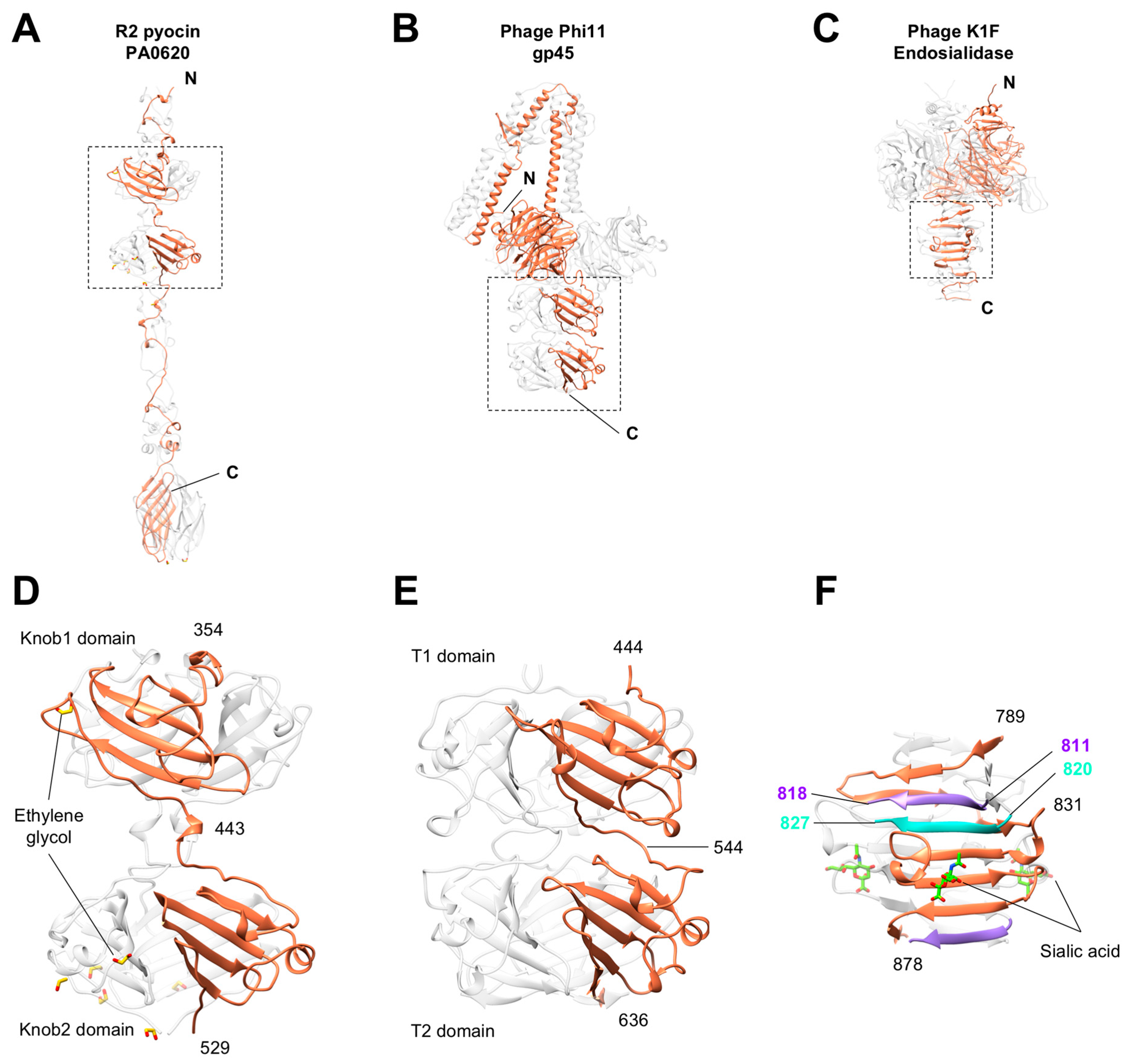
3.5. Knob-Like Domains Are Found in Saccharide-Binding Tail Fibers and Tailspikes
3.6. The C-Terminal β-Sandwich Domain Has a Lectin-Like Fold with a Negatively Charged Surface Groove
3.7. The Binding Constant of the Pyocin to the Pseudomonas Cell Surface Is Three Orders of Magnitude Greater than That of the Free Fiber
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nakayama, K.; Takashima, K.; Ishihara, H.; Shinomiya, T.; Kageyama, M.; Kanaya, S.; Ohnishi, M.; Murata, T.; Mori, H.; Hayashi, T. The R-type pyocin of Pseudomonas aeruginosa is related to P2 phage, and the F-type is related to lambda phage. Mol. Microbiol. 2000, 38, 213–231. [Google Scholar] [CrossRef] [PubMed]
- Matsui, H.; Sano, Y.; Ishihara, H.; Shinomiya, T. Regulation of pyocin genes in Pseudomonas aeruginosa by positive (prtN) and negative (prtR) regulatory genes. J. Bacteriol. 1993, 175, 1257–1263. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Kageyama, M.; Egami, F. Studies of a Pyocin. II. Mode of Production of the Pyocin. J. Biochem. 1964, 55, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Kageyama, M. Studies of a Pyocin. I. Physical and Chemical Properties. J. Biochem. 1964, 55, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Kageyama, M.; Ikeda, K.; Egami, F. Studies of a Pyocin. III. Biological Properties of the Pyocin. J. Biochem. 1964, 55, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Leiman, P.G.; Shneider, M.M. Contractile tail machines of bacteriophages. Adv. Exp. Med. Biol. 2012, 726, 93–114. [Google Scholar] [PubMed]
- Uratani, Y.; Hoshino, T. Pyocin R1 inhibits active transport in Pseudomonas aeruginosa and depolarizes membrane potential. J. Bacteriol. 1984, 157, 632–636. [Google Scholar] [PubMed]
- Taylor, N.M.; Prokhorov, N.S.; Guerrero-Ferreira, R.C.; Shneider, M.M.; Browning, C.; Goldie, K.N.; Stahlberg, H.; Leiman, P.G. Structure of the T4 baseplate and its function in triggering sheath contraction. Nature 2016, 533, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Taylor, N.M.I.; van Raaij, M.J.; Leiman, P.G. Contractile injection systems of bacteriophages and related systems. Mol. Microbiol. 2018, 108, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.R.; Gebhart, D.; Martin, D.W.; Scholl, D. Retargeting R-type pyocins to generate novel bactericidal protein complexes. Appl. Environ. Microbiol. 2008, 74, 3868–3876. [Google Scholar] [CrossRef] [PubMed]
- Scholl, D.; Cooley, M.; Williams, S.R.; Gebhart, D.; Martin, D.; Bates, A.; Mandrell, R. An engineered R-type pyocin is a highly specific and sensitive bactericidal agent for the food-borne pathogen Escherichia coli O157:H7. Antimicrob. Agents Chemother. 2009, 53, 3074–3080. [Google Scholar] [CrossRef] [PubMed]
- Gebhart, D.; Williams, S.R.; Scholl, D. Bacteriophage SP6 encodes a second tailspike protein that recognizes Salmonella enterica serogroups C2 and C3. Virology 2017, 507, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Montag, D.; Henning, U. An open reading frame in the Escherichia coli bacteriophage lambda genome encodes a protein that functions in assembly of the long tail fibers of bacteriophage T4. J. Bacteriol. 1987, 169, 5884–5886. [Google Scholar] [CrossRef] [PubMed]
- Kageyama, M. Bacteriocins and bacteriophages in Pseudomonas aeruginosa. In Microbial Drug Resistance; Mitsuhashi, S., Hashimoto, H., Eds.; University of Tokyo Press: Tokyo, Japan, 1975; pp. 291–305. [Google Scholar]
- Ito, S.; Kageyama, M.; Egami, F. Isolation and Characterization of Pyocins from Several Strains of Pseudomonas-Aeruginosa. J. Gen. Appl. Microbiol. 1970, 16, 205–214. [Google Scholar] [CrossRef]
- Scholl, D.; Martin, D.W., Jr. Antibacterial efficacy of R-type pyocins towards Pseudomonas aeruginosa in a murine peritonitis model. Antimicrob. Agents Chemother. 2008, 52, 1647–1652. [Google Scholar] [CrossRef] [PubMed]
- Kohler, T.; Donner, V.; van Delden, C. Lipopolysaccharide as shield and receptor for R-pyocin-mediated killing in Pseudomonas aeruginosa. J. Bacteriol. 2010, 192, 1921–1928. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.; Fiedler, C.; Grassl, R.; Biebl, M.; Rachel, R.; Hermo-Parrado, X.L.; Llamas-Saiz, A.L.; Seckler, R.; Miller, S.; van Raaij, M.J. Structure of the receptor-binding protein of bacteriophage det7: A podoviral tail spike in a myovirus. J. Virol. 2008, 82, 2265–2273. [Google Scholar] [CrossRef] [PubMed]
- Steinbacher, S.; Seckler, R.; Miller, S.; Steipe, B.; Huber, R.; Reinemer, P. Crystal structure of P22 tailspike protein: Interdigitated subunits in a thermostable trimer. Science 1994, 265, 383–386. [Google Scholar] [CrossRef] [PubMed]
- Steinbacher, S.; Baxa, U.; Miller, S.; Weintraub, A.; Seckler, R.; Huber, R. Crystal structure of phage P22 tailspike protein complexed with Salmonella sp. O-antigen receptors. Proc. Natl. Acad. Sci. USA 1996, 93, 10584–10588. [Google Scholar] [CrossRef] [PubMed]
- Prokhorov, N.S.; Riccio, C.; Zdorovenko, E.L.; Shneider, M.M.; Browning, C.; Knirel, Y.A.; Leiman, P.G.; Letarov, A.V. Function of bacteriophage G7C esterase tailspike in host cell adsorption. Mol. Microbiol. 2017, 105, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Leiman, P.G.; Chipman, P.R.; Kostyuchenko, V.A.; Mesyanzhinov, V.V.; Rossmann, M.G. Three-dimensional rearrangement of proteins in the tail of bacteriophage T4 on infection of its host. Cell 2004, 118, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Margolin, W.; Molineux, I.J.; Liu, J. Structural remodeling of bacteriophage T4 and host membranes during infection initiation. Proc. Natl. Acad. Sci. USA 2015, 112, E4919–E4928. [Google Scholar] [CrossRef] [PubMed]
- Van Raaij, M.J.; Schoehn, G.; Burda, M.R.; Miller, S. Crystal structure of a heat and protease-stable part of the bacteriophage T4 short tail fibre. J. Mol. Biol. 2001, 314, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Thomassen, E.; Gielen, G.; Schutz, M.; Schoehn, G.; Abrahams, J.P.; Miller, S.; van Raaij, M.J. The structure of the receptor-binding domain of the bacteriophage T4 short tail fibre reveals a knitted trimeric metal-binding fold. J. Mol. Biol. 2003, 331, 361–373. [Google Scholar] [CrossRef]
- Bartual, S.G.; Otero, J.M.; Garcia-Doval, C.; Llamas-Saiz, A.L.; Kahn, R.; Fox, G.C.; van Raaij, M.J. Structure of the bacteriophage T4 long tail fiber receptor-binding tip. Proc. Natl. Acad. Sci. USA 2010, 107, 20287–20292. [Google Scholar] [CrossRef] [PubMed]
- Granell, M.; Namura, M.; Alvira, S.; Kanamaru, S.; van Raaij, M.J. Crystal Structure of the Carboxy-Terminal Region of the Bacteriophage T4 Proximal Long Tail Fiber Protein Gp34. Viruses 2017, 9, 168. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Doval, C.; van Raaij, M.J. Structure of the receptor-binding carboxy-terminal domain of bacteriophage T7 tail fibers. Proc. Natl. Acad. Sci. USA 2012, 109, 9390–9395. [Google Scholar] [CrossRef] [PubMed]
- Hendrickson, W.A.; Ogata, C.M. Phase determination from multiwavelength anomalous diffraction measurements. Macromol. Crystallogr. Part A 1997, 276, 494–523. [Google Scholar]
- Kabsch, W. XDS. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66 Pt 2, 125–132. [Google Scholar] [CrossRef]
- Kabsch, W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66 Pt 2, 133–144. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Experimental phasing with SHELXC/D/E: Combining chain tracing with density modification. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66 Pt 4, 479–485. [Google Scholar] [CrossRef]
- Pape, T.; Schneider, T.R. HKL2MAP: A graphical user interface for macromolecular phasing with SHELX programs. J. Appl. Crystallogr. 2004, 37, 843–844. [Google Scholar] [CrossRef]
- Terwilliger, T.C. Automated structure solution, density modification and model building. Acta Crystallogr. Sect. D Biol. Crystallogr. 2002, 58, 1937–1940. [Google Scholar] [CrossRef]
- Perrakis, A.; Harkiolaki, M.; Wilson, K.S.; Lamzin, V.S. ARP/wARP and molecular replacement. Acta Crystallogr. Sect. D Biol. Crystallogr. 2001, 57, 1445–1450. [Google Scholar] [CrossRef]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- Murshudov, G.N.; Skubak, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D. Biol. Crystallogr. 2011, 67 Pt 4, 355–367. [Google Scholar] [CrossRef]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66 Pt 2, 213–221. [Google Scholar] [CrossRef]
- Painter, J.; Merritt, E.A. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr. D Biol. Crystallogr. 2006, 62 Pt 4, 439–450. [Google Scholar] [CrossRef]
- Rossmann, M.G. The Molecular Replacement Method: A Collection of Papers on the Use of Non-Crystallographic Symmetry; Gordon and Breach: New York, NY, USA, 1972. [Google Scholar]
- Mccoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011, 67 Pt 4, 235–242. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Dolinsky, T.J.; Czodrowski, P.; Li, H.; Nielsen, J.E.; Jensen, J.H.; Klebe, G.; Baker, N.A. PDB2PQR: Expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007, 35, W522–W555. [Google Scholar] [CrossRef] [PubMed]
- Unni, S.; Huang, Y.; Hanson, R.M.; Tobias, M.; Krishnan, S.; Li, W.W.; Nielsen, J.E.; Baker, N.A. Web servers and services for electrostatics calculations with APBS and PDB2PQR. J. Comput. Chem. 2011, 32, 1488–1491. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Sanner, M.F.; Olson, A.J.; Spehner, J.C. Reduced surface: An efficient way to compute molecular surfaces. Biopolymers 1996, 38, 305–320. [Google Scholar] [CrossRef]
- Soding, J.; Biegert, A.; Lupas, A.N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005, 33, W244–W248. [Google Scholar] [CrossRef] [PubMed]
- Alva, V.; Nam, S.Z.; Soding, J.; Lupas, A.N. The MPI bioinformatics Toolkit as an integrative platform for advanced protein sequence and structure analysis. Nucleic Acids Res. 2016, 44, W410–W415. [Google Scholar] [CrossRef] [PubMed]
- Dereeper, A.; Guignon, V.; Blanc, G.; Audic, S.; Buffet, S.; Chevenet, F.; Dufayard, J.F.; Guindon, S.; Lefort, V.; Lescot, M.; et al. Phylogeny.fr: Robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008, 36, W465–W469. [Google Scholar] [CrossRef] [PubMed]
- Schwarzer, D.; Browning, C.; Stummeyer, K.; Oberbeck, A.; Muhlenhoff, M.; Gerardy-Schahn, R.; Leiman, P.G. Structure and biochemical characterization of bacteriophage phi92 endosialidase. Virology 2015, 477, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Browning, C.; Shneider, M.M.; Bowman, V.D.; Schwarzer, D.; Leiman, P.G. Phage pierces the host cell membrane with the iron-loaded spike. Structure 2012, 20, 326–339. [Google Scholar] [CrossRef] [PubMed]
- Golovin, A.; Henrick, K. MSDmotif: Exploring protein sites and motifs. BMC Bioinform. 2008, 9, 312. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Rossmann, M.G. Structure of bacteriophage phi29 head fibers has a supercoiled triple repeating helix-turn-helix motif. Proc. Natl. Acad. Sci. USA 2011, 108, 4806–4810. [Google Scholar] [CrossRef] [PubMed]
- Cerritelli, M.E.; Wall, J.S.; Simon, M.N.; Conway, J.F.; Steven, A.C. Stoichiometry and domainal organization of the long tail-fiber of bacteriophage T4: A hinged viral adhesin. J. Mol. Biol. 1996, 260, 767–780. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Strelkov, S.V.; Mesyanzhinov, V.V.; Rossmann, M.G. Structure of bacteriophage T4 fibritin: A segmented coiled coil and the role of the C-terminal domain. Structure 1997, 5, 789–798. [Google Scholar] [CrossRef]
- Kanamaru, S.; Gassner, N.C.; Ye, N.; Takeda, S.; Arisaka, F. The C-terminal fragment of the precursor tail lysozyme of bacteriophage T4 stays as a structural component of the baseplate after cleavage. J. Bacteriol. 1999, 181, 2739–2744. [Google Scholar] [PubMed]
- Shneider, M.M.; Buth, S.A.; Ho, B.T.; Basler, M.; Mekalanos, J.J.; Leiman, P.G. PAAR-repeat proteins sharpen and diversify the type VI secretion system spike. Nature 2013, 500, 350–353. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Chordia, M.D.; Cooper, D.R.; Chruszcz, M.; Muller, P.; Sheldrick, G.M.; Minor, W. Validation of metal-binding sites in macromolecular structures with the CheckMyMetal web server. Nat. Protoc. 2014, 9, 156–170. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Cooper, D.R.; Porebski, P.J.; Shabalin, I.G.; Handing, K.B.; Minor, W. CheckMyMetal: A macromolecular metal-binding validation tool. Acta Crystallogr. D Struct. Biol. 2017, 73 Pt 3, 223–233. [Google Scholar] [CrossRef]
- Buth, S.A.; Menin, L.; Shneider, M.M.; Engel, J.; Boudko, S.P.; Leiman, P.G. Structure and Biophysical Properties of a Triple-Stranded Beta-Helix Comprising the Central Spike of Bacteriophage T4. Viruses 2015, 7, 4676–4706. [Google Scholar] [CrossRef] [PubMed]
- Van Raaij, M.J.; Mitraki, A.; Lavigne, G.; Cusack, S. A triple β-spiral in the adenovirus fibre shaft reveals a new structural motif for a fibrous protein. Nature 1999, 401, 935–938. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Wang, F.; Taylor, N.M.I.; Guerrero-Ferreira, R.C.; Leiman, P.G.; Egelman, E.H. Refined Cryo-EM Structure of the T4 Tail Tube: Exploring the Lowest Dose Limit. Structure 2017, 25, 1436–1441 e2. [Google Scholar] [CrossRef] [PubMed]
- Holm, L.; Laakso, L.M. Dali server update. Nucleic Acids Res. 2016, 44, W351–W355. [Google Scholar] [CrossRef] [PubMed]
- Koc, C.; Xia, G.; Kuhner, P.; Spinelli, S.; Roussel, A.; Cambillau, C.; Stehle, T. Structure of the host-recognition device of Staphylococcus aureus phage varphi11. Sci. Rep. 2016, 6, 27581. [Google Scholar] [CrossRef] [PubMed]
- Stummeyer, K.; Dickmanns, A.; Muhlenhoff, M.; Gerardy-Schahn, R.; Ficner, R. Crystal structure of the polysialic acid-degrading endosialidase of bacteriophage K1F. Nat. Struct. Mol. Biol. 2005, 12, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Schulz, E.C.; Neumann, P.; Gerardy-Schahn, R.; Sheldrick, G.M.; Ficner, R. Structure analysis of endosialidase NF at 0.98 A resolution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66 Pt 2, 176–180. [Google Scholar] [CrossRef]
- Thompson, J.E.; Pourhossein, M.; Waterhouse, A.; Hudson, T.; Goldrick, M.; Derrick, J.P.; Roberts, I.S. The K5 lyase KflA combines a viral tail spike structure with a bacterial polysaccharide lyase mechanism. J. Biol. Chem. 2010, 285, 23963–23969. [Google Scholar] [CrossRef] [PubMed]
- Harada, K.; Yamashita, E.; Nakagawa, A.; Miyafusa, T.; Tsumoto, K.; Ueno, T.; Toyama, Y.; Takeda, S. Crystal structure of the C-terminal domain of Mu phage central spike and functions of bound calcium ion. Biochim. Biophys. Acta 2013, 1834, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Kanamaru, S.; Leiman, P.G.; Kostyuchenko, V.A.; Chipman, P.R.; Mesyanzhinov, V.V.; Arisaka, F.; Rossmann, M.G. Structure of the cell-puncturing device of bacteriophage T4. Nature 2002, 415, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Scholl, D.; Merril, C. The genome of bacteriophage K1F, a T7-like phage that has acquired the ability to replicate on K1 strains of Escherichia coli. J. Bacteriol. 2005, 187, 8499–8503. [Google Scholar] [CrossRef] [PubMed]
- Lescar, J.; Sanchez, J.F.; Audfray, A.; Coll, J.L.; Breton, C.; Mitchell, E.P.; Imberty, A. Structural basis for recognition of breast and colon cancer epitopes Tn antigen and Forssman disaccharide by Helix pomatia lectin. Glycobiology 2007, 17, 1077–1083. [Google Scholar] [CrossRef] [PubMed]
- Aragao, K.S.; Satre, M.; Imberty, A.; Varrot, A. Structure determination of Discoidin II from Dictyostelium discoideum and carbohydrate binding properties of the lectin domain. Proteins 2008, 73, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Muller, J.J.; Barbirz, S.; Heinle, K.; Freiberg, A.; Seckler, R.; Heinemann, U. An intersubunit active site between supercoiled parallel β helices in the trimeric tailspike endorhamnosidase of Shigella flexneri Phage Sf6. Structure 2008, 16, 766–775. [Google Scholar] [CrossRef] [PubMed]
- Ricagno, S.; Campanacci, V.; Blangy, S.; Spinelli, S.; Tremblay, D.; Moineau, S.; Tegoni, M.; Cambillau, C. Crystal structure of the receptor-binding protein head domain from Lactococcus lactis phage bIL170. J. Virol. 2006, 80, 9331–9335. [Google Scholar] [CrossRef] [PubMed]
- Kitov, P.I.; Bundle, D.R. On the nature of the multivalency effect: A thermodynamic model. J. Am. Chem. Soc. 2003, 125, 16271–16284. [Google Scholar] [CrossRef] [PubMed]
- Barbirz, S.; Muller, J.J.; Uetrecht, C.; Clark, A.J.; Heinemann, U.; Seckler, R. Crystal structure of Escherichia coli phage HK620 tailspike: Podoviral tailspike endoglycosidase modules are evolutionarily related. Mol. Microbiol. 2008, 69, 303–316. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crystal | R1 Fiber Native | R2 Fiber Native | R2 Fiber SeMet |
|---|---|---|---|
| Data collection | |||
| Wavelength (Å) | 1.0000 | 1.0000 | 0.9797 |
| Number of frames | 720 | 780 | 1440 |
| Frame width (°) | 0.25 | 0.25 | 0.25 |
| Space group | P212121 | P212121 | P212121 |
| Cell dimensions | |||
| a, b, c (Å) | 144.08, 154.46, 198.83 | 56.12, 126.35, 431.25 | 56.20, 126.82, 433.43 |
| Resolution (Å) | 50.0–2.3 (2.46–2.32) | 50.0–1.9 (2.01–1.90) | 50.0–2.4 (2.54–2.40) |
| Rmeas (%) | 10.5 (61.4) | 13.0 (55.0) | 7.1 (18.0) |
| I/σI | 8.8 (2.1) | 8.8 (2.6) | 20.8 (8.2) |
| CC ½ (%) | 99.5 (81.0) | 99.4 (81.3) | 99.8(98.0) |
| Completeness (%) | 99.2 (96.2) | 99.6 (98.5) | 99.3 (96.1) |
| Redundancy | 3.4 (3.2) | 3.8 (3.8) | 6.7 (6.2) |
| Anomalous signal * | 0.77 (0.65) | 0.81 (0.75) | 1.42 (0.78) |
| Refinement | NA ** | ||
| Resolution | 47.69–2.32 | 49.96–1.90 | |
| No. reflections used in refinement | 192,247 | 246,066 | |
| No. atoms (non-H) | 18,886 | 20,843 | |
| Protein | 16,848 | 16,720 | |
| Ligand/ion | 10 | 98 | |
| Water | 2028 | 4025 | |
| Rwork/Rfree | 0.169/0.206 | 0.160/0.208 | |
| Average B factor (Å2) | 60.7 | 24.8 | |
| Protein | 61.3 | 22.2 | |
| Ligand/Ion | 91.1 | 29.5 | |
| Water | 56.1 | 35.7 | |
| R.m.s. deviations | |||
| Bond lengths (Å) | 0.003 | 0.009 | |
| Bond angles (°) | 0.600 | 0.886 | |
| Ramachandran plot statistics | |||
| Favored (%) | 97.49 | 97.24 | |
| Allowed (%) | 2.51 | 2.76 | |
| Outliers (%) | 0.00 | 0.00 | |
| Protein Data Bank accession code | 6CL5 | 6CL6 |
| Metal Ion | B-Factor (Å2) | Ligands | Valence | nVECSUM | Geometry | gRMSD (°) | Vacancy |
|---|---|---|---|---|---|---|---|
| R1 fiber | |||||||
| Fe2+ 1 (ABC) | 81.0 (102.7) | N6 | 1.8 | 0.05 | OH | 5.7 | 0 |
| Fe2+ 2 (DEF) | 68.7 (79.7) | N6 | 1.9 | 0.10 | OH | 4.2 | 0 |
| Mg2+ 3 (ABC) | 153.5 (87.5) | O6 | 1.6 | 0.15 | OH | 5.8 | 0 |
| Mg2+ 4 (DEF) | 89.8 (63.9) | O6 | 1.5 | 0.10 | OH | 11.5 | 0 |
| Na+ 5 (ABC) | 65.6 (78.0) | O5 | 0.9 | 0.18 | OH | 13.0 | 16% |
| Na+ 6 (ABC) | 75.8 (83.8) | O5 | 1.0 | 0.25 | TB | 13.2 | 0 |
| Na+ 7 (ABC) | 77.9 (94.7) | O6 | 0.9 | 0.18 | OH | 23.0 | 0 |
| Na+ 8 (DEF) | 84.6 (66.2) | O5 | 0.9 | 0.17 | OH | 14.0 | 16% |
| Na+ 9 (DEF) | 138.4 (98.4) | O5 | 1.2 | 0.08 | OH | 16.1 | 16% |
| Na+ 10 (DEF) | 75.5 (91.1) | O5 | 0.9 | 0.21 | OH | 17.3 | 16% |
| R2 fiber | |||||||
| Fe2+ 1 (ABC) | 41.6 (33.3) | N6 | 1.5 | 0.04 | OH | 4.4 | 0 |
| Fe2+ 2 (DEF) | 34.7 (35.2) | N6 | 1.6 | 0.11 | OH | 4.2 | 0 |
| Mg2+ 3 (ABC) | 23.5 (22.8) | O6 | 2.0 | 0.07 | OH | 9.0 | 0 |
| Mg2+ 4 (DEF) | 26.1 (21.2) | O6 | 1.8 | 0.07 | OH | 9.2 | 0 |
| Ca2+ 5 (ABC) | 16.7 (20.4) | O7 | 2.1 | 0.11 | PB | 11.4 | 0 |
| Ca2+ 6 (ABC) | 13.9 (16.8) | O7 | 2.2 | 0.11 | PB | 9.7 | 0 |
| Ca2+ 7 (ABC) | 15.1 (20.0) | O7 | 2.4 | 0.09 | PB | 9.9 | 0 |
| Ca2+ 8 (DEF) | 19.7 (22.9) | O7 | 1.9 | 0.16 | PB | 11.5 | 0 |
| Ca2+ 9 (DEF) | 18.0 (19.3) | O7 | 1.9 | 0.11 | PB | 10.4 | 0 |
| Ca2+ 10 (DEF) | 19.5 (21.6) | O7 | 2.0 | 0.11 | PB | 10.2 | 0 |
| PDB Code | Z-Score | RMSD (Å) | Lali | # res | % id | Hit Description |
|---|---|---|---|---|---|---|
| 4MTM | 11.2/12.6 | 2.3/2.0 | 90/91 | 137 | 26/30 | Tail fiber of bacteriophage AP22 |
| 3WMP | 8.0/7.6 | 2.7/2.2 | 82/73 | 94 | 13/15 | SLL-2, galactose-binding lectin |
| 2VME | 7.8/7.8 | 4.1/3.7 | 88/80 | 256 | 17/13 | Discoidin-2; oligosaccharide-binding lectin |
| 2CGZ | 7.8/6.5 | 2.6/2.1 | 81/71 | 101 | 20/15 | Helix pomatia agglutinin; oligosaccharide-binding lectin |
| 4Y9V | 6.9/6.5 | 3.3/3.1 | 87/80 | 603 | 13/13 | Tailspike of bacteriophage AP22 |
| 2VBK | 5.9/6.1 | 3.3/3.2 | 79/75 | 511 | 13/8 | Tailspike of bacteriophage SF6 |
| 2FSD | 4.7/5.5 | 3.2/3.0 | 82/83 | 110 | 10/11 | Tail fiber of bacteriophage bIL170 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buth, S.A.; Shneider, M.M.; Scholl, D.; Leiman, P.G. Structure and Analysis of R1 and R2 Pyocin Receptor-Binding Fibers. Viruses 2018, 10, 427. https://doi.org/10.3390/v10080427
Buth SA, Shneider MM, Scholl D, Leiman PG. Structure and Analysis of R1 and R2 Pyocin Receptor-Binding Fibers. Viruses. 2018; 10(8):427. https://doi.org/10.3390/v10080427
Chicago/Turabian StyleButh, Sergey A., Mikhail M. Shneider, Dean Scholl, and Petr G. Leiman. 2018. "Structure and Analysis of R1 and R2 Pyocin Receptor-Binding Fibers" Viruses 10, no. 8: 427. https://doi.org/10.3390/v10080427
APA StyleButh, S. A., Shneider, M. M., Scholl, D., & Leiman, P. G. (2018). Structure and Analysis of R1 and R2 Pyocin Receptor-Binding Fibers. Viruses, 10(8), 427. https://doi.org/10.3390/v10080427