Biodiversity, Evolution and Ecological Specialization of Baculoviruses: A Treasure Trove for Future Applied Research


Abstract
1. Introduction
2. Materials and Methods
2.1. Virus Isolate Sequence Database
2.2. Host Ecology Database
2.3. Baculovirus Core-Genome Phylogeny
2.4. Baculovirus Isolate Phylogeny
2.5. Species Delimitation
2.6. Baculovirus Species Phylogeny
2.7. Phylogeny-Trait Correlation and Ancestral State Estimations
2.8. Host-Virus Cophylogeny
3. Results
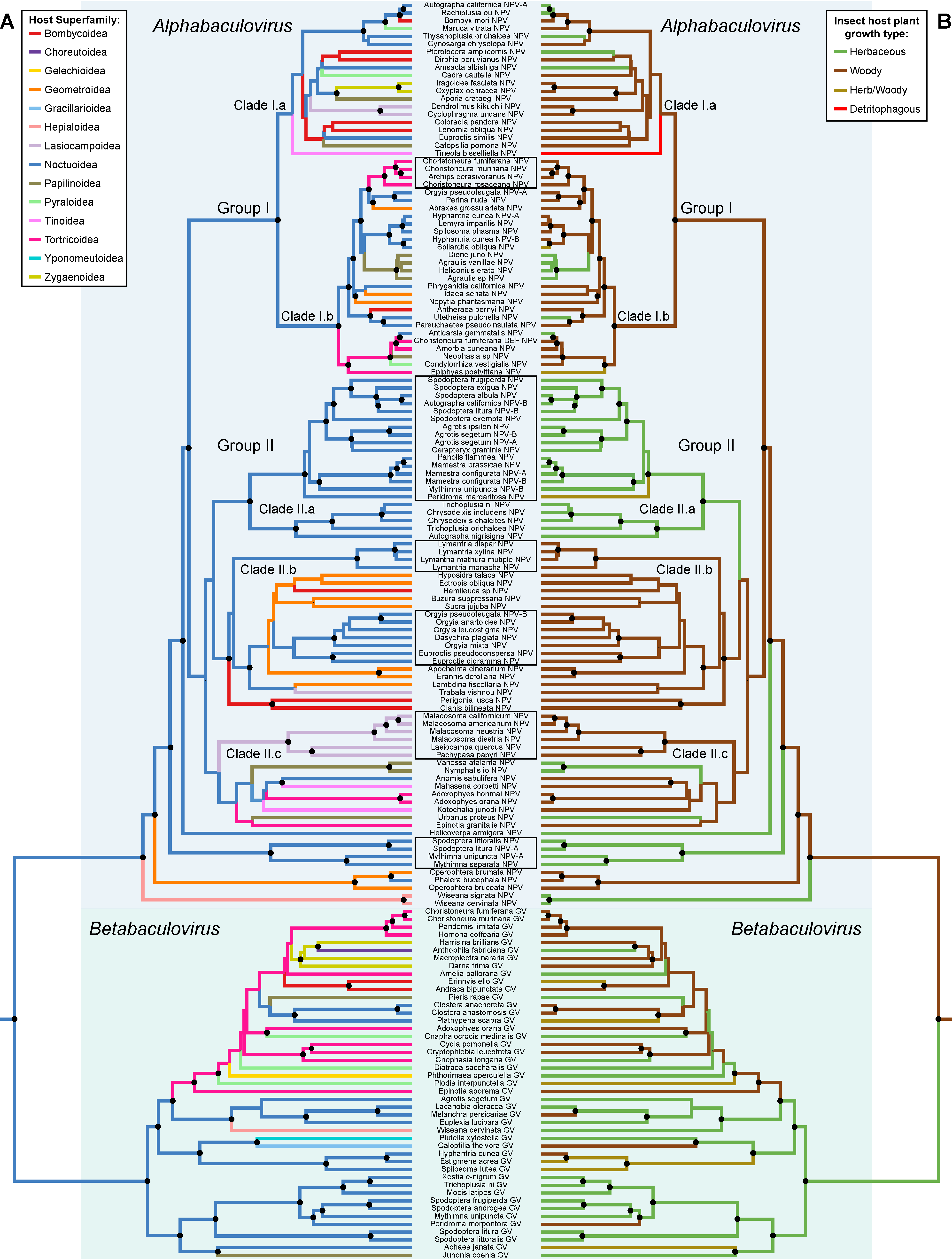
3.1. Baculovirus Phylogeny
3.2. Species Delimitation
3.3. Phylogenetic Conservatism and Host Shifts
3.4. Cophylogeny between Baculoviruses and Their Lepidopteran Hosts
4. Discussion
4.1. Reconstructing Phylogenies and Delineating Species
4.2. Evolution of Host Use and Taxa Sampling
4.3. Cophylogeny and Host Shifts
4.4. Ecological Specialization
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Van Oers, M.M.; Pijlman, G.P.; Vlak, J.M. Thirty years of baculovirus-insect cell protein expression: From dark horse to mainstream technology. J. Gen. Virol. 2015, 96, 6–23. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Kajikawa, M.; Maenaka, K.; Park, E.Y. Silkworm expression system as a platform technology in life science. Appl. Microbiol. Biotechnol. 2010, 85, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.J.; Lefkowitz, E.J.; King, A.M.Q.; Carstens, E.B. Recently agreed changes to the International Code of Virus Classification and Nomenclature. Arch. Virol. 2013, 158, 2633–2639. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Lin, X.-D.; Tian, J.-H.; Chen, L.-J.; Chen, X.; Li, C.-X.; Qin, X.-C.; Li, J.; Cao, J.-P.; Eden, J.-S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Peterson, A.T. Defining viral species: Making taxonomy useful. Virol. J. 2014, 11, 131. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Adams, M.J.; Benko, M.; Breitbart, M.; Brister, J.R.; Carstens, E.B.; Davison, A.J.; Delwart, E.; Gorbalenya, A.E.; Harrach, B.; et al. Consensus statement: Virus taxonomy in the age of metagenomics. Nat. Rev. Microbiol. 2017, 15, 161. [Google Scholar] [CrossRef] [PubMed]
- Jehle, J.A.; Lange, M.; Wang, H.; Hu, Z.; Wang, Y.; Hauschild, R. Molecular identification and phylogenetic analysis of baculoviruses from Lepidoptera. Virology 2006, 346, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, T.; Barraclough, T.G. Delimiting Species Using Single-Locus Data and the Generalized Mixed Yule Coalescent Approach: A Revised Method and Evaluation on Simulated Data Sets. Syst. Biol. 2013, 62, 707–724. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Kapli, P.; Pavlidis, P.; Stamatakis, A. A general species delimitation method with applications to phylogenetic placements. Bioinformatics 2013, 29, 2869–2876. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, D.; Francke, O.F. Two DNA barcodes and morphology for multi-method species delimitation in Bonnetina tarantulas (Araneae: Theraphosidae). Mol. Phylogenet. Evol. 2016, 101, 176–193. [Google Scholar] [CrossRef] [PubMed]
- Ratnasingham, S.; Hebert, P.D.N. A DNA-based registry for all animal species: The barcode index number (BIN) system. PLoS ONE 2013, 8, e66213. [Google Scholar] [CrossRef] [PubMed]
- Barraclough, T.G.; Hughes, M.; Ashford-Hodges, N.; Fujisawa, T. Inferring evolutionarily significant units of bacterial diversity from broad environmental surveys of single-locus data. Biol. Lett. 2009, 5, 425–428. [Google Scholar] [CrossRef] [PubMed]
- Martignoni, M.E.; Iwai, P.J. A catalogue of viral diseases of insects, mites and ticks. In Microbial Control of Pests and Plant Diseases 1970–1980; Burges, H.D., Ed.; Academic Press: London, UK, 1981; pp. 897–911. [Google Scholar]
- Herniou, E.A.; Arif, B.M.; Becnel, J.J.; Bissard, G.W.; Bonning, B.; Harrison, R.; Jehle, J.A.; Theilmann, D.A.; Vlak, J.M. Baculoviridae. In Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses; King, A.M., Lefkowitz, E., Adams, M.J., Carstens, E.B., Eds.; Elsevier: Amsterdam, The Netherlands, 2011; pp. 163–173. [Google Scholar]
- Herniou, E.A.; Olszewski, J.A.; O’Reilly, D.R.; Cory, J.S. Ancient coevolution of baculoviruses and their insect hosts. J. Virol. 2004, 78, 3244–3251. [Google Scholar] [CrossRef] [PubMed]
- Herniou, E.A.; Luque, T.; Chen, X.; Vlak, J.M.; Winstanley, D.; Cory, J.S.; O’Reilly, D.R. Use of whole genome sequence data to infer baculovirus phylogeny. J. Virol. 2001, 75, 8117–8126. [Google Scholar] [CrossRef] [PubMed]
- Herniou, E.A.; Jehle, J.A. Baculovirus phylogeny and evolution. Curr. Drug Targets 2007, 8, 1043–1050. [Google Scholar] [CrossRef] [PubMed]
- Thézé, J.; Bézier, A.; Periquet, G.; Drezen, J.-M.; Herniou, E.A. Paleozoic origin of insect large dsDNA viruses. Proc. Natl. Acad. Sci. USA 2011, 108, 15931–15935. [Google Scholar] [CrossRef] [PubMed]
- Hamblin, M.; Van Beek, N.A.; Hughes, P.R.; Wood, H.A. Co-occlusion and persistence of a baculovirus mutant lacking the polyhedrin gene. Appl. Environ. Microbiol. 1990, 56, 3057–3062. [Google Scholar] [PubMed]
- Hughes, D.S.; Possee, R.D.; King, L.A. Evidence for the presence of a low-level, persistent baculovirus infection of Mamestra brassicae insects. J. Gen. Virol. 1997, 78, 1801–1805. [Google Scholar] [CrossRef] [PubMed]
- Burden, J.P.; Nixon, C.P.; Hodgkinson, A.E.; Possee, R.D.; Sait, S.M.; King, L.A.; Hails, R.S. Covert infections as a mechanism for long-term persistence of baculoviruses. Ecol. Lett. 2003, 6, 524–531. [Google Scholar] [CrossRef]
- Vilaplana, L.; Wilson, K.; Redman, E.M.; Cory, J.S. Pathogen persistence in migratory insects: High levels of vertically-transmitted virus infection in field populations of the African armyworm. Evol. Ecol. 2010, 24, 147–160. [Google Scholar] [CrossRef]
- Thézé, J.; Cabodevilla, O.; Palma, L.; Williams, T.; Caballero, P.; Herniou, E.A. Genomic diversity in European Spodoptera exigua multiple nucleopolyhedrovirus isolates. J. Gen. Virol. 2014, 95, 2297–2309. [Google Scholar] [CrossRef] [PubMed]
- Cory, J.S. Insect virus transmission: Different routes to persistence. Curr. Opin. Insect Sci. 2015, 8, 130–135. [Google Scholar] [CrossRef]
- Cory, J.S.; Hoover, K. Plant-mediated effects in insect-pathogen interactions. Trends Ecol. Evol. 2006, 21, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, D.J.; Vanbergen, A.J.; Hartley, S.E.; Hails, R.S.; Cory, J.S. Differential selection of baculovirus genotypes mediated by different species of host food plant. Ecol. Lett. 2002, 5, 512–518. [Google Scholar] [CrossRef]
- Cory, J.S.; Myers, J.H. Adaptation in an insect host-plant pathogen interaction. Ecol. Lett. 2004, 7, 632–639. [Google Scholar] [CrossRef]
- Shikano, I. Evolutionary Ecology of Multitrophic Interactions between Plants, Insect Herbivores and Entomopathogens. J. Chem. Ecol. 2017, 43, 586–598. [Google Scholar] [CrossRef] [PubMed]
- Robinson, G.S.; Ackery, P.R.; Kitching, I.J.; Beccaloni, G.W.; Hernández, L.M. HOSTS—A Database of the World’s Lepidopteran Hostplants; Natural History Museum: London, UK, 2010; Available online: http://www.nhm.ac.uk/hosts (accessed on 15 April 2018).
- Ratnasingham, S.; Hebert, P.D.N. BOLD: The Barcode of Life Data System (http://www.barcodinglife.org). Mol. Ecol. Notes 2007, 7, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Van Nieukerken, E.J.; Kaila, L.; Kitching, I.J.; Kristensen, N.P.; Lees, D.C.; Minet, J.; Mitter, C.; Mutanen, M.; Regier, J.C.; Simonsen, T.J.; et al. Order Lepidoptera Linnaeus, 1758. In Animal Biodiversity: An Outline of Higher-Level Classification and Survey of Taxonomic Richness; Zhang, Z.Q., Ed.; Zootaxa: Auckland, New Zealand, 2011; Volume 3148, pp. 212–221. [Google Scholar]
- Garavaglia, M.J.; Miele, S.A.B.; Iserte, J.A.; Belaich, M.N.; Ghiringhelli, P.D. The ac53, ac78, ac101, and ac103 genes are newly discovered core genes in the family Baculoviridae. J. Virol. 2012, 86, 12069–12079. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Kapli, P.; Lutteropp, S.; Zhang, J.; Kobert, K.; Pavlidis, P.; Stamatakis, A.; Flouri, T. Multi-rate Poisson tree processes for single-locus species delimitation under maximum likelihood and Markov chain Monte Carlo. Bioinformatics 2017, 33, 1630–1638. [Google Scholar] [CrossRef] [PubMed]
- Masters, B.C.; Fan, V.; Ross, H.A. Species Delimitation—A Geneious plugin for the exploration of species boundaries. Mol. Ecol. Resour. 2011, 11, 154–157. [Google Scholar] [CrossRef] [PubMed]
- Paradis, E.; Claude, J.; Strimmer, K. APE: Analyses of phylogenetics and evolution in R language. Bioinformatics 2004, 20, 289–290. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.; Rambaut, A.; Pybus, O.G. Correlating viral phenotypes with phylogeny: Accounting for phylogenetic uncertainty. Infect. Genet. Evol. 2008, 8, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Charleston, M.A. TreeMap 3B, Build 1234. 2011. Available online: sites.google.com/site/cophylogeny/software (accessed on 15 April 2018).
- Charleston, M.A.; Robertson, D.L. Preferential host switching by primate lentiviruses can account for phylogenetic similarity with the primate phylogeny. Syst. Biol. 2002, 51, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Mutanen, M.; Wahlberg, N.; Kaila, L. Comprehensive gene and taxon coverage elucidates radiation patterns in moths and butterflies. Proc. R. Soc. B 2010, 277, 2839–2848. [Google Scholar] [CrossRef] [PubMed]
- Regier, J.C.; Mitter, C.; Zwick, A.; Bazinet, A.L.; Cummings, M.P.; Kawahara, A.Y.; Sohn, J.-C.; Zwickl, D.J.; Cho, S.; Davis, D.R.; et al. A large-scale, higher-level, molecular phylogenetic study of the insect order Lepidoptera (moths and butterflies). PLoS ONE 2013, 8, e58568. [Google Scholar] [CrossRef] [PubMed]
- Wahlberg, N.; Wheat, C.W.; Peña, C. Timing and patterns in the taxonomic diversification of lepidoptera (butterflies and moths). PLoS ONE 2013, 8, e80875. [Google Scholar] [CrossRef] [PubMed]
- Toussaint, E.F.A.; Condamine, F.L.; Kergoat, G.J.; Capdevielle-Dulac, C.; Barbut, J.; Silvain, J.-F.; Le Ru, B.P. Palaeoenvironmental shifts drove the adaptive radiation of a noctuid stemborer tribe (Lepidoptera, Noctuidae, Apameini) in the miocene. PLoS ONE 2012, 7, e41377. [Google Scholar] [CrossRef] [PubMed]
- DeWaard, J.R.; Mitchell, A.; Keena, M.A.; Gopurenko, D.; Boykin, L.M.; Armstrong, K.F.; Pogue, M.G.; Lima, J.; Floyd, R.; Hanner, R.H.; et al. Towards a global barcode library for Lymantria (Lepidoptera: Lymantriinae) tussock moths of biosecurity concern. PLoS ONE 2010, 5, e14280. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wahlberg, N.; Holloway, J.D.; Bergsten, J.; Fan, X.; Janzen, D.H.; Hallwachs, W.; Wen, L.; Wang, M.; Nylin, S. Molecular phylogeny of Lymantriinae (Lepidoptera, Noctuoidea, Erebidae) inferred from eight gene regions. Cladistics 2015, 31, 579–592. [Google Scholar] [CrossRef]
- Zolotuhin, V.V.; Efimov, R.V.; Anikin, V.V.; Demin, A.G.; Knushevitskaya, M.V. Changes in the suprageneric classification of Lasiocampidae (Lepidoptera) based on the nucleotide sequence of gene EF-1α. Entmol. Rev. 2012, 92, 531–547. [Google Scholar] [CrossRef]
- Kergoat, G.J.; Prowell, D.P.; Le Ru, B.P.; Mitchell, A.; Dumas, P.; Clamens, A.-L.; Condamine, F.L.; Silvain, J.-F. Disentangling dispersal, vicariance and adaptive radiation patterns: A case study using armyworms in the pest genus Spodoptera (Lepidoptera: Noctuidae). Mol. Phylogenet. Evol. 2012, 65, 855–870. [Google Scholar] [CrossRef] [PubMed]
- Virus Taxonomy: The Classification and Nomenclature of Viruses. The Online (10th) Report of the ICTV. Available online: https://talk.ictvonline.org/ictv-reports/ictv_online_report/ (accessed on 1 July 2018).
- Cory, J. Use of baculoviruses as biological insecticides. Mol. Biotechnol. 1997, 7, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Sanjuán, R.; Nebot, M.R.; Chirico, N.; Mansky, L.M.; Belshaw, R. Viral mutation rates. J. Virol. 2010, 84, 9733–9748. [Google Scholar] [CrossRef] [PubMed]
- Esselstyn, J.A.; Evans, B.J.; Sedlock, J.L.; Anwarali Khan, F.A.; Heaney, L.R. Single-locus species delimitation: A test of the mixed Yule-coalescent model, with an empirical application to Philippine round-leaf bats. Proc. R. Soc. B 2012, 279, 3678–3686. [Google Scholar] [CrossRef] [PubMed]
- Fontaneto, D.; Herniou, E.A.; Boschetti, C.; Caprioli, M.; Melone, G.; Ricci, C.; Barraclough, T.G. Independently evolving species in asexual bdelloid rotifers. PLoS Biol. 2007, 5, e87. [Google Scholar] [CrossRef] [PubMed]
- Monaghan, M.T.; Wild, R.; Elliot, M.; Fujisawa, T.; Balke, M.; Inward, D.J.G.; Lees, D.C.; Ranaivosolo, R.; Eggleton, P.; Barraclough, T.G.; et al. Accelerated species inventory on Madagascar using coalescent-based models of species delineation. Syst. Biol. 2009, 58, 298–311. [Google Scholar] [CrossRef] [PubMed]
- Tehrani, J.J.; Collard, M.; Shennan, S.J. The cophylogeny of populations and cultures: Reconstructing the evolution of Iranian tribal craft traditions using trees and jungles. Philos. Trans. R. Soc. B 2010, 365, 3865–3874. [Google Scholar] [CrossRef] [PubMed]
- Hafner, M.S.; Page, R.D.M. Molecular phylogenies and host-parasite cospeciation: Gophers and lice as a model system. Philos. Trans. R. Soc. B 1995, 349, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Brooks, D.R.; McLennan, D.A. Phylogeny, Ecology, and Behavior: A Research Program in Comparative Biology; University of Chicago Press: Chicago, IL, USA, 1991. [Google Scholar]
- Page, R.D.M. Tangled Trees: Phylogeny, Cospeciation and Coevolution; Page, R.D.M., Ed.; University of Chicago Press: Chicago, IL, USA, 2003. [Google Scholar]
- Myers, J.H.; Cory, J.S. Ecology and evolution of pathogens in natural populations of Lepidoptera. Evol. Appl. 2016, 9, 231–247. [Google Scholar] [CrossRef] [PubMed]
- Cory, J.S. Ecological impacts of virus insecticides: Host range and non-target organisms. In Environmental Impacts of Microbial Insecticides; Hokkanen, H., Hajek, A., Eds.; Springer: Dordrecht, The Netherlands, 2003; pp. 73–91. [Google Scholar]
- Markov, P.V.; Pepin, J.; Frost, E.; Deslandes, S.; Labbe, A.C.; Pybus, O.G. Phylogeography and molecular epidemiology of hepatitis C virus genotype 2 in Africa. J. Gen. Virol. 2009, 90, 2086–2096. [Google Scholar] [CrossRef] [PubMed]
- May, F.J.; Davis, C.T.; Tesh, R.B.; Barrett, A.D.T. Phylogeography of West Nile virus: From the cradle of evolution in Africa to Eurasia, Australia, and the Americas. J. Virol. 2011, 85, 2964–2974. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, S.D.; Cory, J.S.; Wilson, K.R.; Sait, S.M.; Hails, R.S. Modified Behavior in Baculovirus-Infected Lepidopteran Larvae and Its Impact on the Spatial Distribution of Inoculum. Biol. Control 1996, 7, 299–306. [Google Scholar] [CrossRef]
- Hoover, K.; Grove, M.; Gardner, M.; Hughes, D.P.; McNeil, J.; Slavicek, J. A gene for an extended phenotype. Science 2011, 333, 1401. [Google Scholar] [CrossRef] [PubMed]
- Thézé, J.; Takatsuka, J.; Nakai, M.; Arif, B.; Herniou, E.A. Gene Acquisition Convergence between Entomopoxviruses and Baculoviruses. Viruses 2015, 7, 1960–1974. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Traits | AI 1 | PS 1 | PD 1 | NT 1 | NR 1 | UniFrac 1 | MC 2 |
|---|---|---|---|---|---|---|---|
| Host superfamily | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001/21 |
| Host family | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001/21 |
| Host subfamily | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001/7 |
| Host biogeography distribution (ecozone) | <0.001 | <0.001 | <0.001 | <0.001 | 0.58 | 0.029 | 0.019/4 |
| Insect host plant growth type (Herb/Woody) | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001/22 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thézé, J.; Lopez-Vaamonde, C.; Cory, J.S.; Herniou, E.A. Biodiversity, Evolution and Ecological Specialization of Baculoviruses: A Treasure Trove for Future Applied Research. Viruses 2018, 10, 366. https://doi.org/10.3390/v10070366
Thézé J, Lopez-Vaamonde C, Cory JS, Herniou EA. Biodiversity, Evolution and Ecological Specialization of Baculoviruses: A Treasure Trove for Future Applied Research. Viruses. 2018; 10(7):366. https://doi.org/10.3390/v10070366
Chicago/Turabian StyleThézé, Julien, Carlos Lopez-Vaamonde, Jenny S. Cory, and Elisabeth A. Herniou. 2018. "Biodiversity, Evolution and Ecological Specialization of Baculoviruses: A Treasure Trove for Future Applied Research" Viruses 10, no. 7: 366. https://doi.org/10.3390/v10070366
APA StyleThézé, J., Lopez-Vaamonde, C., Cory, J. S., & Herniou, E. A. (2018). Biodiversity, Evolution and Ecological Specialization of Baculoviruses: A Treasure Trove for Future Applied Research. Viruses, 10(7), 366. https://doi.org/10.3390/v10070366