1. Introduction
An increase in the occurrence of antibiotic-resistant bacteria has sparked an interest in phage–host interactions [
1,
2]. As bacteriophages have evolved multiple mechanisms to take over their hosts, hosts have responded with mechanisms to thwart takeover. An understanding of these mechanisms would be beneficial to the development of new antibacterial strategies. However, the lack of information about the functions of a large portion of phage genes [
3] has hampered this investigation.
The lytic bacteriophage T4 infects
Escherichia coli, resulting in rapid cell lysis after ~20 min at 37 °C. Despite the use of T4 as a model organism for decades, the functions of many genes, especially those expressed early during infection, remain unknown [
4]. As T4 does not encode its own RNA polymerase (RNAP), it must use the host’s RNAP to program its temporal pattern of early, middle, and late gene expression (reviewed in [
5]). T4 directs RNAP to early, middle and late promoters by encoding factors that change the specificity of RNAP as infection proceeds. Early RNAs, which are expressed immediately after infection, arise from T4 early promoters (Pe’s). Pe’s, whose activation does not require phage-encoded factors, contain a very strong match to the consensus sequences of host promoters, allowing them to compete with host DNA for the same pool of RNAP. T4 middle transcripts are expressed approximately 1 to 2 min post-infection at 37 °C from middle promoters (Pm’s) and through the extension of early transcription into downstream middle genes. Activation of Pm’s requires two T4 proteins that modify RNAP, MotA and AsiA, resulting in the recognition of promoters that have a new DNA motif [
6]. T4 late transcripts, which are expressed ~5 min after infection, use late promoters (Pl’s) that contain a novel −10 sequence known as the TATA box. The T4-encoded late σ factor, σ
55 (gene product [gp]55), recognizes this element, facilitating late transcription along with the coactivator, gp33, and the sliding clamp, gp45 (reviewed in [
7]).
The histone-like protein H-NS and its less abundant homolog StpA are known to protect bacteria from the expression of foreign genes. H-NS is a DNA-binding protein that targets AT-rich DNA sequences, condensing genomic DNA through the formation of ordered and looped structures that typically repress transcription at the affected region (reviewed in [
8]). As phage genomes and xenogeneic sequences acquired from horizontal gene transfer often display a high AT content, H-NS can repress expression of this DNA (reviewed in [
9]). Given the AT-richness of T4 DNA (65.5%), it would not be surprising for H-NS binding to exert such an inhibitory effect on T4 gene expression. In fact, previous work has shown that the T4 Arn protein, an early gene product that structurally mimics DNA, can bind H-NS, preventing its interaction with DNA and formation of higher order structures [
10]. However, Arn is not essential [
11] and whether its absence affects T4 gene expression is not known. Specific H-NS antagonists have also been identified for T7 [
12], Luz24 [
13], and Mu [
14,
15].
Like
arn,
motB is an early gene that is not essential. It is located in a dispensable region of the T4 genome [
16], where a subset of genes have been shown to modulate gene expression and/or host functions under certain growth conditions [
17]. For instance,
mrh and
srh affect late T4 transcription under heat shock (42 °C) and in certain
rpoH (σ
32) mutants. In addition,
69,
modA, and
srd impair
E. coli growth when heterologously expressed [
17]. Previously,
motB has been implicated in the optimal expression of certain T4 middle genes [
16] and was given the name
motB for modifier of transcription B. In addition, MotB protein, identified as p17.6, was isolated in a prereplicative complex containing DNA, RNAP, MotA, AsiA, and other polymerase-associated and DNA-binding proteins [
18]. However, its function has remained unknown.
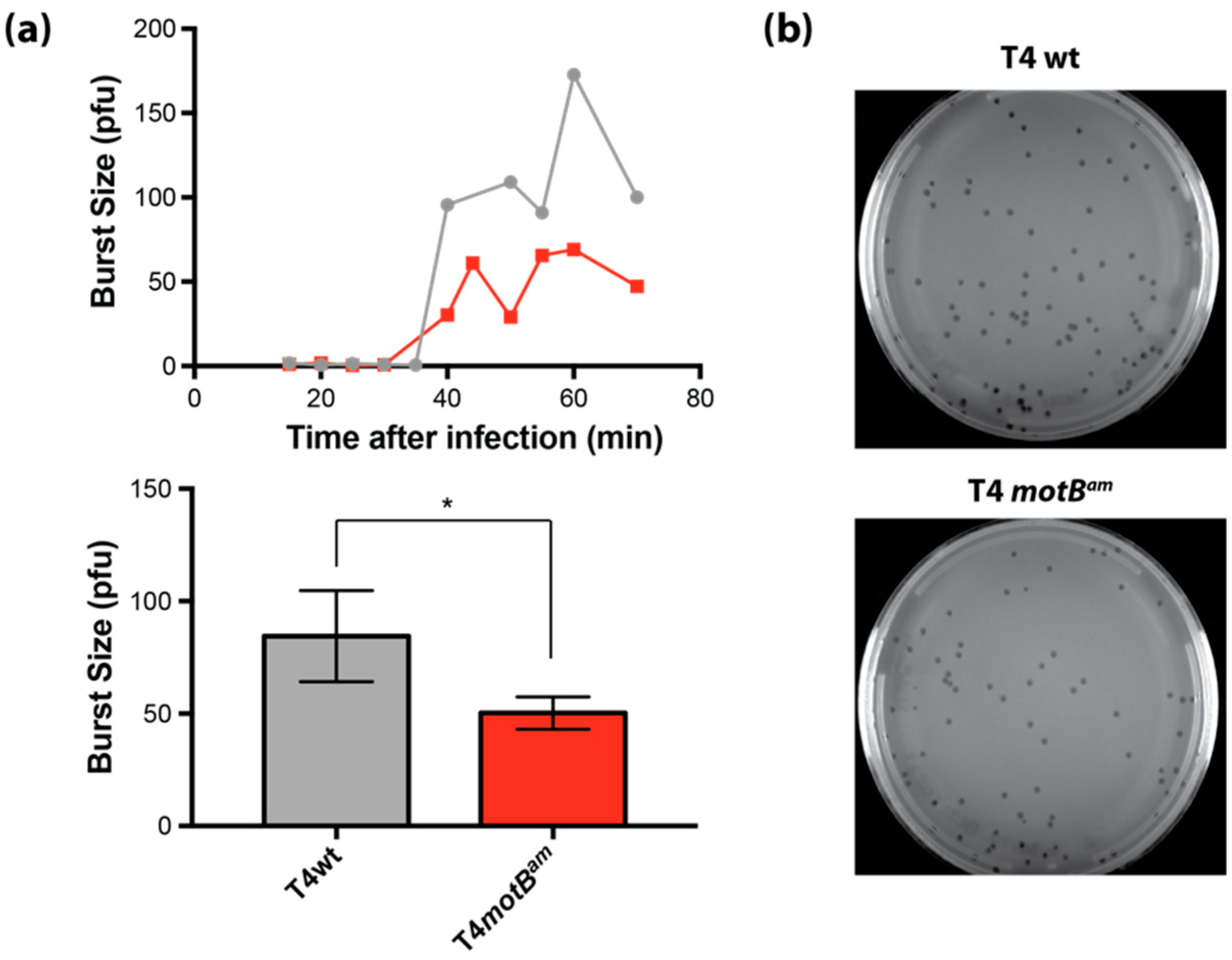
We show here that the presence of motB increases T4 burst size twofold, and heterologous production of MotB is highly toxic to E. coli, resulting in cell death or growth arrest depending on the strain. Our transcriptome analyses of T4 wild type (wt) RNA vs. T4 motBam RNA reveal only mild impairment of a few late genes at 5 min post-infection, while early and middle genes are not significantly affected. Consequently, it seems unlikely that MotB is directly involved in T4 gene expression. We have purified MotB and demonstrated that it binds to unmodified and modified [(glucosylated, hydroxymethylated-5 cytosine, (GHme-C)] DNA fragments with no detectable sequence specificity. Interestingly, MotB copurifies with the host histone-like proteins, H-NS and StpA, either directly or through cobinding to DNA, and we find that H-NS also binds T4 modified DNA. We speculate that MotB may alter how H-NS interacts with host DNA, T4 DNA, or both, thereby improving T4 growth.
2. Materials and Methods
2.1. DNA
pTE103-
motBam was prepared by cloning the 489 base pair (bp) sequence preceding and following the codon for
motB residue S12, mutated from TCT to TAG, between the BamHI and SalI sites. pNW129-MotB was constructed by cloning
motB, whose sequence was optimized for codon usage, between KpnI and SalI sites of the kan
r, pACYC-derived vector pNW129 [
19]. In this arrangement,
motB is appropriately downstream of a ribosome-binding site and is under the control of the arabinose inducible promoter P
BAD. pNW129-MotB-His was constructed as described for pNW129-MotB except the native stop codon was omitted so that the C-terminal His
6-tag was transcribed. pTXB1-MotB was obtained by cloning the native sequence minus its stop codon between the NdeI and SapI sites of pTXB1 (New England BioLabs, Ipswich, MA, USA). The resulting plasmid produces MotB fused to an auto-cleavable C-terminal intein with a chitin-binding domain (CBD) under the control of the isopropyl β-
d-thiogalactopyranoside (IPTG) inducible T7 promoter. pTXB1-HNS was constructed as described for pTXB1-MotB. GenScript (Piscataway Township, NJ, USA) performed gene synthesis, plasmid construction, and DNA sequencing for the plasmids used in this study.
T4 DNA was purified from phage extraction with phenol, phenol:chloroform:isoamyl alcohol (25:24:1), and chloroform:isoamyl alcohol (24:1) (two extractions with each solvent). DNA was then dialyzed into 10 mM Tris-HCl (pH 8.0) and 1 mM ethylenediaminetetraacetic acid (EDTA) at 4 °C. λ DNA was purchased from New England BioLabs. Where indicated, T4 and λ DNA were treated with SspI (New England BioLabs) and HindIII (New England BioLabs), respectively, purified by phenol extraction/ethanol precipitation, and dissolved in nuclease-free water.
Oligonucleotides used for primer extensions, RT-qPCR, and gel retardation assays were synthesized by Integrated DNA Technologies (Coralville, IA, USA); sequences are available upon request. Radiolabeled oligonucleotides were prepared by treating the top strand oligonucleotide with OptiKinase (Affymetrix, Santa Clara, CA, USA) in the presence of [γ-32P]ATP. 32P-labeled single stranded (ss) DNA, purified by phenol extraction/butanol precipitation, was resuspended in 1× TE (Quality Biological, Gaithersburg, MD, USA). Double-stranded (ds) oligonucleotides were prepared by mixing the 32P-labeled top strand with the complementary bottom strand in 1× OptiKinase Reaction Buffer (Affymetrix, Santa Clara, CA, USA), heating at 92 °C for 2 min, and slowly cooling to room temperature. The 32P-labeled dsDNA was subsequently purified using a G-25 microspin column (GE Healthcare, Little Chalfont, UK).
The 32P-labeled PCR product for DNase I footprinting was obtained using Pfu Turbo polymerase (Stratagene, San Diego, CA, USA), upstream and downstream primers that annealed from positions −143 to +75 of the T4 late promoter for gp8 (Pl8), and purified T4 DNA. The top strand (nontemplate) primer was treated with T4 polynucleotide kinase (Affymetrix) in the presence of [γ-32P]ATP prior to PCR. The PCR product was isolated after gel electrophoresis using an Elutrap (GE Healthcare) and ethanol precipitated.
2.2. Bacterial and Bacteriophage Strains
E. coli strains TOP10F’ (Invitrogen, Carlsbad, CA, USA), BL21(DE3), and BL21(DE3)/pLysE [
20] were used for expression studies. NapIV suppressing (NapIV S) and NapIV wt (non-suppressing, NapIV NS) [
21]
E. coli were used for T4 infections. Unless otherwise noted, cells were grown at 37 °C with shaking at 250 rpm.
Wild-type T4D
+ (T4 wt), T4
amG1 (T4
motAam) [
22], and T4
motBam were used for infections. T4
motBam was obtained by recombination of pTE103-
motBam into the T4 genome. BL21(DE3)/pTE103-
motBam was infected with T4 wt at a multiplicity of infection (MOI) of 1 during exponential phase and incubated for 75 min. Cells were lysed with chloroform and resulting phage were titered on NapIV S. Plaques were screened for the presence of the amber mutation by hybridization of a
32P-labeled probe containing either the wt or mutant sequence as described [
23]. Potential mutant plaques were used to generate phage stocks by infecting NapIV S. Phage stocks containing the amber mutation were subjected to a subsequent round of single plaque selection to ensure a homogenous stock of T4
motBam. To confirm the presence of the amber mutation, the
motB gene was amplified by PCR then sequenced by Macrogen (Rockville, MD, USA).
2.3. MotB Toxicity Assay
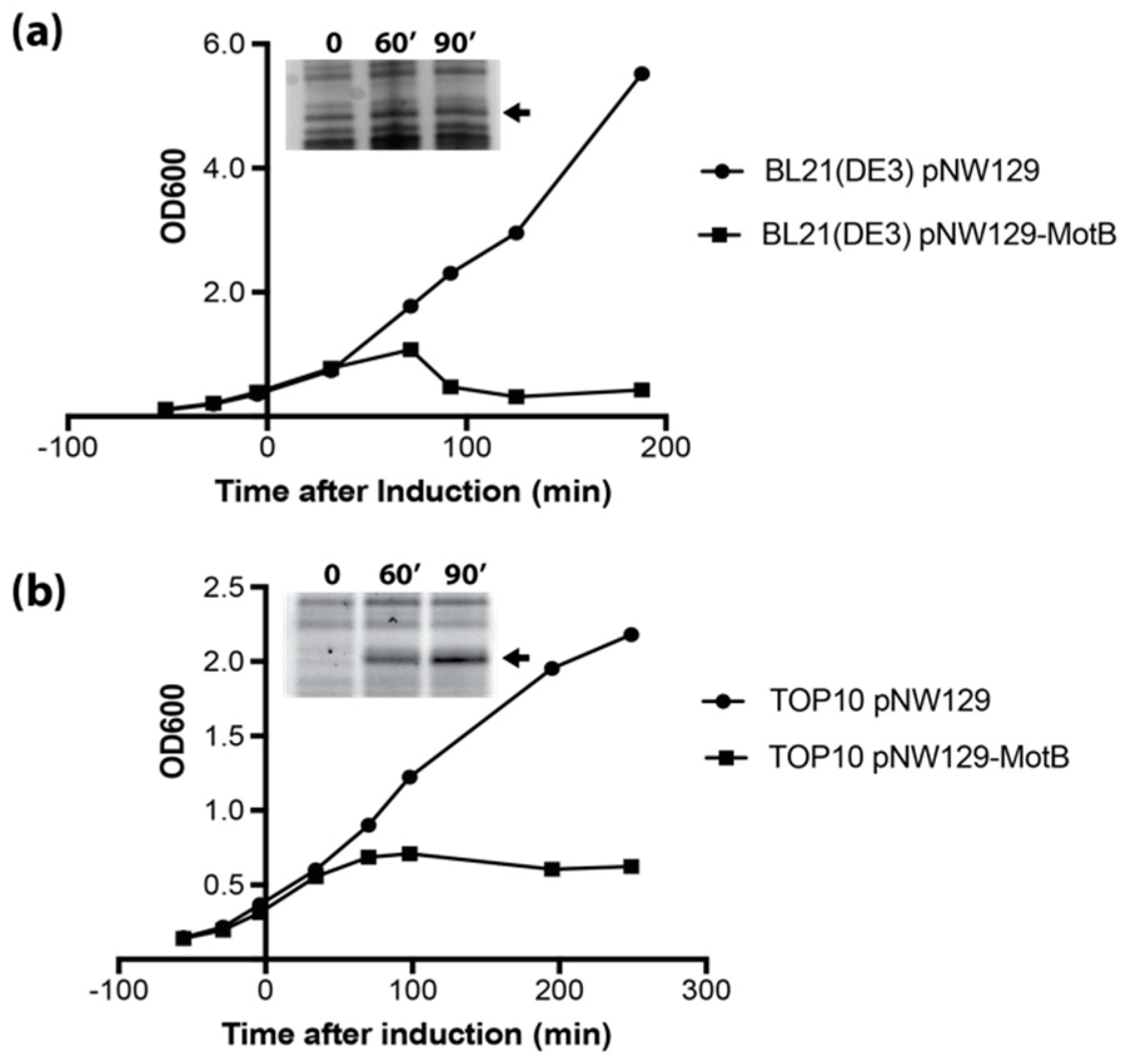
BL21(DE3) and TOP10F’ containing pNW129 (empty vector) or pNW129-MotB were plated on 1.5% (w/v) LB agar (Quality Biological or Sigma, St. Louis, MO, USA) containing 40 µg/mL kanamycin, 12 µg/mL tetracycline (TOP10F’ only), and 0.5% (w/v) glucose. Overnight cultures were grown in LB (Quality Biological) containing 40 µg/mL kanamycin, 12 µg/mL tetracycline (TOP10F’ only), and 0.025% (w/v) glucose. Overnight cultures were diluted to an OD600 of 0.1 with LB containing 40 µg/mL kanamycin and 12 µg/mL tetracycline (TOP10F’ only). At an OD600 of approximately 0.3, 0.2% (w/v) arabinose (final concentration) was added to each sample. At the indicated times samples were taken and electrophoresed on SDS-PAGE gels to monitor MotB production.
2.4. Protein Purification
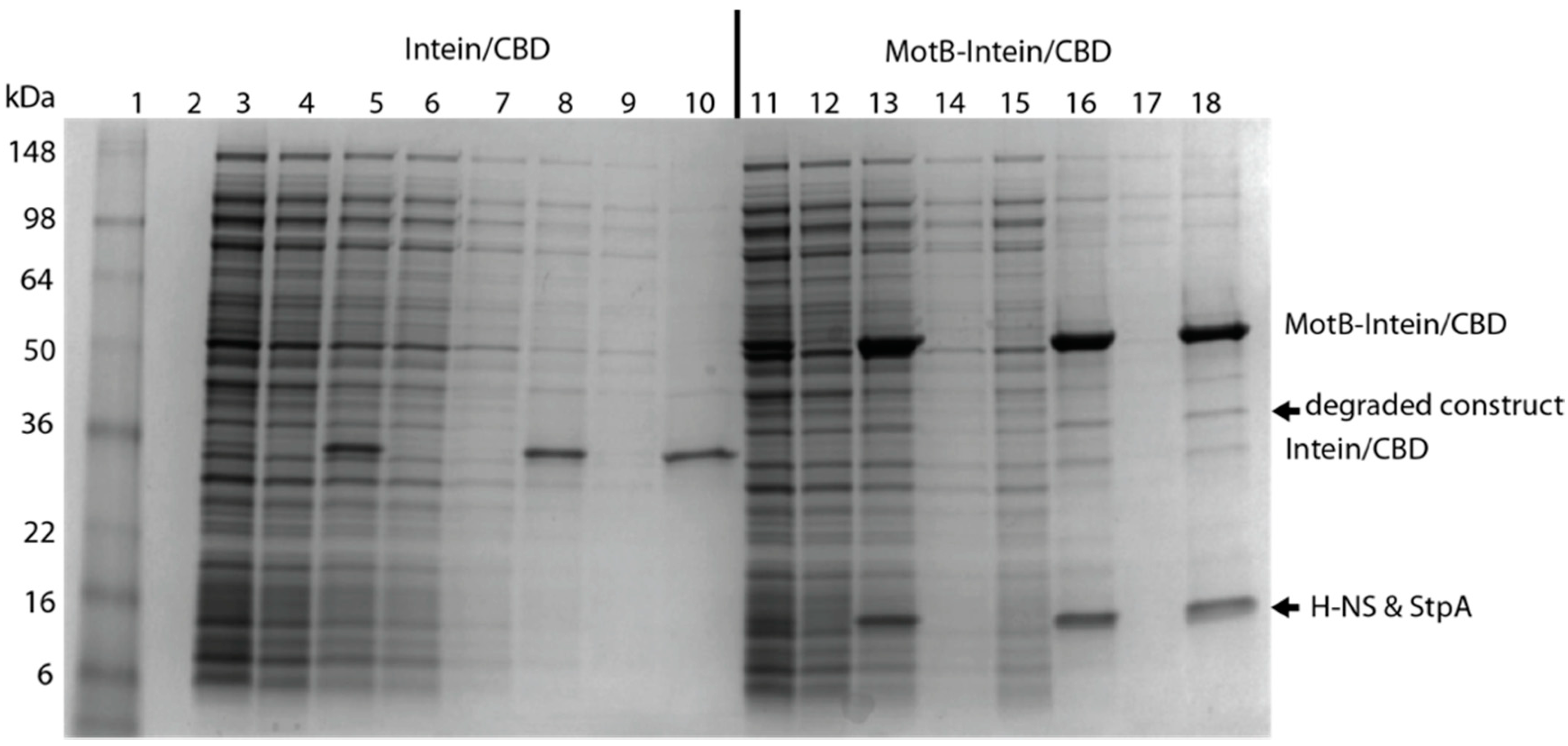
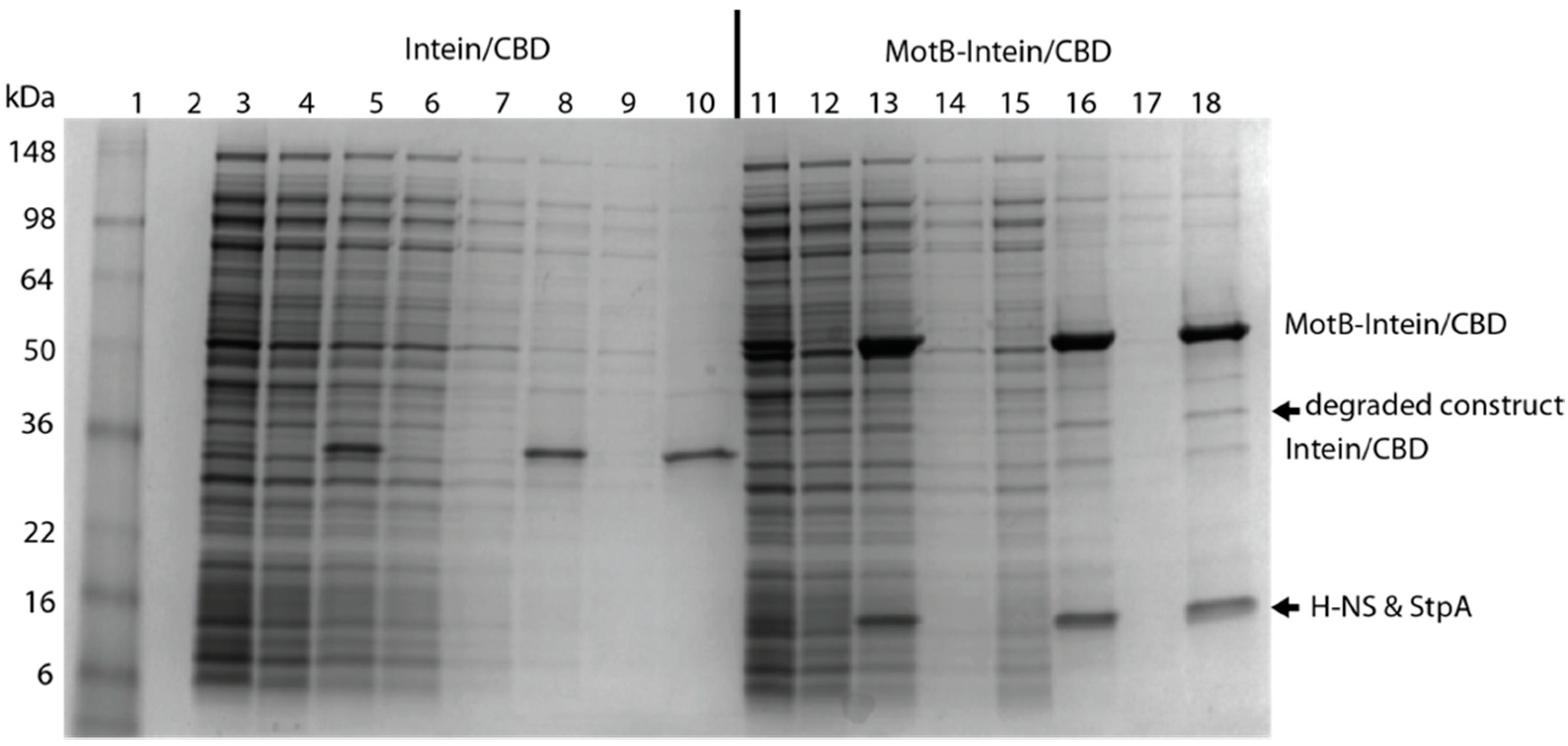
2.4.1. MotB Purification (Method I)
MotB containing a C-terminal intein tag with a chitin binding domain (MotB-Intein/CBD) was isolated and purified from BL21(DE3)/pLysE containing pTXB1-MotB. Overnight cultures were grown in LB containing 25 µg/mL chloramphenicol and 100 µg/mL carbenicillin. Cells were diluted to an OD
600 ~0.1 in LB containing 25 µg/mL chloramphenicol and 100 µg/mL carbenicillin and then grown at 25 °C with shaking at 250 rpm. At an OD
600 between 0.3 and 0.4, synthesis of MotB-Intein/CBD was induced by the addition of 4 mM IPTG (final concentration) for 2 h. Cells were harvested by centrifugation at 13,000×
g for 10 min and stored at −80 °C. Unless otherwise noted, the following procedures were performed on ice or at 4 °C. Cells were resuspended in CB Buffer (20 mM HEPES-OH (pH 8.5), 50 mM NaCl, 1 mM EDTA, 0.01% (
v/
v) Triton X-100) containing 1 mM benzamidine, then lysed by sonication until OD
600 was reduced ~3-fold. Clarified supernatant was obtained by centrifugation at 15,000×
g for 30 min followed by filtration through a 0.4 µm syringe filter. Chitin resin (New England BioLabs) equilibrated with CB Buffer (5 mL slurry per 125 mL starting culture) was added to the supernatant, and the suspension was gently rocked overnight. Resin was transferred to a 30 mL disposable column (Bio-Rad, Hercules, CA, USA) and then washed with ~25 column volumes of CB Buffer containing 1 M NaCl at a flow rate of <2 mL/min (gravity drip) until the A
260 reached a minimum. The resin was subsequently washed with an additional 7 column volumes of CB Buffer containing 500 mM NaCl. On-column cleavage of the Intein/CBD-tag was initiated by quickly washing the column with 3 column volumes of CB Buffer containing 500 mM NaCl and 1 mM dithiothreitol (DTT). After ≥40 h, MotB was eluted by the addition of 2 column volumes of CB Buffer containing 500 mM NaCl and 2 mL fractions were collected. Fractions containing MotB were identified by SDS-PAGE and then dialyzed into Buffer A (25 mM Tris-HCl (pH 8.0), 50 mM NaCl, 6 M deionized Urea). The denatured MotB was isolated after passage through a 1 mL HiTrap Q column (GE healthcare), in which MotB was in the flow-through fraction while residual nucleic acid bound to the column. MotB was then refolded using the dialysis procedure previously described for
E. coli σ
70 renaturation [
24] and stored at −20 °C in MotB storage buffer (50 mM Tris-HCl (pH 8.0), 1 mM EDTA, 50 mM NaCl, 0.01%
v/
v Triton X-100, 50%
v/
v glycerol, 0.1 mM DTT).
2.4.2. MotB Purification (Method II)
MotB containing a C-terminal His6-tag (MotB-His) was isolated and purified from TOP10F’ containing pNW129-MotB-His. Overnight cultures were grown in LB containing 12 µg/mL tetracycline, 40 µg/mL kanamycin, and 0.05% (w/v) glucose. Cells were diluted to an OD600 ~0.1 in LB containing 12 µg/mL tetracycline and 40 µg/mL kanamycin and grown to an OD600 around 0.3, when synthesis of MotB-His was induced by the addition of 0.2% (w/v) arabinose (final concentration) for 1 h. Cells were harvested by centrifugation at 13,000× g for 10 min and stored at −80 °C. Unless otherwise noted, the following procedures were performed on ice or at 4 °C. Cells were resuspended in 40 mL of Lysis Buffer (25 mM Tris-HCl (pH 8.0), 50 mM NaCl, 5 mM imidazole, 0.01% (v/v) Triton X-100, 1 mM benzamidine) per 1 L culture, lysed by sonication as described above and then centrifuged at 17,500× g for 20 min. The pellet, containing MotB-His, was resuspended in Lysis Buffer and centrifuged again at 17,500× g for 15 min. To recover MotB-His, the pellet was resuspended in 20 mL Binding Buffer (20 mM Tris-HCl (pH 8.0), 500 mM NaCl, 5 mM imidazole, 6 M urea, 1 mM benzamidine, pH adjusted to 7.9) per 1 L culture, incubated for 1 h with gentle mixing, and then centrifuged at 17,500× g for 15 min. The supernatant, containing MotB-His, was then incubated with gentle mixing for a minimum of 1 h with 5 mL (per 1 L culture) Ni-NTA His-binding resin (Millipore, Burlington, MA, USA) that was pre-equilibrated with Binding Buffer. Resin was transferred to a 30 mL disposable column (Bio-Rad) then washed with four column volumes of Binding Buffer followed by six column volumes of Wash Buffer (25 mM Tris-HCl (pH 8.0), 500 mM NaCl, 60 mM imidazole, 6 M urea, pH adjusted to 7.9) at a flow rate of <2 mL/min (gravity drip). The major fraction of MotB-His eluted with 2 column volumes of Elution Buffer 1 (20 mM Tris-HCl (pH 8.0), 500 mM NaCl, 200 mM imidazole, 6 M urea, pH adjusted to 7.9). Residual MotB-His was subsequently eluted with 2 column volumes of Elution Buffer 2 (20 mM Tris-HCl (pH 8.0), 500 mM NaCl, 400 mM imidazole, 6 M urea, pH adjusted to 7.9). For both elution conditions, 2.5 mL fractions were collected. Fractions containing MotB-His, identified by SDS-PAGE, were refolded as described in Method I.
2.4.3. H-NS Purification
H-NS containing a C-terminal intein tag with a chitin binding domain (H-NS-Intein/CBD) was isolated and purified from BL21(DE3)/pLysE containing pTXB1-H-NS. Overnight cultures were grown in LB containing 25 µg/mL chloramphenicol and 100 µg/mL carbenicillin. Cells were diluted to an OD600 ~0.1 in LB containing 25 µg/mL chloramphenicol and 100 µg/mL carbenicillin and then grown at 25 °C with shaking at 250 rpm. At an OD600 between 0.3 and 0.4, synthesis of H-NS-Intein/CBD was induced by the addition of 4 mM IPTG (final concentration) for 2 h. Cells were harvested by centrifugation at 13,000× g for 10 min and stored at −80 °C. Unless otherwise noted, the following procedures were performed on ice or at 4 °C. Cells were resuspended in 30 mL of H-NS Sonication Buffer (20 mM Tris-HCl (pH 8.0), 750 mM NaCl, 1 mM EDTA, 0.01% (v/v) Triton X-100, 1 mM Benzamidine) then lysed by sonication until OD600 was reduced ~3-fold. Clarified supernatant was obtained by centrifugation at 15,000× g for 30 min followed by filtration through a 0.45 μm syringe filter. Chitin resin (New England BioLabs) equilibrated with H-NS Sonication Buffer (5 mL slurry per 250 mL starting culture) was added to the supernatant and rocked overnight. Resin/cell lysate was transferred to a 30 mL disposable column (Bio-Rad) and washed with approximately 20 column volumes of CCB buffer (20 mM Tris-HCl (pH 8.0), 500 mM NaCl, 1 mM EDTA, 0.01% (v/v) Triton X-100) at a flow rate of <2 mL/min (gravity drip). On-column cleavage of the Intein/CBD-tag was initiated by quickly washing the column with 3 column volumes of CCB buffer containing 50 mM DTT. After ≥40 h, H-NS was eluted by the addition of 3 column volumes of CCB buffer and 1 mL fractions were collected. Fractions containing H-NS were identified by SDS-PAGE and pooled. As this pool contained a small amount of intein tagged protein, it was passed over a new column containing 5 mL of fresh, equilibrated chitin slurry, and fractions from the flow-through plus 2 column volume washes were collected. The now pure H-NS was dialyzed into H-NS Storage Buffer (10 mM potassium phosphate (pH 7.5), 200 mM NaCl, 0.1 mM EDTA, 50% (v/v) glycerol) and stored at −20 °C.
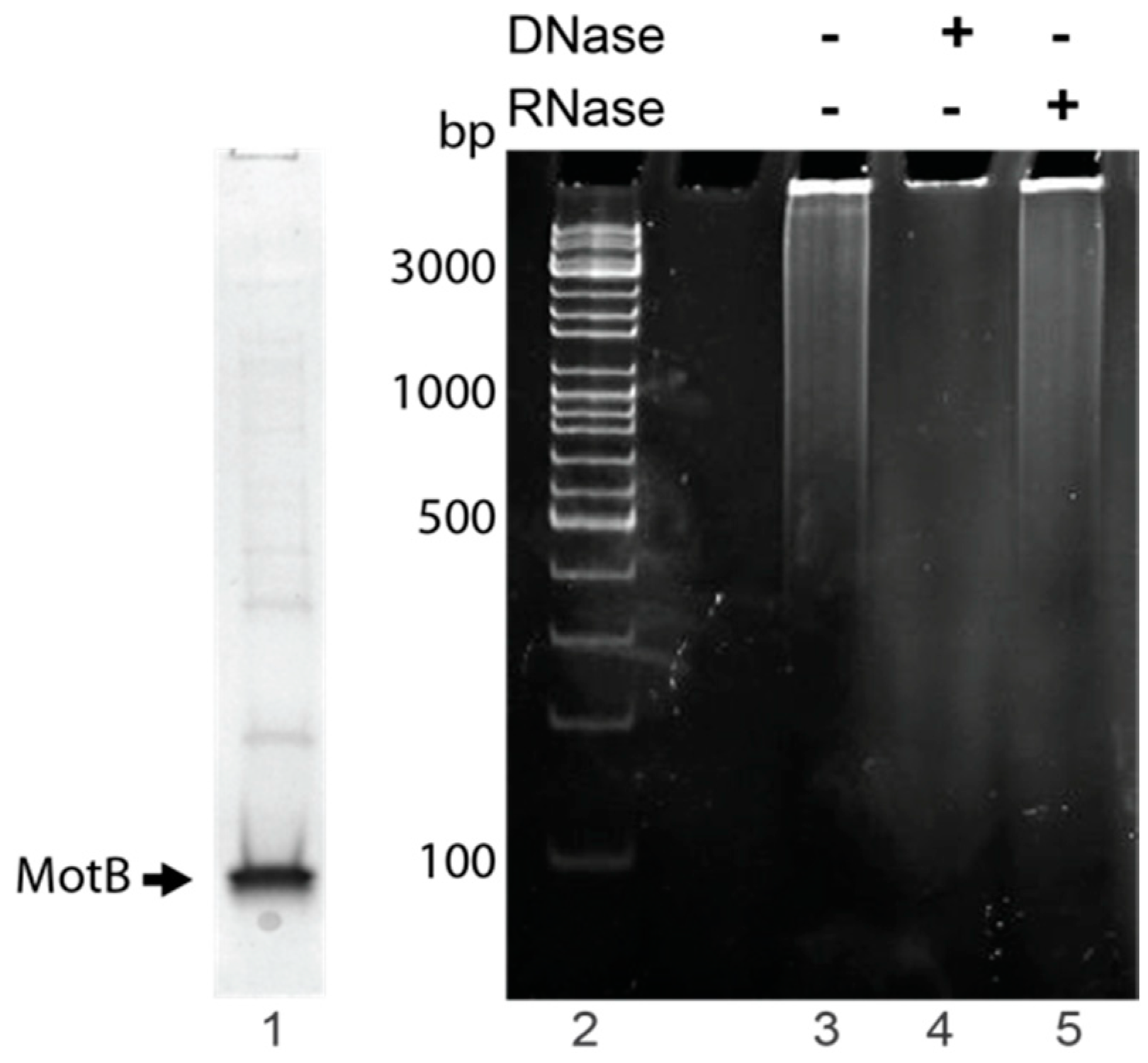
2.5. Identification of Nucleic Acid Copurifying with MotB
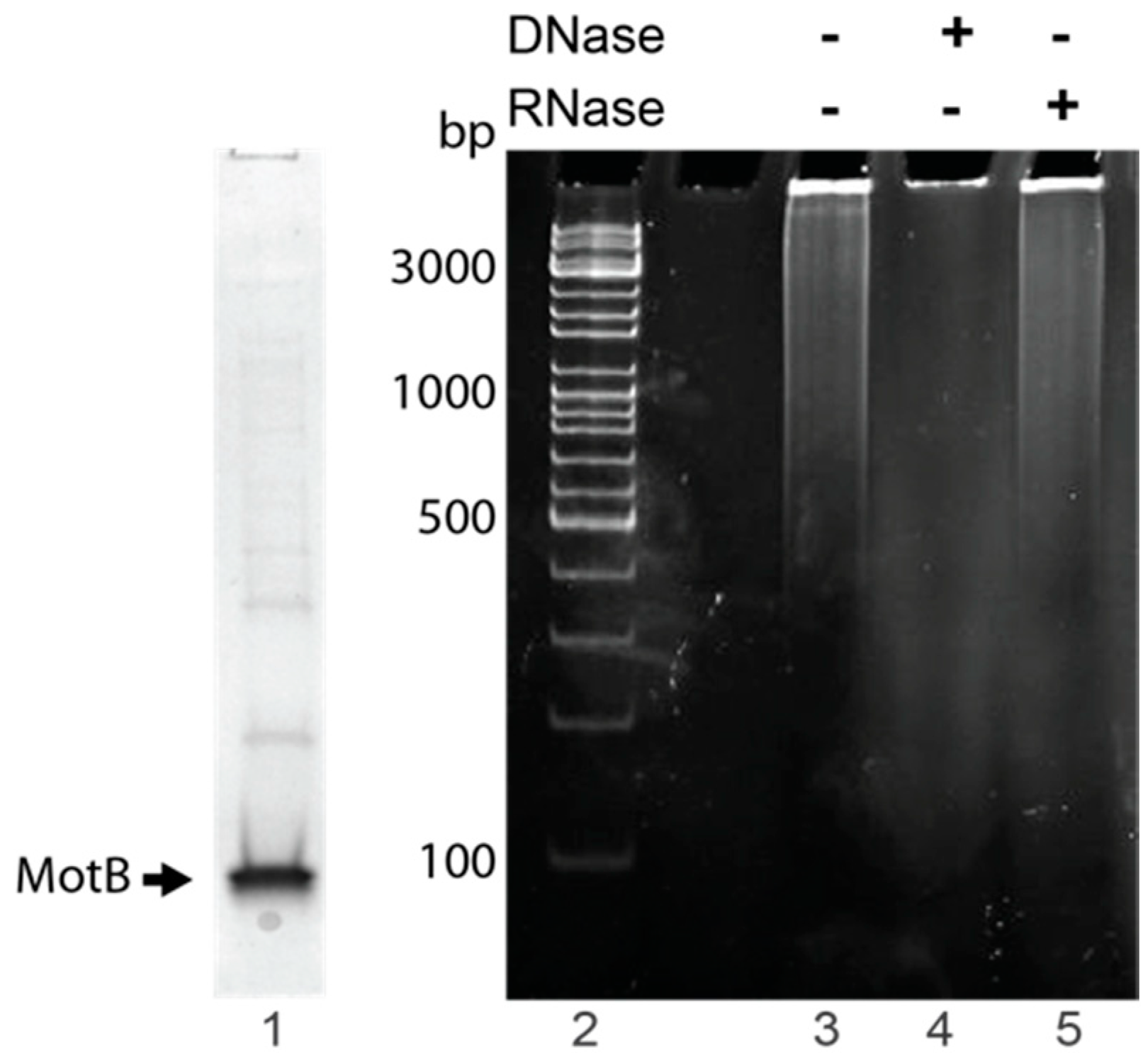
MotB samples used to identify copurifying nucleic acid were purified using MotB purification Method I with the following modifications. Elution fractions from the chitin column that contained MotB as determined by SDS-PAGE were directly dialyzed into MotB Storage Buffer and stored at −80 °C. Protein was removed by chloroform:isoamyl alcohol (24:1) extraction, and sodium acetate and glycogen (Thermo Scientific, Waltham, MA, USA) were added to the aqueous phase at a final concentration of 496 mM and 0.17 mg/mL, respectively. The nucleic acid was then ethanol precipitated, resuspended in nuclease-free water, and quantified by NanoDrop 2000c (Thermo Scientific).
To identify nucleic acid, 1× DNase buffer (Ambion, Carlsbad, CA, USA) was added to ~300–400 ng nucleic acid followed by the addition of either 2 units of RNase-free DNase I (Ambion) or 100 µg of DNase-free RNase A and incubation at 37 °C for 10 min. Treated samples and the untreated control were electrophoresed on 0.8% (w/v) agarose gels at 120 V for 45 min in 1× TAE (Quality Biological) at room temperature. Gels were stained with ethidium bromide for 1 min and visualized using UV illumination. Two independent replicates were performed.
2.6. Electrophoretic Mobility Shift Assays (EMSAs)
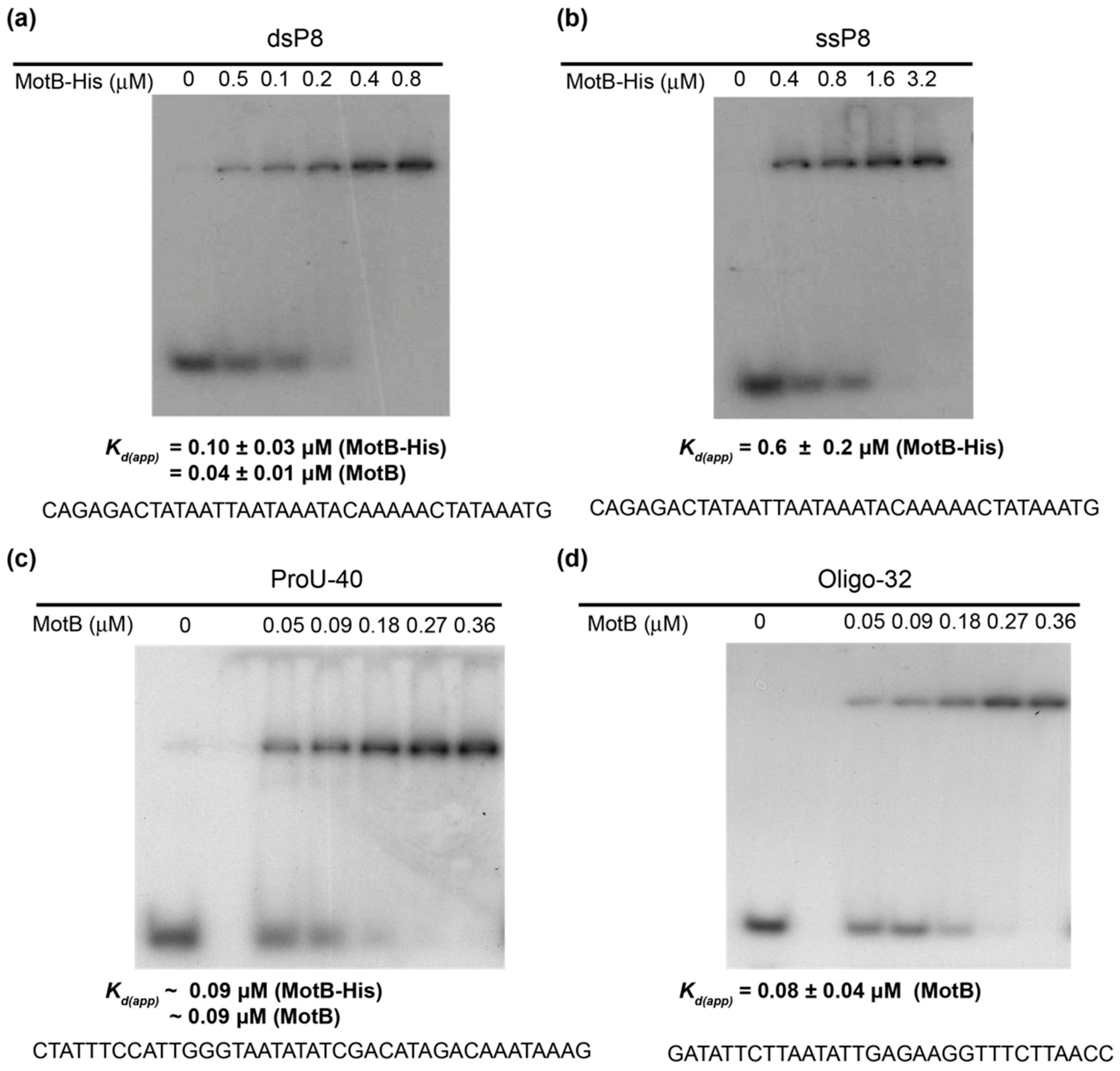
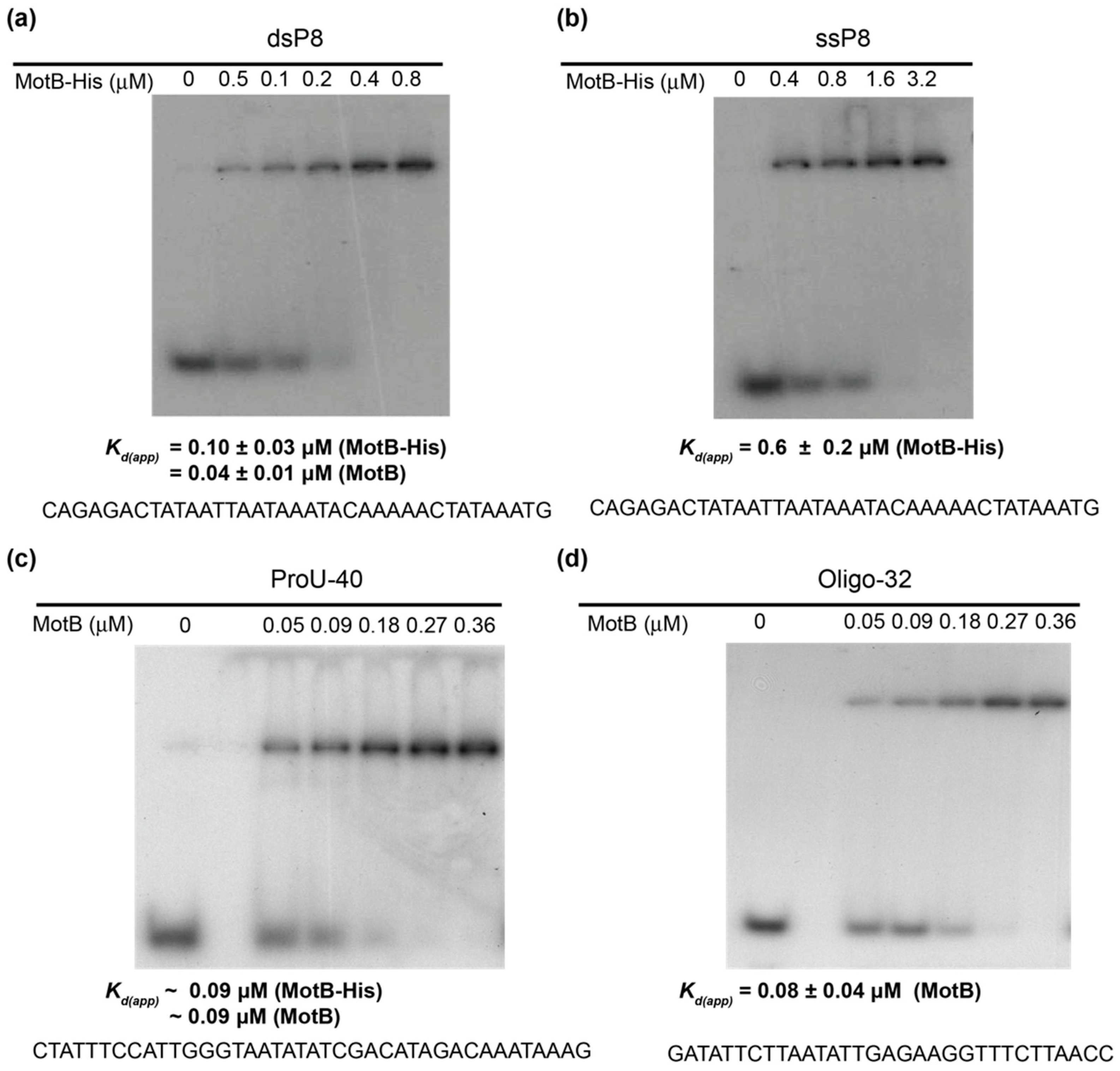
EMSAs with oligonucleotides (10 µL) contained the indicated amount of MotB, 0.01 pmol of 32P-labeled DNA, 40 mM Tris acetate (pH 7.9), 20 mM Tris-HCl (pH 8.0), 2 mM potassium phosphate (pH 7.5), 60 mM NaCl, 150 mM potassium glutamate, 4 mM magnesium acetate, 0.52 mM EDTA, 0.14 mM DTT, 0.1 mg/mL Bovine Serum Albumin (BSA), 0.004% (v/v) Triton X-100, and 30% (v/v) glycerol. In the case of the oligonucleotide ProU-40, the solution contained 40 mM Tris acetate (pH 7.9), 20 mM Tris-HCl (pH 8.0), 20 mM Tris-HCl (pH 7.6), 70 mM NaCl, 150 mM potassium glutamate, 4 mM magnesium acetate, 2 mM magnesium chloride, 0.5 mM EDTA, 0.14 mM DTT, 0.1 mg/mL BSA, 0.004% (v/v) Triton X-100, and 20% (v/v) glycerol. Solutions were incubated for 10 min at 37 °C, collected on ice, and electrophoresed on 12% (w/v) polyacrylamide (37.5:1, acrylamide:bis) gels in 1× TBE at 150 V for 3 h at room temperature. Gels were visualized by autoradiography, and the films were scanned using a GS-800 calibrated densitometer (Bio-Rad). Quantification was performed using Quantity One software (Bio-Rad). Kd(app) was determine as the protein concentration needed to shift 50% of the 32P-labeled DNA.
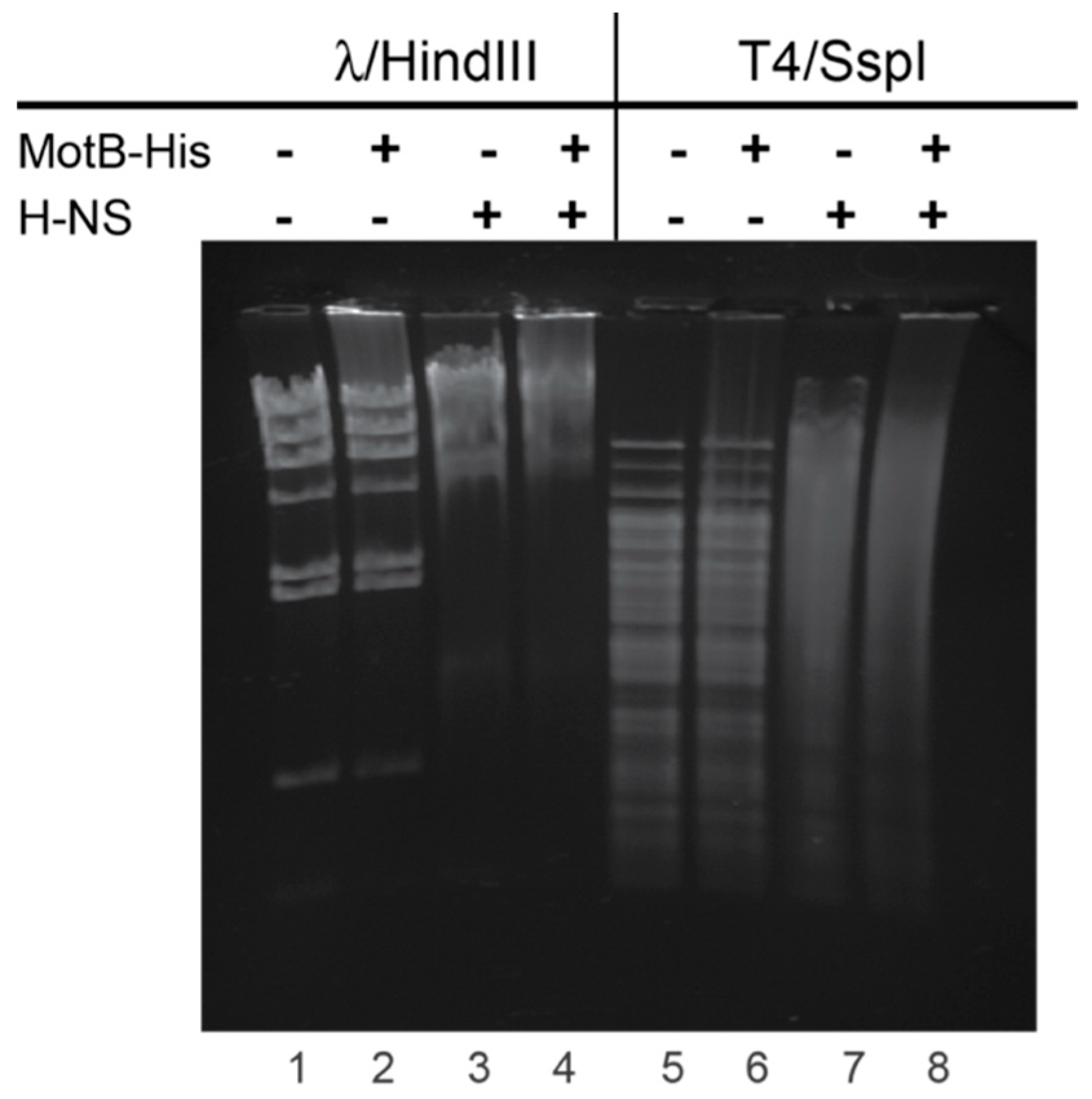
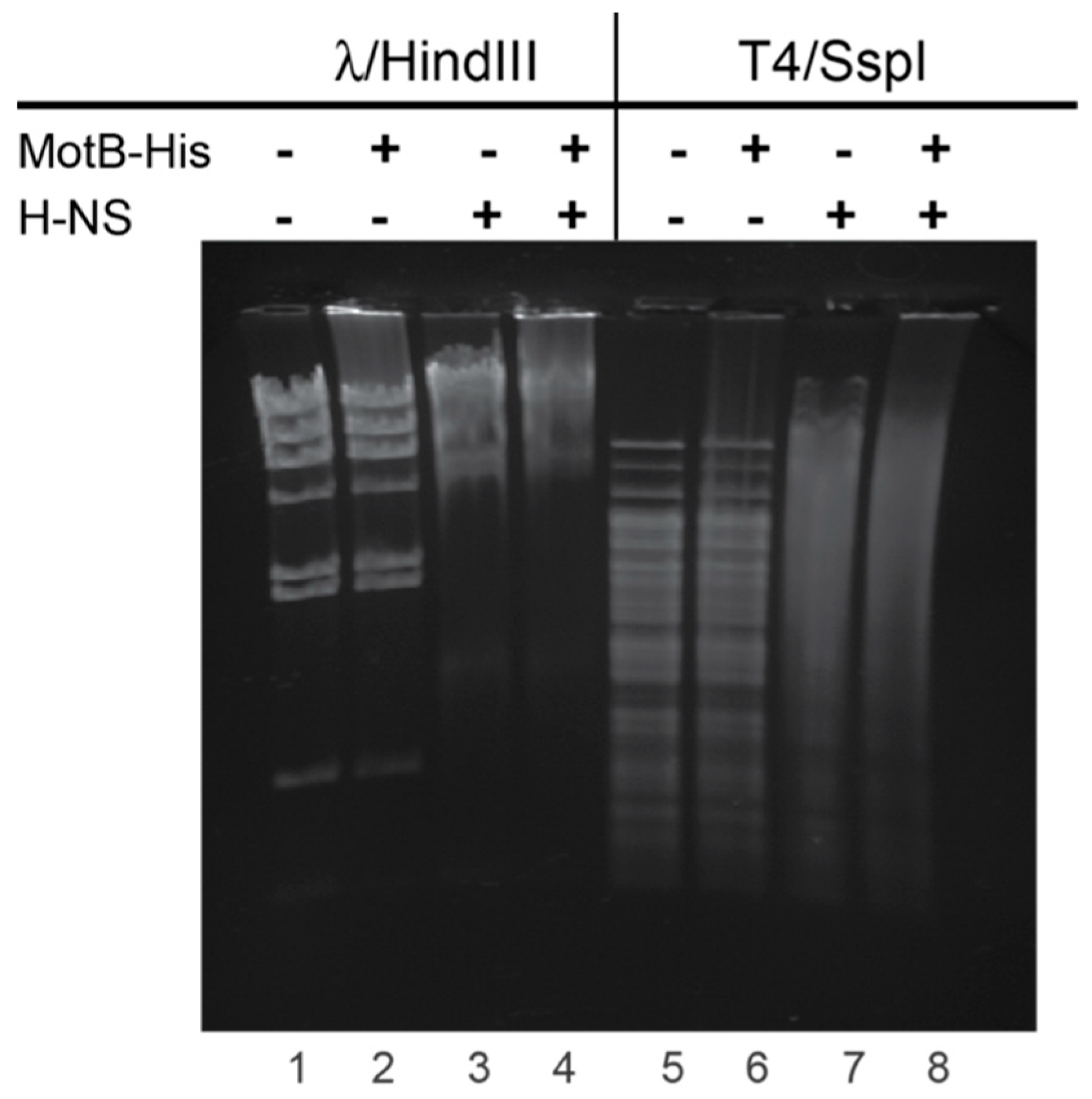
For gel shift assays with T4 or λ DNA, 60 pmol MotB-His and/or H-NS were incubated with 500 ng of either T4 DNA treated with SspI or λ DNA treated with HindIII in a reaction mixture (5 µL) containing 20 mM Tris-HCl (pH 8), 2 mM potassium phosphate (pH 7.5), 60 mM NaCl, 0.42 mM EDTA, 0.04 mM DTT, 0.004% (v/v) Triton X-100, and 30% (v/v) glycerol at 37 °C for 10 min. Samples were electrophoresed on 0.8% (w/v) agarose gels at 70 V for 90 min in 1× TAE at 4 °C. Gels were stained with ethidium bromide for 1 min and visualized using UV illumination.
2.7. DNase I Footprinting
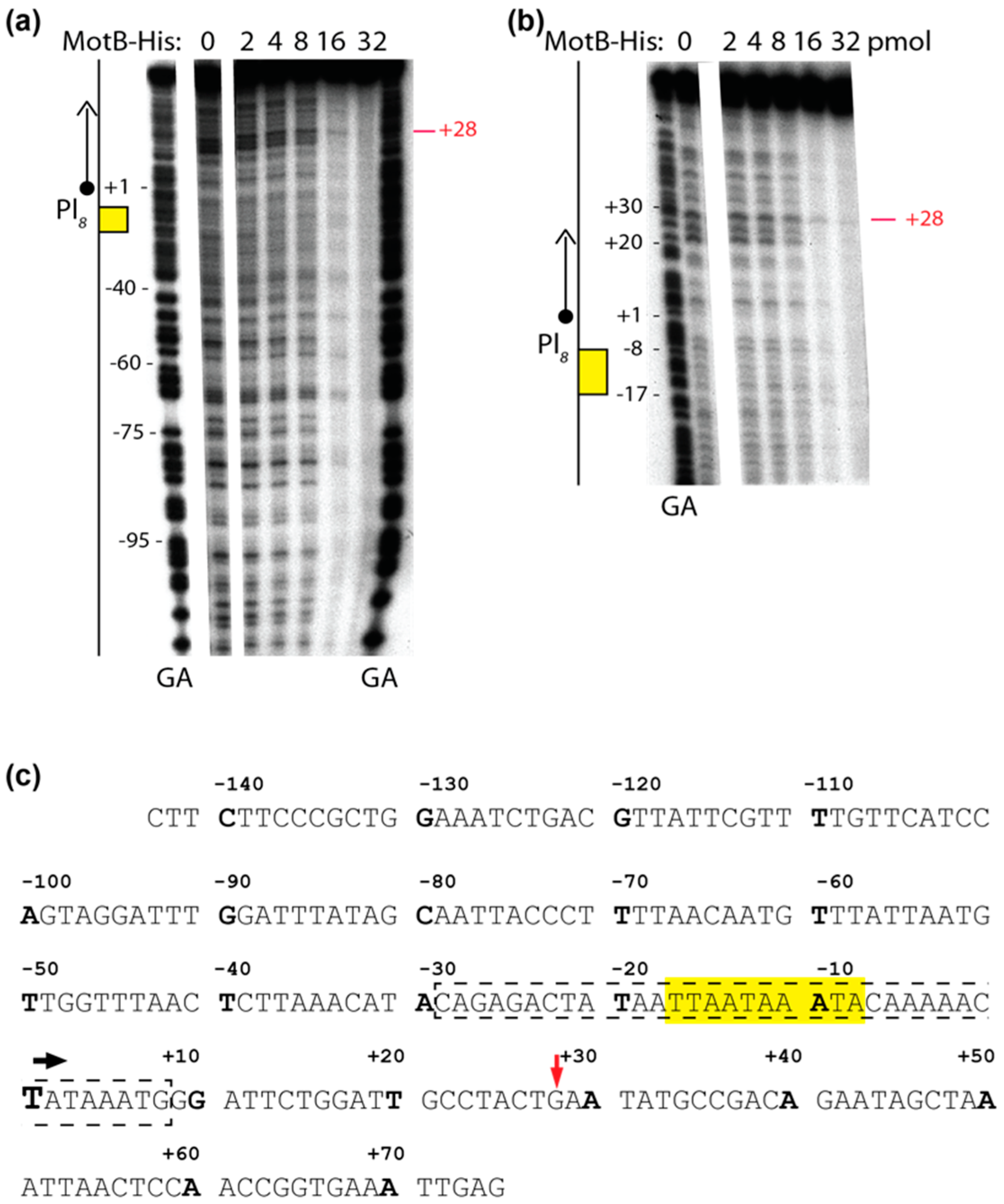
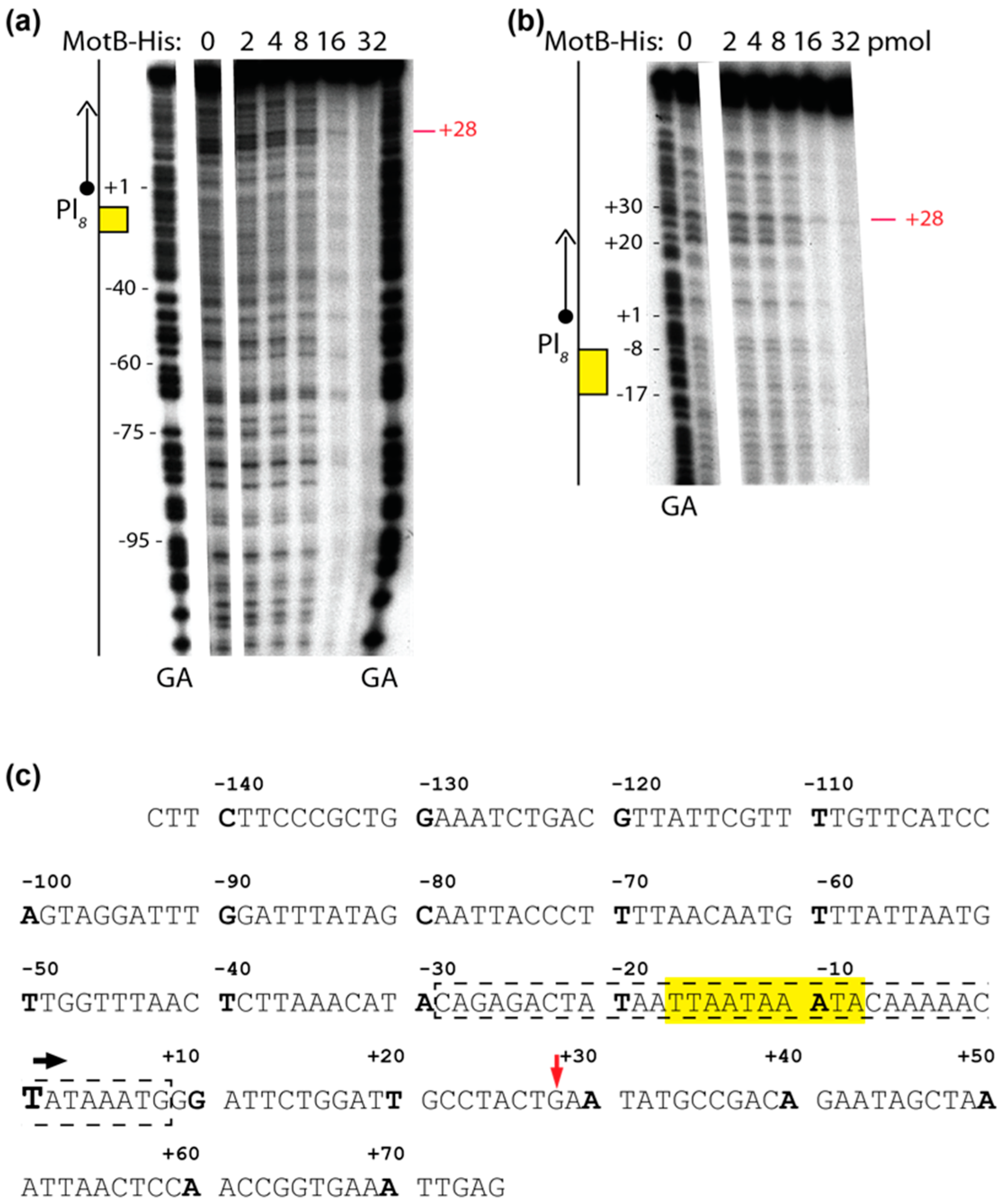
Solutions were assembled in a total volume of 10 µL using 0.005 μM DNA (1 μL) and 0 to 3.2 μM of MotB (4 μL), as indicated, in the same buffer used for gel shift assays with oligonucleotides as described above. After 10 min of incubation at 37 °C, 0.8 units of DNase I (1.1 μL) was added to the reaction. Solutions were incubated for an additional 45 s at 37 °C and quenched with equal volumes of phenol. DNA was extracted, ethanol precipitated, and electrophoresed on 5% (w/v) polyacrylamide (19:1, acrylamide:bis), 7 M urea denaturing gels for 2500 volt-h in 0.5× TBE. After electrophoresis, gels were visualized by autoradiography and analyzed as described above.
2.8. Pull-Down Assay
BL21(DE3)/pLysE containing pTXB1-MotB or pTXB1 was grown in LB containing 25 μg/mL chloramphenicol and 100 μg/mL carbenicillin at 25 °C. MotB was isolated as described in Method I except that after binding to the chitin column, the resin was washed with 5 column volumes of CB buffer containing 500 mM NaCl followed by 5 column volumes of CB buffer containing 1 M NaCl. Proteins, isolated from the SDS-PAGE gel, were analyzed by mass spectrometry (Whitehead Institute, Cambridge, MA, USA).
2.9. Bacteriophage Fitness Assays
Overnight cultures were diluted in fresh LB to an OD
600 ~0.1, grown to early log phase (OD
600 ~0.3), and then transferred to ice until ready to titer. For each titer, ~50–100 phages in 100 μL LB were incubated with 200 μL of culture for 5 min at room temperature. Following addition of 2.5 mL 0.65% (
w/
v) Drake top agar at 45 °C [
25], cells were plated on 1.5% (
w/
v) LB agar (Sigma) and incubated at 37 °C overnight. For infections using cells containing pNW129-MotB or pNW129, cultures were grown to an OD
600 ~0.4 as described above and then protein synthesis was induced by the addition of 0.1% (
w/
v) arabinose (final concentration) for 20 min before collecting on ice.
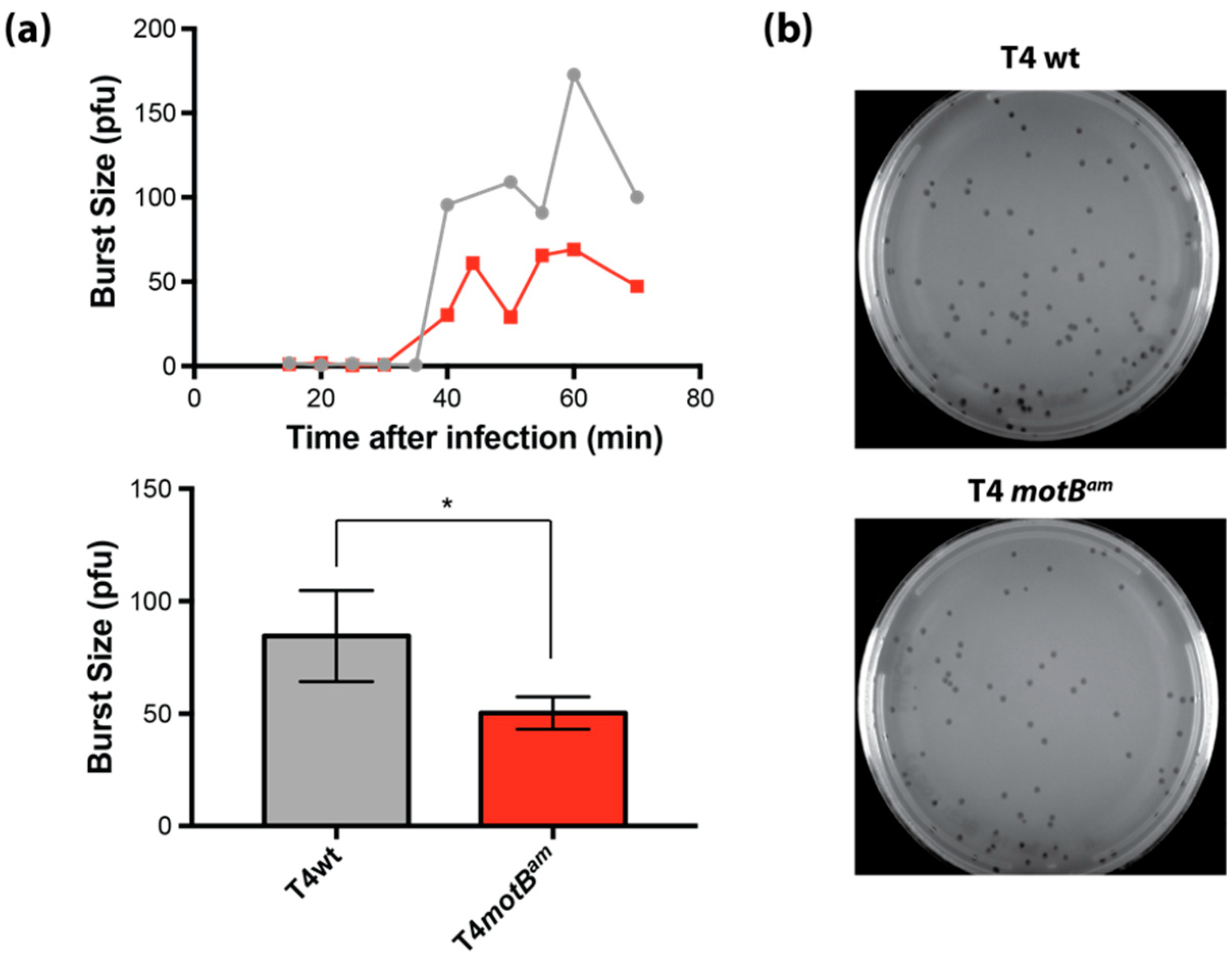
Burst size experiments were performed as described [
26] for single-step growth experiments with the following modifications. NapIV S and NapIV NS were grown to mid log phase (OD
600 ~0.5). The NapIV S culture was transferred to ice for use as the plating strain. NapIV NS (900 μL) was transferred to a 1.5 microcentrifuge tube containing 100 μL of phage (MOI of ~0.004) pre-warmed at 37 °C. At 10 min post-infection, the phage-culture sample was diluted 10,000-fold in LB warmed to 37 °C with gentle vortexing to mix and returned to 37 °C. For pre-burst (≤30 min) and post-burst (35 to 70 min) time points, 100 μL or 10 μL of the diluted phage culture, respectively, were added to 100 μL of NapIV S followed by the addition of 3 mL of 0.65% Drake top agar (at 50 °C). The cells were immediately plated on 1.5% (
w/
v) LB agar and incubated overnight at 37 °C. Burst size was calculated by dividing the average number of plaques for the post-burst time points (50 to 70 min) by the average number of plaques for pre-burst time points (15 to 30 min).
2.10. Purification of Total RNA
NapIV NS were grown to a cell density of ~4 × 10
8 cells/mL (OD
600 ~0.4) and then infected with either T4 wt or T4
motBam at a MOI of 10. As indicated, RNA was isolated at 1, 5 or 10 min post-infection using method II as described [
27]. Briefly, cells were chemically lysed and nucleic acid was isolated by hot phenol extraction followed by ethanol precipitation. Samples were then treated with DNase, and total RNA was isolated by phenol extraction followed by ethanol precipitation. Total RNA used for RNA-seq was run on a Bioanalyzer using the Agilent RNA 6000 Nano Kit (Agilent Technologies, Santa Clara, CA, USA) to evaluate the sample quality [
28]. RNA used for RT-qPCR and primer extensions was evaluated using a 0.8% (
w/
v) agarose gel stained with ethidium bromide.
2.11. Transcriptome Analysis
rRNA subtraction was performed with the bacterial RiboMinus Kit (Ambion) according to manufacturer instructions. cDNA was prepared using the NEBNext strand specific kit (New England BioLabs) according to manufacturer instructions for libraries with 300–450 bp insert size with the following modifications. Illumina adaptor sequences based on TruSeq HT Sample Prep Kits were purchased from Integrated DNA Technologies and used in the ligation step. TruSeq-1 and TruSeq-2 primer were used for PCR enrichment of adaptor ligated DNA. Library size was verified with a Bioanalyzer using an Agilent High Sensitivity DNA kit (Agilent Technologies). The concentration of each library was determined using the KAPA Library Quantification Kit (Roche, Basel, Switzerland) for Illumina platforms. Sequencing was performed by the NIDDK Genomics Core facility using a MiSeq system with the MiSeq 2 × 250 bp Sequencing Kit (Illumina, San Diego, CA, USA).
Quality trimming (max ambiguity = 2) and trimming of 15 nucleotides from the 5’ terminal of all sequences were performed with CLC Genomics Workbench (version 8.5, Qiagen, Hilden, Germany). The quality of raw sequencing data was accessed in CLC Biomedical Workbench software (Qiagen). A bimodal distribution of GC-content was observed in the data set with peak maximums at 36% GC and 55% GC (
Figure S1). This distribution is consistent with the presence of both host-encoded (~55% GC) and T4-encoded (34.5% GC) transcripts within the data set.
Trimmed data were aligned to the
E. coli reference genome (NC_012971.2) and an unmapped reads file was generated. Approximately 44–54% of total reads mapped to the host genome. A significant number of host-encoded transcripts is consistent with other transcriptome analysis of bacteriophage infections during early and middle stages of temporal expression [
29,
30]. The unmapped reads file, presumably containing sequences derived from the T4 genome, was subsequently aligned to the bacteriophage T4 reference genome (NC_000866.4). To mitigate the occurrence of multi-mapping reads, overlapping annotations of genes with alternative start sites or introns were resolved to a single annotation in the T4 reference genome to enable unique gene assignment by CLC Biomedical Workbench software. Differential expression experiments consisted of 3 replicates of NapIV NS infected with either T4 wt or T4
motBam. Statistical analysis was performed in CLC Genomics Workbench using the Baggerley’s Test with False Discovery Rate (FDR) corrected
p values. RNA-seq data is available in the NCBI database (GEO number GSE113425) and in
Table S1.
2.12. RT-qPCR
RNA was reverse transcribed into cDNA using the iScript cDNA Synthesis Kit (BioRad), and target genes were amplified with iTaq Universal SYBR green supermix (BioRad) in a BioRad CFX95. The relative gene expression was determined with the comparative
Ct method using CFX manager software (Bio-Rad), also known as 2
−ΔΔCt [
31]. Genes
frd.2,
nrdD, and
motA, whose expression was unaffected in RNA-seq analysis, were used as endogenous controls. Primers were designed using Primer3web [
32,
33]; sequences are available upon request.
2.13. Primer Extension
Primer extension was performed using Avian Myeloblastosis Virus (AMV) reverse transcriptase (Life Sciences, Inc., St. Petersburg, FL, USA) as previously described [
34,
35,
36]. Oligonucleotides were designed to anneal ~150 nucleotides (nt) downstream of the predicted 5’ end unless otherwise stated; primer sequences are available upon request. Total RNA (4 μg) was added to 1 pmol of 5’
32P-labeled primer for each analysis. Primer extension products were separated on 5% (
w/
v) denaturing, polyacrylamide (19:1, acrylamide:bis) gels. Gels were visualized by autoradiography and analyzed as described above.
4. Discussion
Although many aspects of bacteriophage T4 biology have been extensively characterized, dozens of T4’s ~300 genes encode proteins whose specific functions remain unknown. Most of these are nonessential genes that are expressed early, leading to the presumption that they are helpful for host takeover, particularly under certain conditions. In this study, we have characterized one such early gene,
motB, located in a large dispensable region of the T4 genome, called Δ12 (from
69 to
39; map units 16,280 to 3778) [
17]. Other genes located in Δ12 have been shown to be important for phage fitness under certain environmental conditions or in certain host strains and/or to be toxic when expressed in
E. coli [
17,
43]. The toxicity of
motB when it is heterologously expressed in
E. coli and the phage burst increase in its presence lead to the conclusion that MotB is needed for optimal phage growth, perhaps through a host takeover function.
Previous work indicated that MotB can be isolated in a pre-replicative complex containing DNA, RNAP, and other polymerase-associated and DNA-binding proteins [
18]. Other work reported that the expression of particular middle genes was enhanced by the presence of
motB in a
motA knockdown infection [
16]. Taken together, these results suggested that MotB might be needed for optimal middle gene expression, hence the name, modifier of transcription B. However, our transcriptome analyses indicate that at 5 min after infection, when middle gene expression is robust, neither early nor middle gene RNA levels are affected in a T4
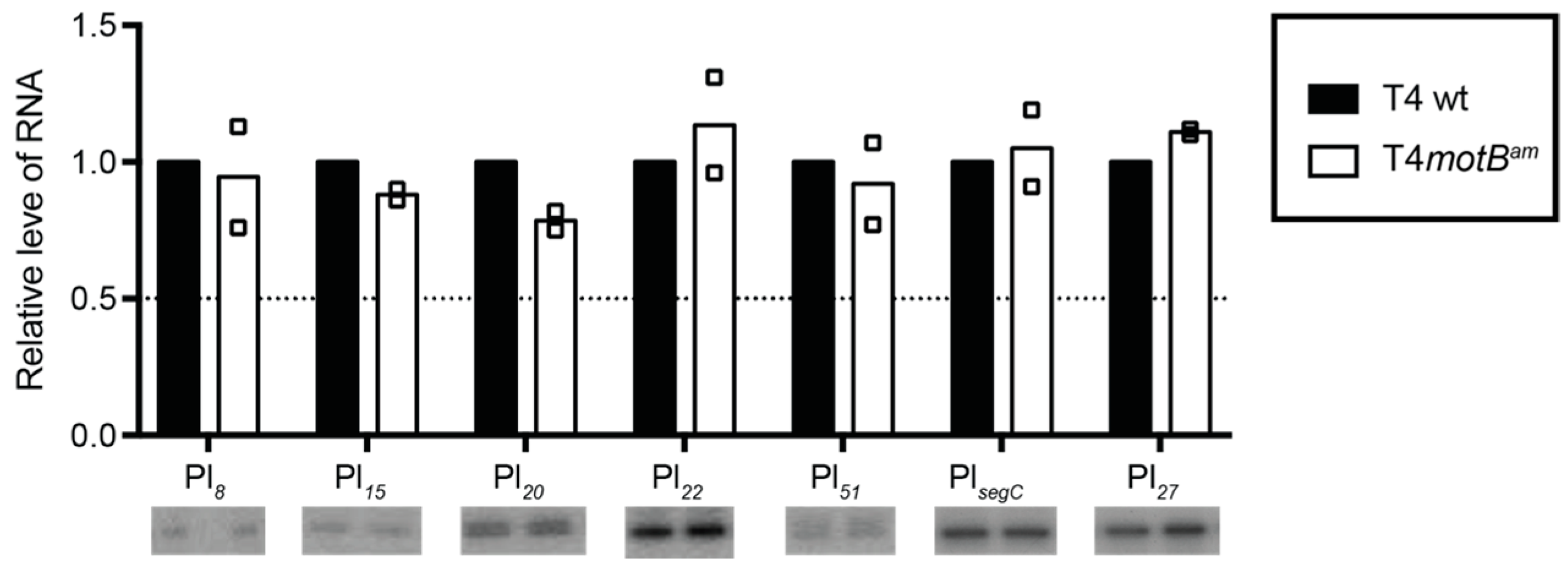
motBam infection. Rather only the expression of a few late genes is lowered at 5 min post-infection. By 10 min when late transcripts are highly expressed, transcription of these genes has recovered. Therefore, it seems unlikely then that MotB directly regulates T4 gene expression unless this function is redundant within the genome.
Our biochemical analyses indicate that MotB is a protein that binds to both unmodified and T4 wt GHme-modified DNA. It binds very tightly to host DNA when produced heterologously in vivo, it binds with ~100 nanomolar affinity to various ds oligonucleotides, and in EMSAs it retards each of the λ/HindIII fragments. The DNase I footprint of MotB on an unmodified 218 bp fragment, which includes the T4 late promoter P8, does not reveal a high affinity binding site despite the down-regulation of late gene 8 in the T4 motBam infection. Instead, we observe MotB protection of the entire region of the DNA once a sufficient protein concentration is reached. This type of binding profile is indicative of a protein that can “spread” along the DNA with little, if any sequence specificity, perhaps through protein–protein cooperativity. However, we cannot eliminate the possibility that MotB has a high affinity site that was not present in the tested DNAs.
Curiously, MotB copurifies with H-NS and StpA,
E. coli histone-like proteins that also can spread along the DNA with limited DNA specificity. H-NS, and its less abundant homolog StpA, target AT-rich DNA sequences, condensing genomic DNA through the formation of ordered and looped structures, and typically repress transcription at the affected region (reviewed in [
8]). Our gel retardation assays indicate that H-NS binds to T4 wt GHMe-modified DNA and that the retardation of either unmodified or GHMe-modified DNA increases in the presence of both MotB and H-NS over that observed with either alone (
Figure 6). At this point it is unclear whether both H-NS and MotB bind to the same region of DNA simultaneously. Furthermore, despite multiple efforts to show a direct interaction between H-NS and MotB, we have not observed an interaction in the absence of DNA.
Given the high predicted pKa of MotB (9.23) and its presence in the T4 dispensable region Δ12, we can determine its relative abundance during infection from previous 2-D gels of radiolabeled T4 proteins at various times after infection [
44]. We identify a highly basic protein, which migrates as predicted for MotB’s size and predicted pKa and is missing in the T4 Δ12 infection. The level of this protein is high when T4 proteins are labeled from 1 to 3 min post-infection and remains high even with 7–9 min post-infection labeling. This identification is supported by another finding that MotB (designated as p17.6) production initiates early after infection (0–4 min) and continues as infection progresses (7–12 min) [
18]. It is known that T4 factors begin to degrade the host DNA shortly after infection, but degradation of host DNA to acid-soluble products is still less than 50% at 15 min post-infection [
45]. It seems reasonable then to conclude that the high levels of MotB would be able to bind host and/or T4 DNA throughout the time it is present. Given the copurification of MotB with H-NS and StpA, two proteins that would be present on the host DNA at the time of infection and could interact with T4 wt DNA, it is tempting to speculate that MotB binding to the DNA might work to counteract H-NS/StpA binding. However, additional work is needed to determine the localization of MotB and H-NS relative to host and phage DNA. Nonetheless, it is clear that heterologous expression of
motB is highly toxic, resulting in rapid cell lysis of BL21(DE3) cells and growth arrest of TOP10F’ cells. The bactericidal properties of phage proteins when expressed in their host is one of the driving forces of phage research. The mechanisms by which proteins such as MotB inhibit bacterial growth could provide insight for the development of future antibiotics.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}