Host Long Noncoding RNA lncRNA-PAAN Regulates the Replication of Influenza A Virus


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Plasmids and Antibodies
2.2. Cells and Virus
2.3. Cell Transfection
2.4. IAV Infection and Virus Titer Assay
2.5. RNA Isolation and Quantitative RT-PCR (qRT-PCR)
2.6. Gluc Acitivity Assay
2.7. Co-Immunoprecipitation (Co-IP) and Western Blot
2.8. Immunofluorescence (IF)
2.9. Fluorescence In Situ Hybridization (FISH)
2.10. Cell Viability Assay
2.11. Subcellular Fractionation
2.12. Native RNA Immunoprecipitation (RIP) and Cross-Linked RIP
2.13. RNA Pulldown
2.14. Statistical Analysis
3. Results
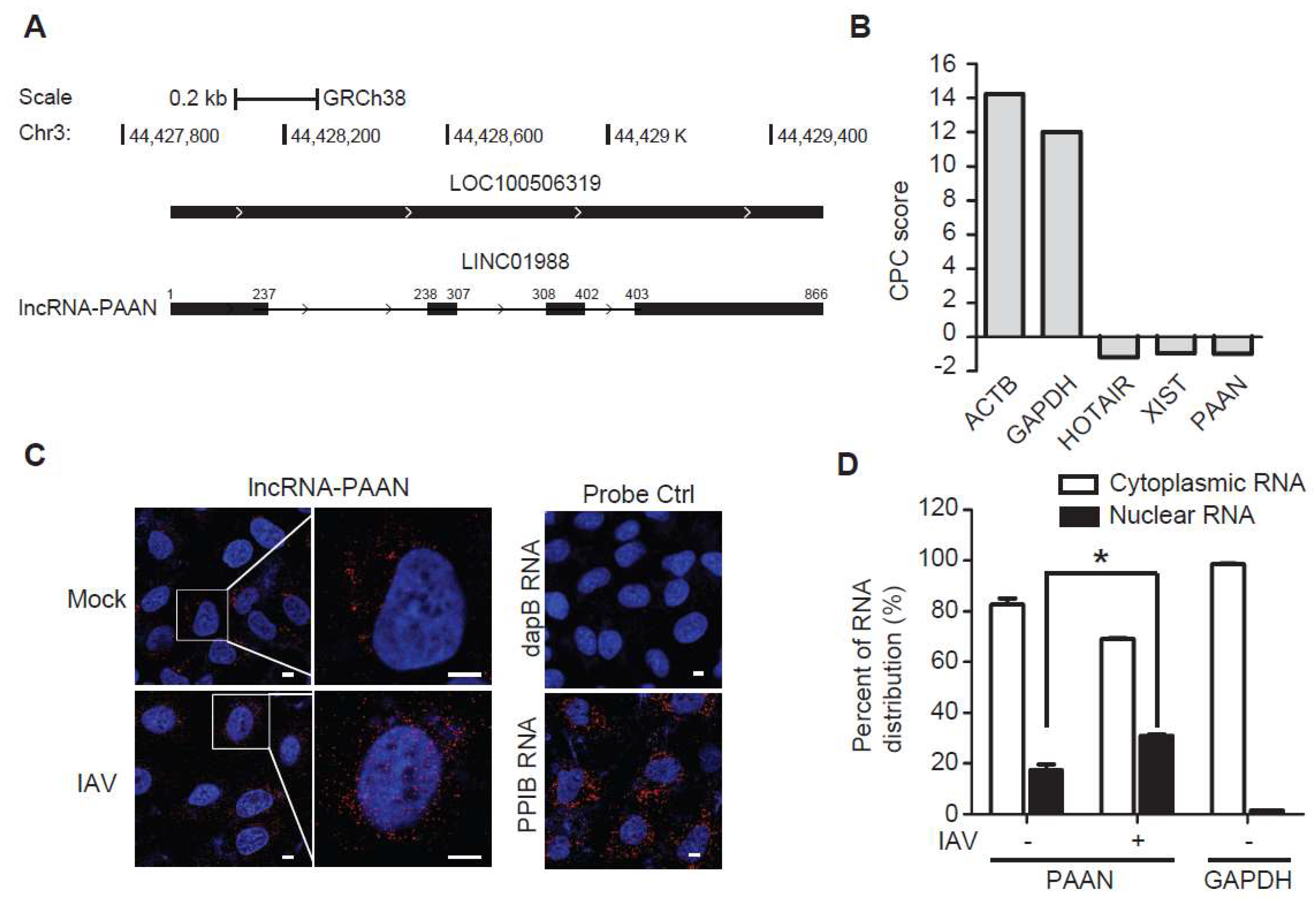
3.1. Characterization of the lncRNA-PAAN
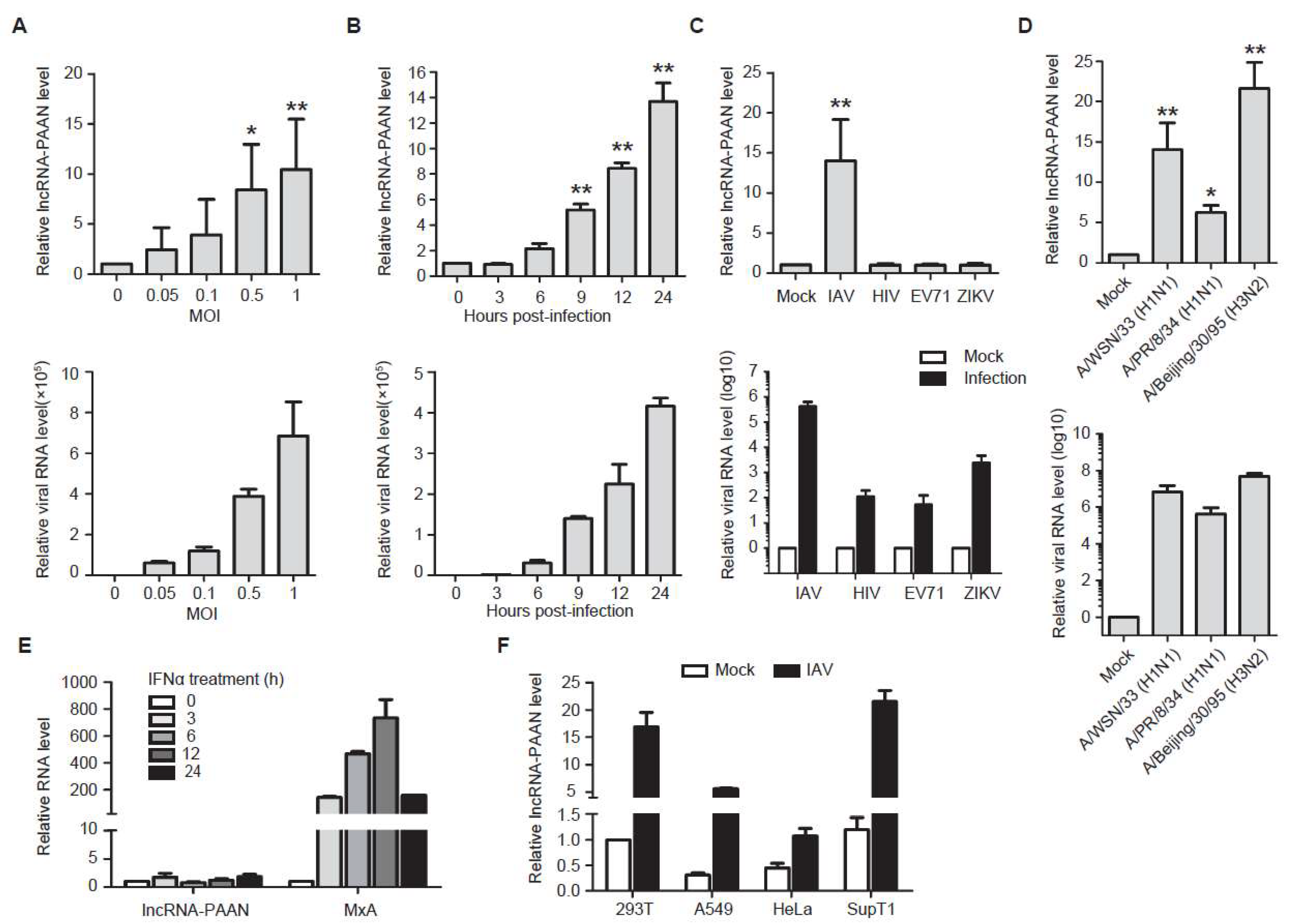
3.2. lncRNA-PAAN is Induced by IAV Infection Independent of Interferon
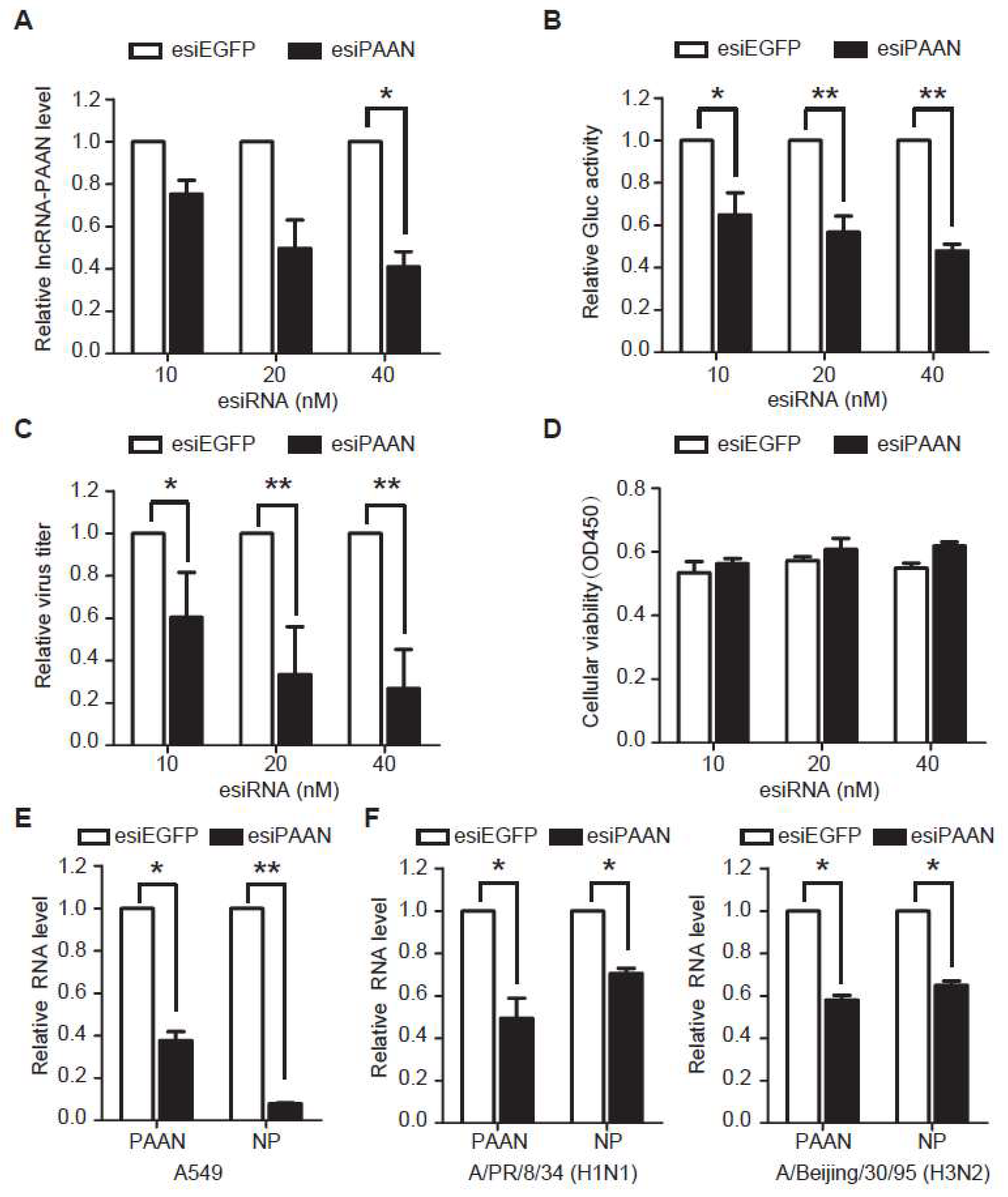
3.3. Downregulation of lncRNA-PAAN Inhibits IAV Replication
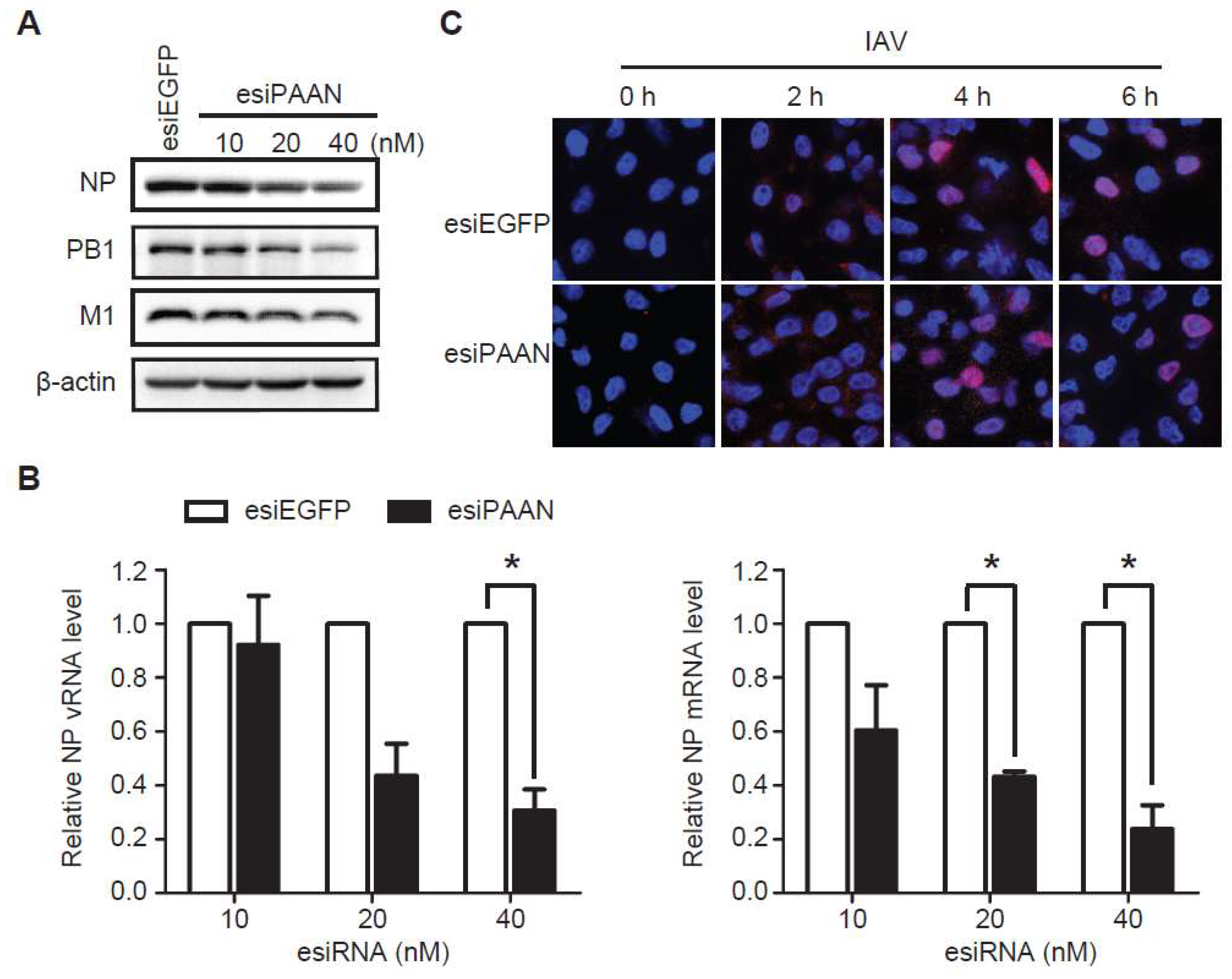
3.4. LncRNA-PAAN Knockdown Reduces Viral RNA Transcription and Replication
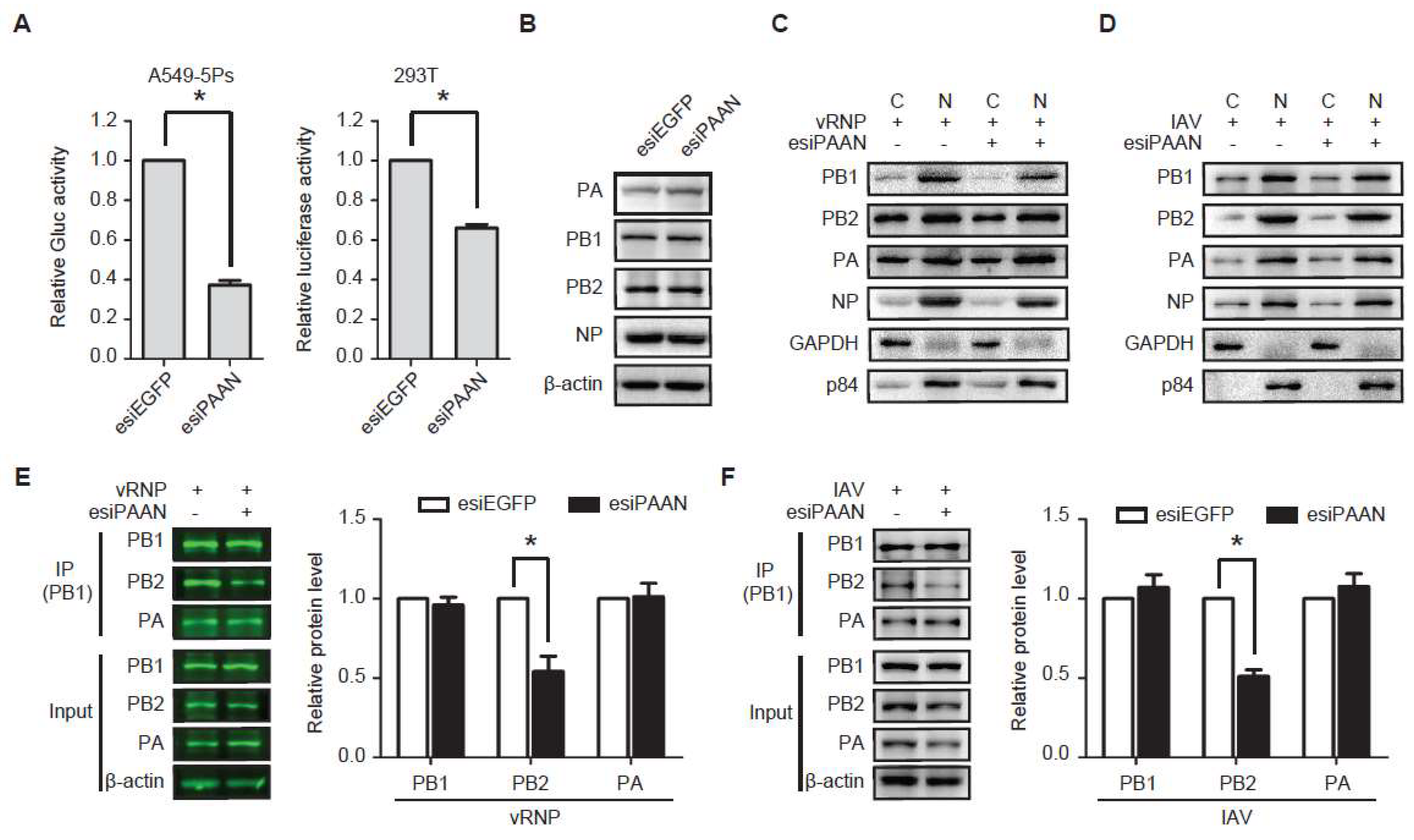
3.5. LncRNA-PAAN Knockdown Impairs IAV Polymerase Activity and the PB1-PB2 Interaction
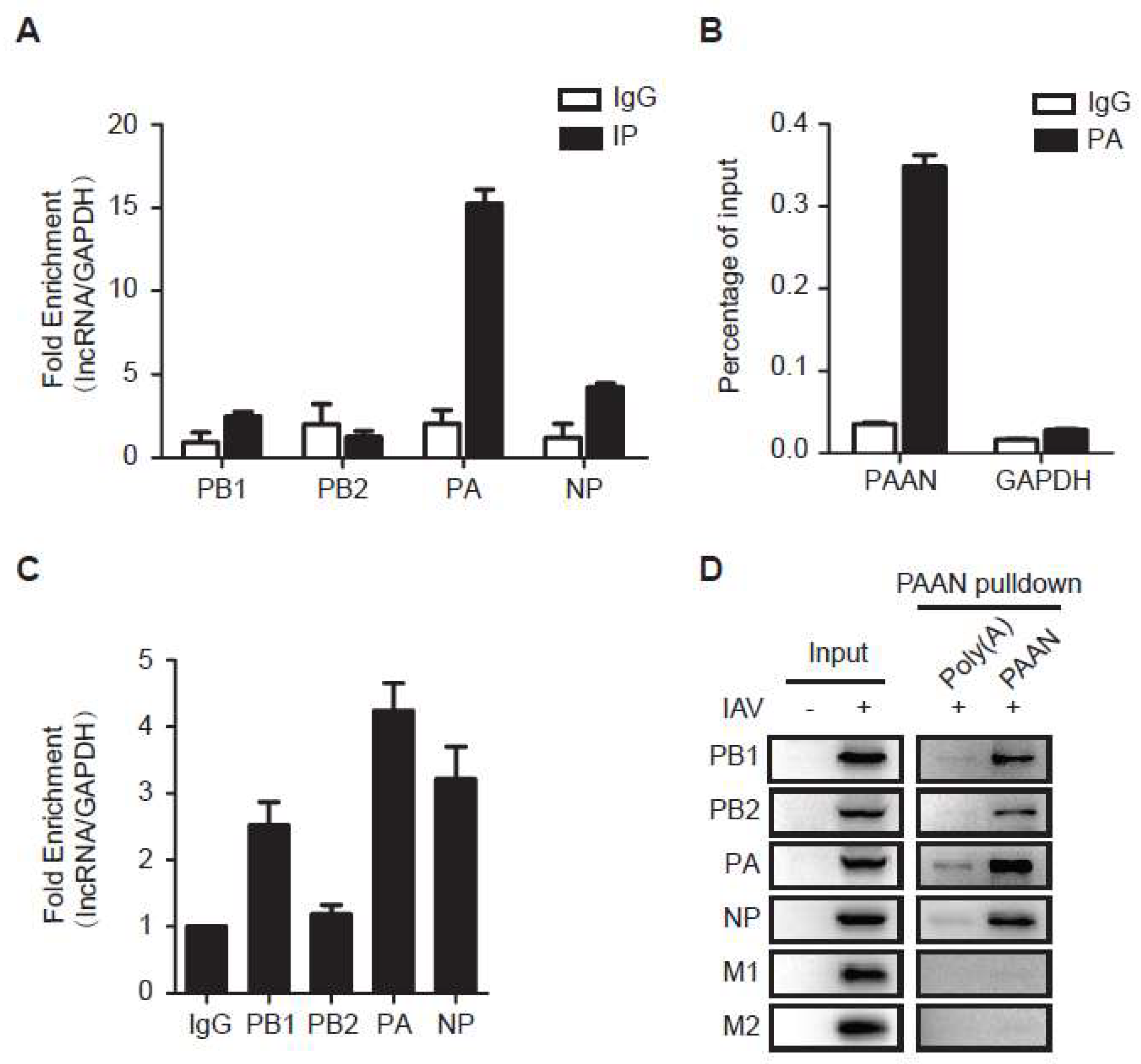
3.6. LncRNA-PAAN Interacts with IAV PA
4. Discussion
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Neumann, G.; Noda, T.; Kawaoka, Y. Emergence and pandemic potential of swine-origin H1N1 influenza virus. Nature 2009, 459, 931–939. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Kawaoka, Y. Pathogenesis of the 1918 pandemic influenza virus. PLoS Pathog. 2011, 7, e1001218. [Google Scholar] [CrossRef] [PubMed]
- Taubenberger, J.K.; Morens, D.M. The pathology of influenza virus infections. Annu. Rev. Pathol. 2008, 3, 499–522. [Google Scholar] [CrossRef] [PubMed]
- Eisfeld, A.J.; Neumann, G.; Kawaoka, Y. At the centre: Influenza A virus ribonucleoproteins. Nat. Rev. Microbiol. 2015, 13, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Te Velthuis, A.J.; Fodor, E. Influenza virus rna polymerase: Insights into the mechanisms of viral RNA synthesis. Nat. Rev. Microbiol. 2016, 14, 479–493. [Google Scholar] [CrossRef] [PubMed]
- Stubbs, T.M.; Te Velthuis, A.J. The RNA-dependent RNA polymerase of the influenza A virus. Future Virol. 2014, 9, 863–876. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, Y.; Matsumae, H.; Katoh, M.; Eisfeld, A.J.; Neumann, G.; Hase, T.; Ghosh, S.; Shoemaker, J.E.; Lopes, T.J.; Watanabe, T.; et al. A comprehensive map of the influenza A virus replication cycle. BMC Syst. Biol. 2013, 7, 97. [Google Scholar] [CrossRef] [PubMed]
- Boivin, S.; Cusack, S.; Ruigrok, R.W.; Hart, D.J. Influenza A virus polymerase: Structural insights into replication and host adaptation mechanisms. J. Biol. Chem. 2010, 285, 28411–28417. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yu, M.; Zheng, W.; Liu, W. Nucleocytoplasmic shuttling of influenza A virus proteins. Viruses 2015, 7, 2668–2682. [Google Scholar] [CrossRef] [PubMed]
- Konig, R.; Stertz, S.; Zhou, Y.; Inoue, A.; Hoffmann, H.H.; Bhattacharyya, S.; Alamares, J.G.; Tscherne, D.M.; Ortigoza, M.B.; Liang, Y.; et al. Human host factors required for influenza virus replication. Nature 2010, 463, 813–817. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.Y.; Luo, Y.; Anwar, M.N.; Sun, Y.; Gao, Y.; Zhang, H.; Munir, M.; Qiu, H.J. Long non-coding RNAs: Emerging and versatile regulators in host-virus interactions. Front. Immunol. 2017, 8, 1663. [Google Scholar] [CrossRef] [PubMed]
- Gottwein, E.; Cullen, B.R. Viral and cellular micrornas as determinants of viral pathogenesis and immunity. Cell Host Microbe 2008, 3, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Morris, K.V.; Mattick, J.S. The rise of regulatory RNA. Nat. Rev. Genet. 2014, 15, 423–437. [Google Scholar] [CrossRef] [PubMed]
- Ernst, C.; Morton, C.C. Identification and function of long non-coding RNA. Front. Cell. Neurosci. 2013, 7, 168. [Google Scholar] [CrossRef] [PubMed]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The gencode V7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef] [PubMed]
- Dey, B.K.; Mueller, A.C.; Dutta, A. Long non-coding rnas as emerging regulators of differentiation, development, and disease. Transcription 2014, 5, e944014. [Google Scholar] [CrossRef] [PubMed]
- Ulitsky, I.; Bartel, D.P. Lincrnas: Genomics, evolution, and mechanisms. Cell 2013, 154, 26–46. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.L.; Carmichael, G.G. Decoding the function of nuclear long non-coding RNAs. Curr. Opin. Cell. Biol. 2010, 22, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.L.; Carmichael, G.G. Long noncoding RNAs in mammalian cells: What, where, and why? Wiley Interdiscip. Rev. RNA 2010, 1, 2–21. [Google Scholar] [CrossRef] [PubMed]
- Winterling, C.; Koch, M.; Koeppel, M.; Garcia-Alcalde, F.; Karlas, A.; Meyer, T.F. Evidence for a crucial role of a host non-coding rna in influenza A virus replication. RNA Biol. 2014, 11, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, J.; Zhu, X.; Chen, Y.; Wei, H.; Chen, Q.; Chi, X.; Qi, B.; Zhang, L.; Zhao, Y.; Gao, G.F.; et al. Nrav, a long noncoding RNA, modulates antiviral responses through suppression of interferon-stimulated gene transcription. Cell Host Microbe 2014, 16, 616–626. [Google Scholar] [CrossRef] [PubMed]
- Landeras-Bueno, S.; Ortin, J. Regulation of influenza virus infection by long non-coding RNAs. Virus Res. 2016, 212, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Xu, J.; Wang, Y.; Cao, X. An interferon-independent lncRNA promotes viral replication by modulating cellular metabolism. Science 2017, 358, 1051–1055. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, E.; Neumann, G.; Kawaoka, Y.; Hobom, G.; Webster, R.G. A DNA transfection system for generation of influenza A virus from eight plasmids. Proc. Natl. Acad. Sci. USA 2000, 97, 6108–6113. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Wang, Z.; Liu, Z.; Li, X.; Zhang, Y.; Zhang, Z.; Cen, S. A cell-based high-throughput approach to identify inhibitors of influenza A virus. Acta Pharm. Sin. B 2014, 4, 301–306. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, Z.; Zhao, F.; Gao, Q.; Liu, Z.; Zhang, Y.; Li, X.; Li, Y.; Ma, W.; Deng, T.; Zhang, Z.; et al. Establishment of a high-throughput assay to monitor influenza A virus rna transcription and replication. PLoS ONE 2015, 10, e0133558. [Google Scholar] [CrossRef] [PubMed]
- Mi, Z.; Ding, J.; Zhang, Q.; Zhao, J.; Ma, L.; Yu, H.; Liu, Z.; Shan, G.; Li, X.; Zhou, J.; et al. A small molecule compound IMB-LA inhibits HIV-1 infection by preventing viral VPU from antagonizing the host restriction factor BST-2. Sci. Rep. 2015, 5, 18499. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.X.; Huang, Y.M.; Li, Q.J.; Li, X.Y.; Zhou, Y.D.; Guo, F.; Zhou, J.M.; Cen, S. A highly conserved amino acid in VP1 regulates maturation of enterovirus 71. PLoS Pathog. 2017, 13, e1006625. [Google Scholar] [CrossRef] [PubMed]
- Reed, L.; Muench, H. A simple method for estimating fifty percent endpoints. Am. J. Hyg. 1938, 27, 493–497. [Google Scholar]
- Tannous, B.A. Gaussia luciferase reporter assay for monitoring biological processes in culture and in vivo. Nat. Protoc. 2009, 4, 582–591. [Google Scholar] [CrossRef] [PubMed]
- Knuckles, P.; Vogt, M.A.; Lugert, S.; Milo, M.; Chong, M.M.; Hautbergue, G.M.; Wilson, S.A.; Littman, D.R.; Taylor, V. Drosha regulates neurogenesis by controlling neurogenin 2 expression independent of microRNAs. Nat. Neurosci. 2012, 15, 962–969. [Google Scholar] [CrossRef] [PubMed]
- Lerner, R.A.; Hodge, L.D. Nonpermissive infections of mammalian cells: Synthesis of inluenza virus genome in hela cells. Proc. Natl. Acad. Sci. USA 1969, 64, 544–551. [Google Scholar] [CrossRef] [PubMed]
- Taniue, K.; Kurimoto, A.; Sugimasa, H.; Nasu, E.; Takeda, Y.; Iwasaki, K.; Nagashima, T.; Okada-Hatakeyama, M.; Oyama, M.; Kozuka-Hata, H.; et al. Long noncoding RNA UPAT promotes colon tumorigenesis by inhibiting degradation of UHRF1. Proc. Natl. Acad. Sci USA 2016, 113, 1273–1278. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, C.; Ma, X.; Geng, G.; Liu, B.; Zhang, Y.; Zhang, S.; Zhong, F.; Liu, C.; Yin, Y.; et al. Long noncoding RNA nron contributes to HIV-1 latency by specifically inducing TAT protein degradation. Nat. Commun. 2016, 7, 11730. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; Abdelmohsen, K.; Kim, J.; Yang, X.; Martindale, J.L.; Tominaga-Yamanaka, K.; White, E.J.; Orjalo, A.V.; Rinn, J.L.; Kreft, S.G.; et al. Scaffold function of long non-coding RNA hotair in protein ubiquitination. Nat. Commun. 2013, 4, 2939. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Kawakami, E.; Shoemaker, J.E.; Lopes, T.J.; Matsuoka, Y.; Tomita, Y.; Kozuka-Hata, H.; Gorai, T.; Kuwahara, T.; Takeda, E.; et al. Influenza virus-host interactome screen as a platform for antiviral drug development. Cell Host Microbe 2014, 16, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Cao, M.; Guo, Y.; Zhao, L.; Wang, J.; Jia, X.; Li, J.; Wang, C.; Gabriel, G.; Xue, Q.; et al. Fragile X mental retardation protein stimulates ribonucleoprotein assembly of influenza A virus. Nat. Commun. 2014, 5, 3259. [Google Scholar] [CrossRef] [PubMed]
- Mondal, A.; Dawson, A.R.; Potts, G.K.; Freiberger, E.C.; Baker, S.F.; Moser, L.A.; Bernard, K.A.; Coon, J.J.; Mehle, A. Influenza virus recruits host protein kinase C to control assembly and activity of its replication machinery. eLife 2017, 6, e26910. [Google Scholar] [CrossRef] [PubMed]
- Deng, T.; Engelhardt, O.G.; Thomas, B.; Akoulitchev, A.V.; Brownlee, G.G.; Fodor, E. Role of ran binding protein 5 in nuclear import and assembly of the influenza virus RNA polymerase complex. J. Virol. 2006, 80, 11911–11919. [Google Scholar] [CrossRef] [PubMed]
- Deng, T.; Sharps, J.; Fodor, E.; Brownlee, G.G. In vitro assembly of PB2 with a PB1-PA dimer supports a new model of assembly of influenza A virus polymerase subunits into a functional trimeric complex. J. Virol. 2005, 79, 8669–8674. [Google Scholar] [CrossRef] [PubMed]
- Mukaigawa, J.; Nayak, D.P. Two signals mediate nuclear localization of influenza virus (A/WSN/33) polymerase basic protein 2. J. Virol. 1991, 65, 245–253. [Google Scholar] [PubMed]
- Pflug, A.; Guilligay, D.; Reich, S.; Cusack, S. Structure of influenza A polymerase bound to the viral RNA promoter. Nature 2014, 516, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Swale, C.; Monod, A.; Tengo, L.; Labaronne, A.; Garzoni, F.; Bourhis, J.M.; Cusack, S.; Schoehn, G.; Berger, I.; Ruigrok, R.W.; et al. Structural characterization of recombinant iav polymerase reveals a stable complex between viral PA-PB1 heterodimer and host Ranbp5. Sci. Rep. 2016, 6, 24727. [Google Scholar] [CrossRef] [PubMed]
- Hemerka, J.N.; Wang, D.; Weng, Y.; Lu, W.; Kaushik, R.S.; Jin, J.; Harmon, A.F.; Li, F. Detection and characterization of influenza a virus PA-PB2 interaction through a bimolecular fluorescence complementation assay. J. Virol. 2009, 83, 3944–3955. [Google Scholar] [CrossRef] [PubMed]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Wang, Y.; Zhou, R.; Zhao, J.; Zhang, Y.; Yi, D.; Li, Q.; Zhou, J.; Guo, F.; Liang, C.; et al. Host Long Noncoding RNA lncRNA-PAAN Regulates the Replication of Influenza A Virus. Viruses 2018, 10, 330. https://doi.org/10.3390/v10060330
Wang J, Wang Y, Zhou R, Zhao J, Zhang Y, Yi D, Li Q, Zhou J, Guo F, Liang C, et al. Host Long Noncoding RNA lncRNA-PAAN Regulates the Replication of Influenza A Virus. Viruses. 2018; 10(6):330. https://doi.org/10.3390/v10060330
Chicago/Turabian StyleWang, Jing, Yujia Wang, Rui Zhou, Jianyuan Zhao, Yongxin Zhang, Dongrong Yi, Quanjie Li, Jinming Zhou, Fei Guo, Chen Liang, and et al. 2018. "Host Long Noncoding RNA lncRNA-PAAN Regulates the Replication of Influenza A Virus" Viruses 10, no. 6: 330. https://doi.org/10.3390/v10060330
APA StyleWang, J., Wang, Y., Zhou, R., Zhao, J., Zhang, Y., Yi, D., Li, Q., Zhou, J., Guo, F., Liang, C., Li, X., & Cen, S. (2018). Host Long Noncoding RNA lncRNA-PAAN Regulates the Replication of Influenza A Virus. Viruses, 10(6), 330. https://doi.org/10.3390/v10060330