Nanomedicine and Phage Capsids



Abstract
1. Introduction
1.1. A Principle
1.2. The Scientific Environment
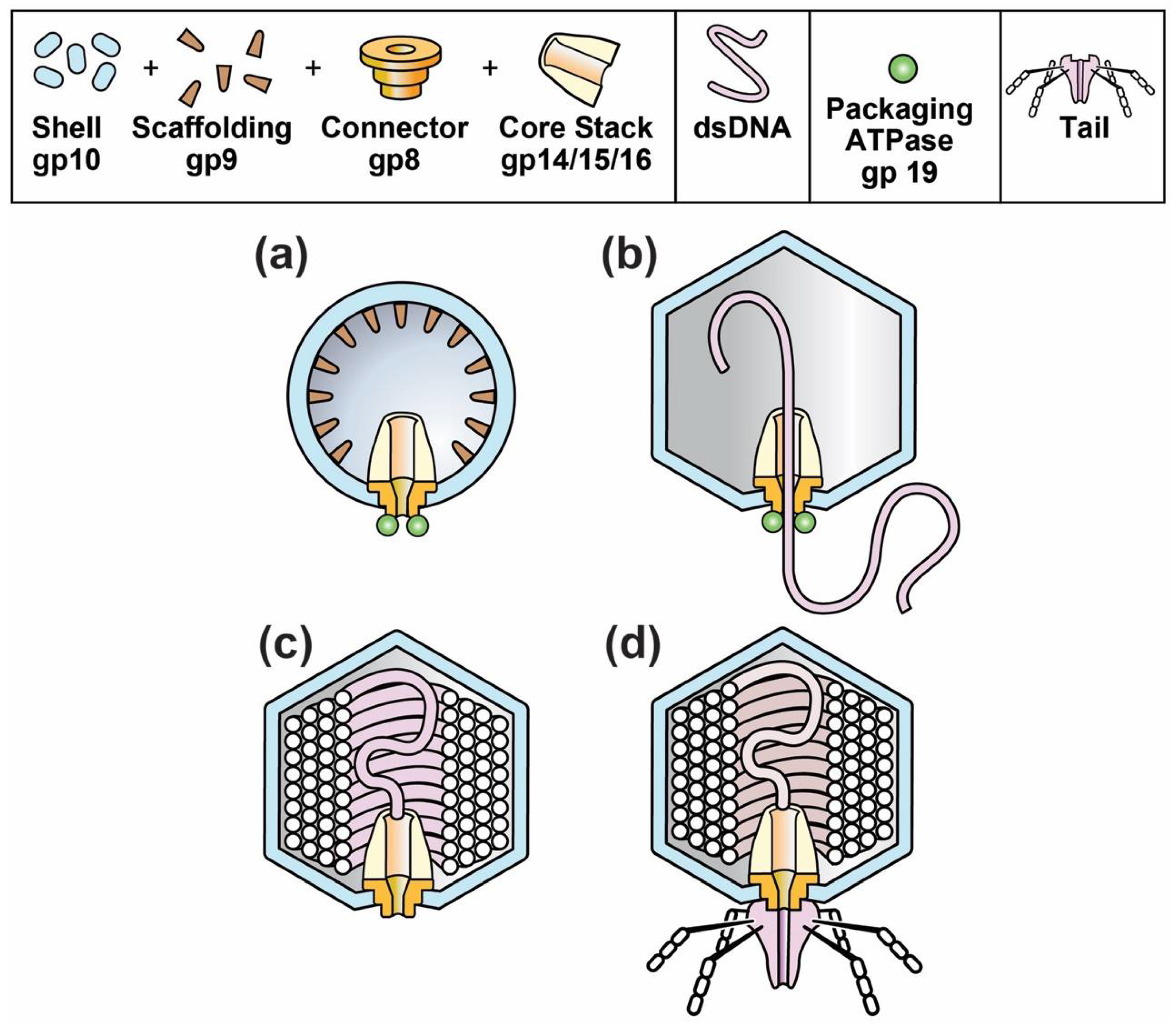
1.3. Phage Assembly Basics
2. Phage Assembly and Dynamic States
2.1. The Stability of the Icosahedral Shell of the Related Phages, T3 and T7
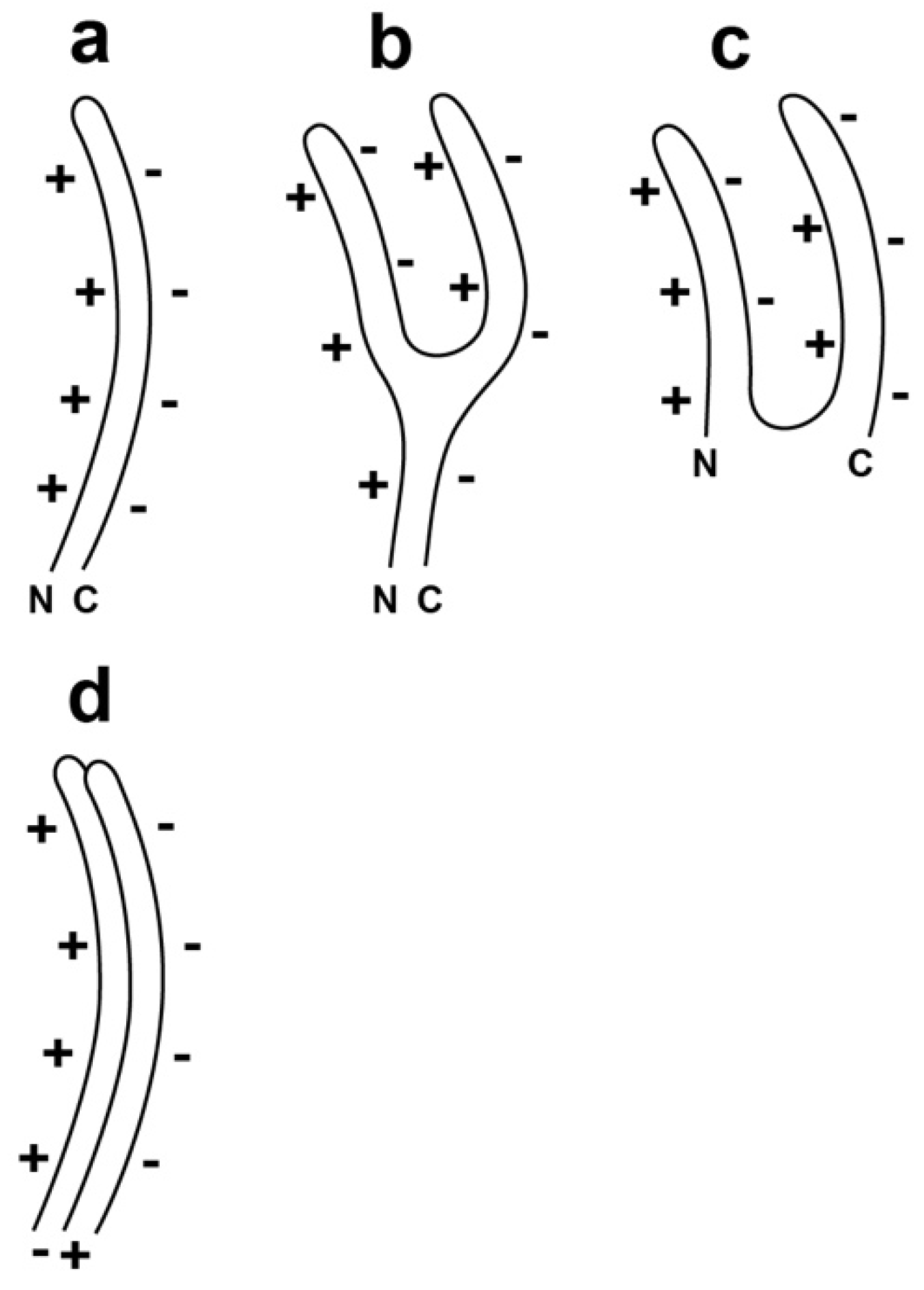
2.2. α-Sheet, Rather than β-Sheet, in Size-Altered Capsid II?
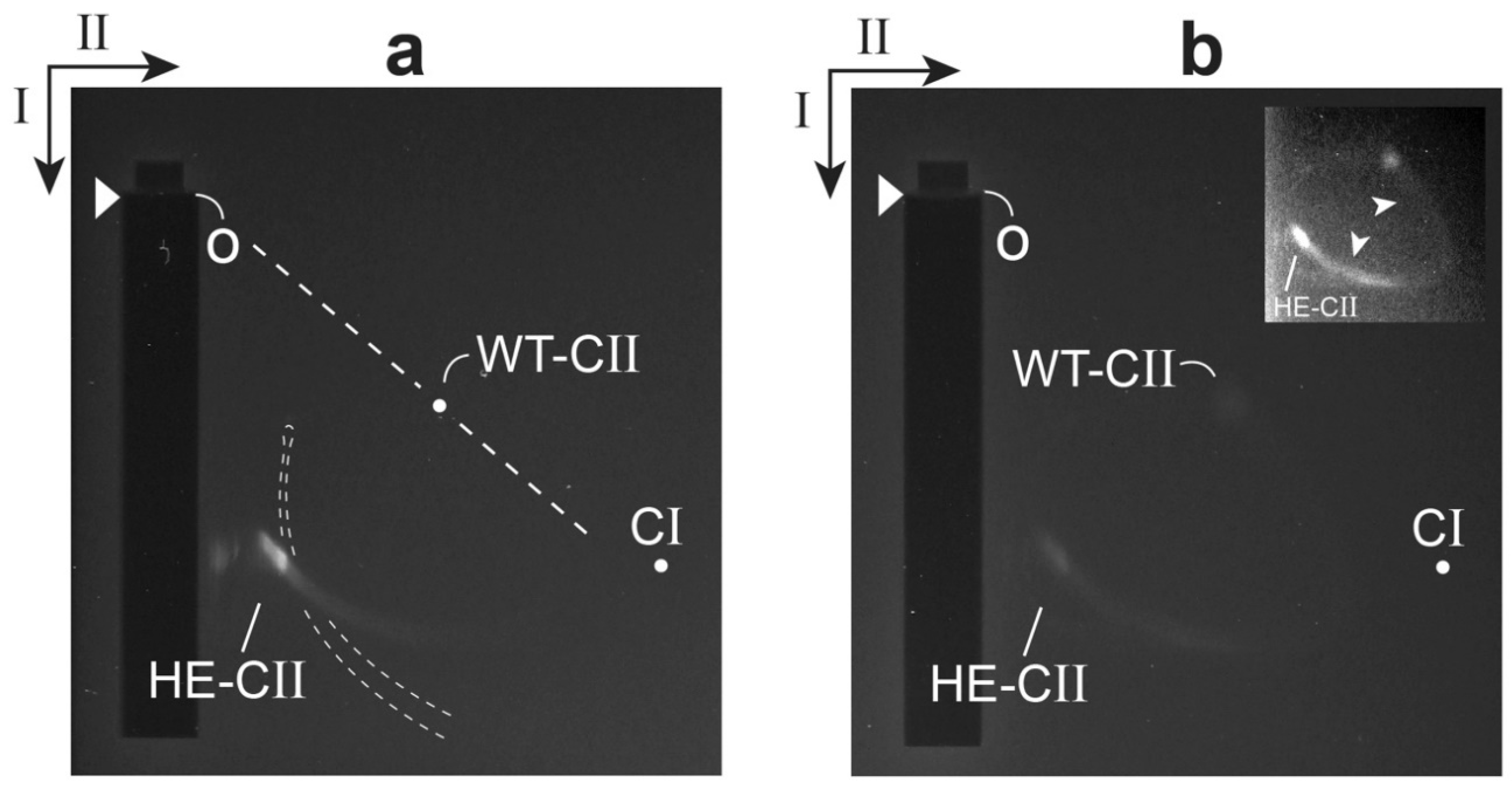
2.3. Test of a Prediction: Surface Charge
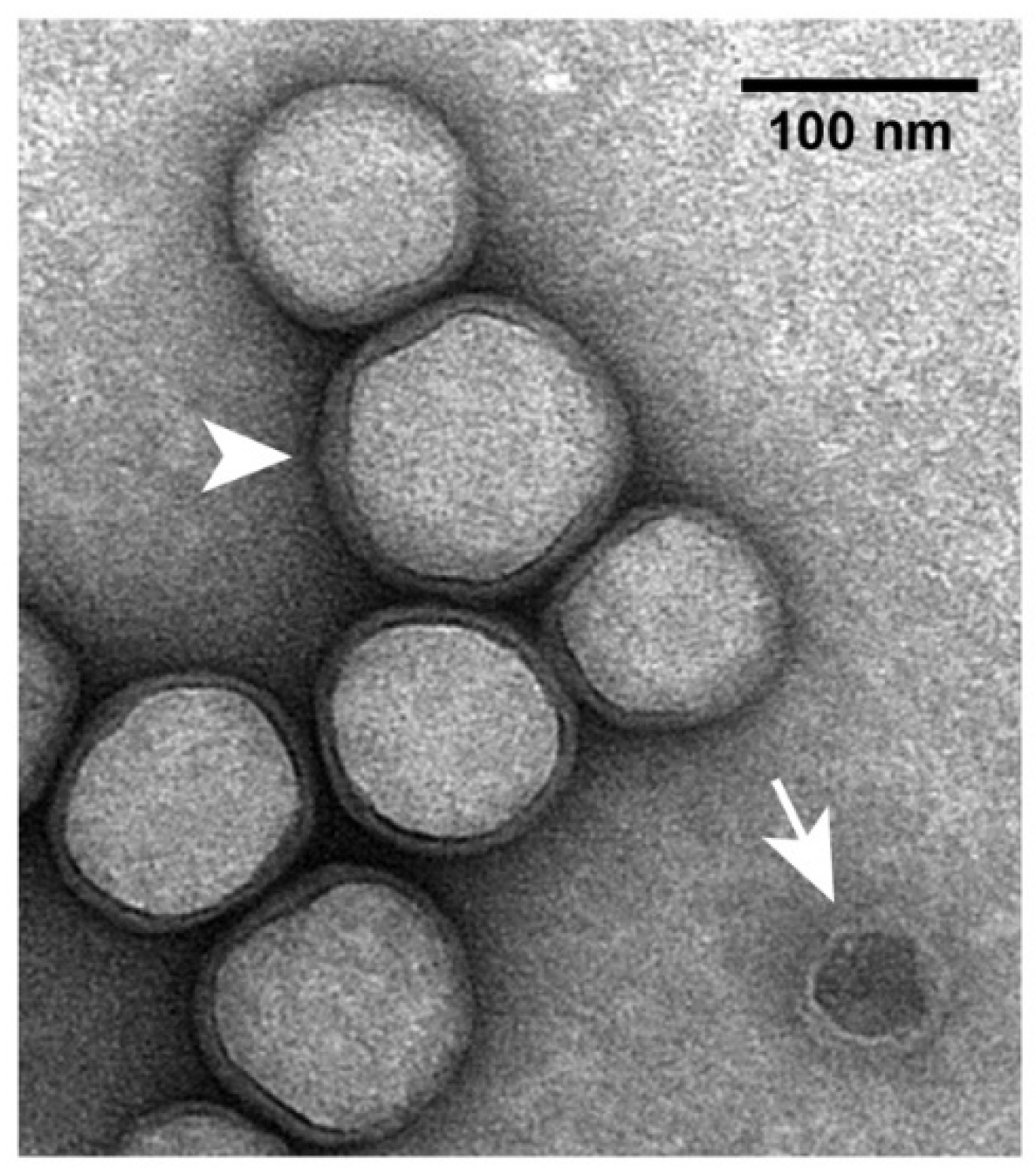
2.4. Electron Microscopy
2.5. Possible Pathway to Anti-Viral Therapeutics
3. Dynamic States and Neurodegenerative Disease
3.1. Some Details
3.2. A Proposal for Reduction to Practice
4. Phage Assembly and Development of Gated, Targeted Drug Delivery Vehicles
4.1. Avoiding Immune Systems
4.2. Targeting Tumors
4.3. Adequate Loading of a Phage Capsid-DDV
5. Prospects for the Future
6. Relationship to Scientific Details
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Riedel, S. Edward Jenner and the history of smallpox and vaccination. Proc. (Baylor Univ. Med. Cent.) 2005, 18, 21–25. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1200696/ (accessed on 10 March 2018). [CrossRef] [PubMed]
- Schwartz, M. The life and works of Louis Pasteur. J. Appl. Microbiol. 2001, 91, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.Y.; Tatsumura, Y. Alexander Fleming (1881–1955): Discoverer of penicillin. Singap. Med. J. 2015, 56, 366–367. [Google Scholar] [CrossRef] [PubMed]
- Kong, K.-F.; Schneper, L.; Mathee, K. Beta-lactam antibiotics: From antibiosis to resistance and bacteriology. APMIS 2010, 118, 1–36. [Google Scholar] [CrossRef] [PubMed]
- Jelić, D.; Antolović, R. From erythromycin to azithromycin and new potential ribosome-binding antimicrobials. Antibiotics 2016, 5, 29. [Google Scholar] [CrossRef] [PubMed]
- Summers, W.C. Felix d’Herelle and the Origins of Molecular Biology; Yale Univ. Press: New Haven, CT, USA, 1999. [Google Scholar]
- Kutter, E.; De Vos, D.; Gvasalia, G.; Alavidze, Z.; Gogokhia, L.; Kuhl, S.; Abedon, S.T. Phage therapy in clinical practice: Treatment of human infections. Curr. Pharm. Biotechnol. 2010, 11, 69–86. [Google Scholar] [CrossRef] [PubMed]
- Dubos, R.J. Louis Pasteur: Free Lance of Science; Scribner: New York, NY, USA, 1976. [Google Scholar]
- Yonath, A. Antibiotics targeting ribosomes: Resistance, selectivity, synergism, and cellular regulation. Annu. Rev. Biochem. 2005, 74, 649–679. [Google Scholar] [CrossRef] [PubMed]
- Graham, W.V.; Bonito-Oliva, A.; Sakmar, T.P. Update on Alzheimer’s disease therapy and prevention strategies. Annu. Rev. Med. 2017, 68, 413–430. [Google Scholar] [CrossRef] [PubMed]
- Piemontese, L. New approaches for prevention and treatment of Alzheimer’s disease: A fascinating challenge. Neural. Regen. Res. 2017, 12, 405–406. [Google Scholar] [CrossRef] [PubMed]
- Gitler, A.D.; Dhillon, P.; Shorter, J. Neurodegenerative disease: Models, mechanisms, and a new hope. Dis. Models Mech. 2017, 10, 499–502. [Google Scholar] [CrossRef] [PubMed]
- Santiago, J.A.; Potashkin, J.A. A network approach to clinical intervention in neurodegenerative diseases. Trends Mol. Med. 2014, 20, 694–703. [Google Scholar] [CrossRef] [PubMed]
- Leaf, C. The Truth in Small Doses; Simon and Schuster: New York, NY, USA, 2013. [Google Scholar]
- Rose, D.W. Friends and Partners: The Legacy of Franklin D. Roosevelt and Basil O’Connor in the History of Polio; Elsevier: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Schooley, R.T.; Biswas, B.; Gill, J.J.; Hernandez-Morales, A.; Lancaster, J.; Lessor, L.; Barr, J.J.; Reed, S.L.; Rohwer, F.; Benler, S.; et al. Development and use of personalized bacteriophage-based therapeutic cocktails to treat a patient with a disseminated resistant Acinetobacter baumannii infection. Antimicrob. Agents Chemother. 2017, 61, e00954-17. [Google Scholar] [CrossRef] [PubMed]
- Serwer, P. Restoring logic and data to phage-cures for infectious disease. AIMS Microbiol. 2017, 3, 706–712. [Google Scholar] [CrossRef]
- Sankar, A.; Merril, C.R.; Biswas, B. Therapeutic and prophylactic applications of bacteriophage components in modern medicine. Cold Spring Harb. Perspect. Med. 2014, 4, a012518. [Google Scholar] [CrossRef]
- Weber, L. Sewage Saved This Man’s Life. Someday It Could Save Yours. Bacteriophages—Viruses Found in Soil, Water and Human Waste—May Be the Cure in a Post—Antibiotic World. HuffPost, US Edition. 2017. Available online: http://www.huffingtonpost.com/entry/antibioticresistant-superbugs-phage-therapy_us_5913414de4b05e1ca203f7d4 (accessed on 10 March 2018).
- Edgar, R.S.; Feynman, R.P.; Klein, S.; Lielausis, I.; Steinberg, C.M. Mapping Experiments with R Mutants of Bacteriophage T4D. Genetics 1962, 47, 179–186. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1210321/ (accessed on 12 March 2018). [CrossRef]
- Pauling, L.; Corey, R.B. The pleated sheet, a new layer configuration of polypeptide chains. Proc. Natl. Acad. Sci. USA 1951, 37, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Pauling, L.; Corey, R.B. The structure of feather rachis keratin. Proc. Natl. Acad. Sci. USA 1951, 37, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Pauling, L.; Corey, R.B. Configurations of polypeptide chains with favored orientations around single bonds: Two new pleated sheets. Proc. Natl. Acad. Sci. USA 1951, 37, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Liu, Z.; Vago, F.; Ren, Y.; Wu, W.; Wright, E.T.; Serwer, P.; Jiang, W. Visualization of uncorrelated, tandem symmetry mismatches in the internal genome packaging apparatus of bacteriophage T7. Proc. Natl. Acad. Sci. USA 2013, 110, 6811–6816. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Liu, Z.; Fang, P.-A.; Zhang, Q.; Wright, E.T.; Wu, W.; Zhang, C.; Vago, F.; Ren, Y.; Jakana, J.; et al. Capsid expansion mechanism of bacteriophage T7 revealed by multistate atomic models derived from cryo-EM reconstructions. Proc. Natl. Acad. Sci. USA 2014, 111, E4606–E4614. [Google Scholar] [CrossRef] [PubMed]
- Serwer, P.; Wright, E.T.; Liu, Z.; Jiang, W. Length quantization of DNA partially expelled from heads of a bacteriophage T3 mutant. Virology 2014, 456–457, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Pajunen, M.I.; Elizondo, M.R.; Skurnik, M.; Kieleczawa, J.; Molineux, I.J. Complete nucleotide sequence and likely recombinatorial origin of bacteriophage T3. J. Mol. Biol. 2002, 319, 1115–1132. [Google Scholar] [CrossRef]
- Serwer, P.; Pichler, M.E. Electrophoresis of bacteriophage T7 and T7 capsids in agarose gels. J. Virol. 1978, 28, 917–928. [Google Scholar] [PubMed]
- Serwer, P.; Hayes, S.J.; Watson, R.H. The structure of a bacteriophage T7 procapsid and its in vivo conversion product probed by digestion with trypsin. Virology 1982, 122, 392–401. [Google Scholar] [CrossRef]
- Wikoff, W.R.; Liljas, L.; Duda, R.L.; Tsuruta, H.; Hendrix, R.W.; Johnson, J.E. Topologically linked protein rings in the bacteriophage HK97 capsid. Science 2000, 289, 2129–2133. [Google Scholar] [CrossRef] [PubMed]
- Duda, R.L.; Ross, P.D.; Cheng, N.; Firek, B.A.; Hendrix, R.W.; Conway, J.F.; Steven, A.C. Structure and energetics of encapsidated DNA in bacteriophage HK97 studied by scanning calorimetry and cryoelectron microscopy. J. Mol. Biol. 2009, 391, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Vago, F.; Zhang, D.; Snyder, J.E.; Yan, R.; Zhang, C.; Benjamin, C.; Jiang, X.; Kuhn, R.J.; Serwer, P.; et al. Single-step antibody-based affinity cryo-electron microscopy for imaging and structural analysis of macromolecular assemblies. J. Struct. Biol. 2014, 187, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Serwer, P.; Wright, E.T.; Demeler, B.; Jiang, W. States of phage T3/T7 capsids: Buoyant density centrifugation and cryo-EM. Biophys. Rev. 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Serwer, P.; Wright, E. Testing a proposed paradigm shift in analysis of phage DNA packaging. Bacteriophage 2016, 6, e1268664. [Google Scholar] [CrossRef] [PubMed]
- Serwer, P.; Wright, E.T. ATP-driven contraction of phage T3 capsids with DNA incompletely packaged in vivo. Viruses 2017, 9, 119. [Google Scholar] [CrossRef] [PubMed]
- Serwer, P. Proposed ancestors of phage nucleic acid packaging motors (and cells). Viruses 2011, 3, 1249–1280. [Google Scholar] [CrossRef] [PubMed]
- Milner-White, E.J.; Russell, M.J. Predicting the conformations of peptides and proteins in early evolution. A review article submitted to Biology Direct. Biol. Direct 2008, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Milner-White, E.J.; Russell, M.J. Functional capabilities of the earliest peptides and the emergence of life. Genes 2011, 2, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Naryzhny, S. Towards the full realization of 2DE power. Proteomes 2016, 4, 33. [Google Scholar] [CrossRef] [PubMed]
- Stroud, R.M.; Serwer, P.; Ross, M.J. Assembly of bacteriophage t7. Dimensions of the bacteriophage and its capsids. Biophys. J. 1981, 36, 743–757. [Google Scholar] [CrossRef]
- Van der Linden, L.; Wolthers, K.C.; van Kuppeveld, F.J.M. Replication and inhibitors of enteroviruses and parechoviruses. Viruses 2015, 7, 4529–4562. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.J.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Van Dam, D.; Vermeiren, Y.; Dekker, A.D.; Naudé, P.J.W.; De Deyn, P.P. Neuropsychiatric disturbances in Alzheimer’s disease: What have we learned from neuropathological studies? Curr. Alzheimer Res. 2016, 13, 1145–1164. [Google Scholar] [CrossRef] [PubMed]
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R.; et al. Huntington Disease. Nat. Rev. Dis. Primers 2015, 1, 15005. Available online: https://www.nature.com/articles/nrdp20155 (accessed on 13 March 2018). [CrossRef] [PubMed]
- Kalia, L.V.; Lang, A.E. Parkinson disease in 2015: Evolving basic, pathological and clinical concepts in PD. Nat. Rev. Neurol. 2016, 12, 65–66. [Google Scholar] [CrossRef] [PubMed]
- Collinge, J. Mammalian prions and their wider relevance in neurodegenerative diseases. Nature 2016, 539, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Daggett, V. Alpha-sheet: The toxic conformer in amyloid diseases? Acc. Chem. Res. 2006, 39, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Armen, R.S.; Alonso, D.O.V.; Daggett, V. Anatomy of an amyloidogenic intermediate: Conversion of β-sheet to α-sheet in transthyretin. Structure 2004, 12, 1847–1863. [Google Scholar] [CrossRef] [PubMed]
- Kellock, J.; Hopping, G.; Caughey, B.; Daggett, V. Peptides composed of alternating L- and D-amino acids inhibit amyloidogenesis in three distinct amyloid systems independent of sequence. J. Mol. Biol. 2016, 428, 2317–2328. [Google Scholar] [CrossRef] [PubMed]
- Bandea, C.I. Aβ, tau, α-synuclein, huntingtin, TDP-43, PrP and AA are members of the innate immune system: A unifying hypothesis on the etiology of AD, PD, HD, ALS, CJD and RSA as innate immunity disorders. bioRχiv 2013. [Google Scholar] [CrossRef]
- Serwer, P. Hypothesis for the cause and therapy of neurodegenerative diseases. Med. Hypotheses 2018, 110, 60–63. [Google Scholar] [CrossRef] [PubMed]
- Vijaya Kumar, D.K.; Choi, S.H.; Washicosky, K.J.; Eimer, W.A.; Tucker, S.; Ghofrani, J.; Lefkowitz, A.; McColl, G.; Goldstein, L.E.; Tanzi, R.E.; et al. Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci. Transl. Med. 2016, 8, 340ra72. [Google Scholar] [CrossRef] [PubMed]
- White, M.R.; Kandel, R.; Hsieh, I.-N.; De Luna, X.; Hartshorn, K.L. Critical role of C-terminal residues of the Alzheimer’s associated β-amyloid protein in mediating antiviral activity and modulating viral and bacterial interactions with neutrophils. PLoS ONE 2018, 13, e0194001. [Google Scholar] [CrossRef] [PubMed]
- Deleidi, M.; Isacson, O. Viral and inflammatory triggers of neurodegenerative diseases. Sci. Transl. Med. 2012, 4, 121ps3. [Google Scholar] [CrossRef] [PubMed]
- Itzhaki, R.F.; Lathe, R.; Balin, B.J.; Ball, M.J.; Bearer, E.L.; Braak, H.; Bullido, M.J.; Carter, C.; Clerici, M.; Cosby, S.L.; et al. Microbes and Alzheimer’s disease. J. Alzheimers Dis. 2016, 51, 979–984. [Google Scholar] [CrossRef] [PubMed]
- Hodyra-Stefaniak, K.; Miernikiewicz, P.; Drapała, J.; Drab, M.; Jończyk-Matysiak, E.; Lecion, D.; Kaźmierczak, Z.; Beta, W.; Majewska, J.; Harhala, M.; et al. Mammalian host-versus-phage immune response determines phage fate in vivo. Sci. Rep. 2015, 5, 14802. [Google Scholar] [CrossRef] [PubMed]
- Aksyuk, A.A.; Rossmann, M.G. Bacteriophage assembly. Viruses 2011, 3, 172–203. [Google Scholar] [CrossRef] [PubMed]
- Fokine, A.; Rossmann, R.G. Molecular architecture of tailed double-stranded DNA phages. Bacteriophage 2014, 4, e28281. [Google Scholar] [CrossRef] [PubMed]
- Hume, D.A. The mononuclear phagocyte system. Curr. Opin. Immunol. 2006, 18, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Yona, S.; Gordon, S. From the reticuloendothelial to mononuclear phagocyte system—The unaccounted years. Front. Immunol. 2015, 6, 328. [Google Scholar] [CrossRef] [PubMed]
- Merril, C.R.; Biswas, B.; Carlton, R.; Jensen, N.C.; Creed, G.J.; Zullo, S.; Adhya, S. Long-circulating bacteriophage as antibacterial agents. Proc. Natl. Acad. Sci. USA 1996, 93, 3188–3192. [Google Scholar] [CrossRef] [PubMed]
- Merril, C.R.; Scholl, D.; Adhya, S.L. The prospect for bacteriophage therapy in Western medicine. Nat. Rev. Drug Discov. 2003, 2, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Serwer, P.; Wright, E.T.; Williams, T.L.; Demeler, B.; Lee, J.C. Phage-based therapies: What is the “real thing”? In Proceedings of the XXV Biennial Conference on Phage/Virus Assembly, Ellicott City, MD, USA, 20–25 August 2017. [Google Scholar]
- Schaue, D.; McBride, W.H. Opportunities and challenges of radiotherapy for treating cancer. Nat. Rev. Clin. Oncol. 2015, 12, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Chabner, B.A.; Roberts, T.G. Chemotherapy and the war on cancer. Nat. Rev. Cancer 2005, 5, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Mehlen, P.; Puisieux, A. Metastasis: A question of life or death. Nat. Rev. Cancer 2006, 6, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Kottschade, L.A. Incidence and management of immune-related adverse events in patients undergoing treatment with immune checkpoint inhibitors. Curr. Oncol. Rep. 2018, 20, 24. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Watanabe, R.; Choyke, P.L. Improving conventional enhanced permeability and retention (EPR) effects; what is the appropriate target? Theranostics 2014, 4, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Fang, J.; Maeda, H. Development of next-generation macromolecular drugs based on the EPR effect: Challenges and pitfalls. Expert Opin. Drug Deliv. 2015, 12, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Weissig, V.; Pettinger, T.K.; Murdock, N. Nanopharmaceuticals (Part 1): Products on the market. Int. J. Nanomed. 2014, 9, 4357–4373. [Google Scholar] [CrossRef] [PubMed]
- Barenholz, Y. Doxil®—The first FDA-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef] [PubMed]
- Anchordoquy, T.J.; Barenholz, Y.; Boraschi, D.; Chorny, M.; Decuzzi, P.; Dobrovolskaia, M.; Farhangrazi, Z.S.; Farrell, D.; Gabizon, A.; Ghandehari, H.; et al. Mechanisms and barriers in cancer nanomedicine: Addressing challenges, looking for solutions. ACS Nano 2017, 11, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Summers, W.C. Bacteriophage therapy. Annu. Rev. Microbiol. 2001, 55, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Serwer, P.; Wright, E.T.; Chang, J.T.; Liu, X. Enhancing and initiating phage-based therapies. Bacteriophage 2014, 4, e961869. [Google Scholar] [CrossRef] [PubMed]
- Boodman, E. To Save a Young Woman Besieged by Superbugs, Scientists Hunt a Killer Virus. Available online: https://www.pbs.org/newshour/health/to-save-a-young-woman-besieged-by-superbugs-scientists-hunt-a-killer-virus (accessed on 13 November 2017).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serwer, P.; Wright, E.T. Nanomedicine and Phage Capsids. Viruses 2018, 10, 307. https://doi.org/10.3390/v10060307
Serwer P, Wright ET. Nanomedicine and Phage Capsids. Viruses. 2018; 10(6):307. https://doi.org/10.3390/v10060307
Chicago/Turabian StyleSerwer, Philip, and Elena T. Wright. 2018. "Nanomedicine and Phage Capsids" Viruses 10, no. 6: 307. https://doi.org/10.3390/v10060307
APA StyleSerwer, P., & Wright, E. T. (2018). Nanomedicine and Phage Capsids. Viruses, 10(6), 307. https://doi.org/10.3390/v10060307