Burkholderia cenocepacia Prophages—Prevalence, Chromosome Location and Major Genes Involved


Abstract
1. Introduction
2. Materials and Methods
2.1. PHASTER Automated Annotation
2.2. Manual Annotation
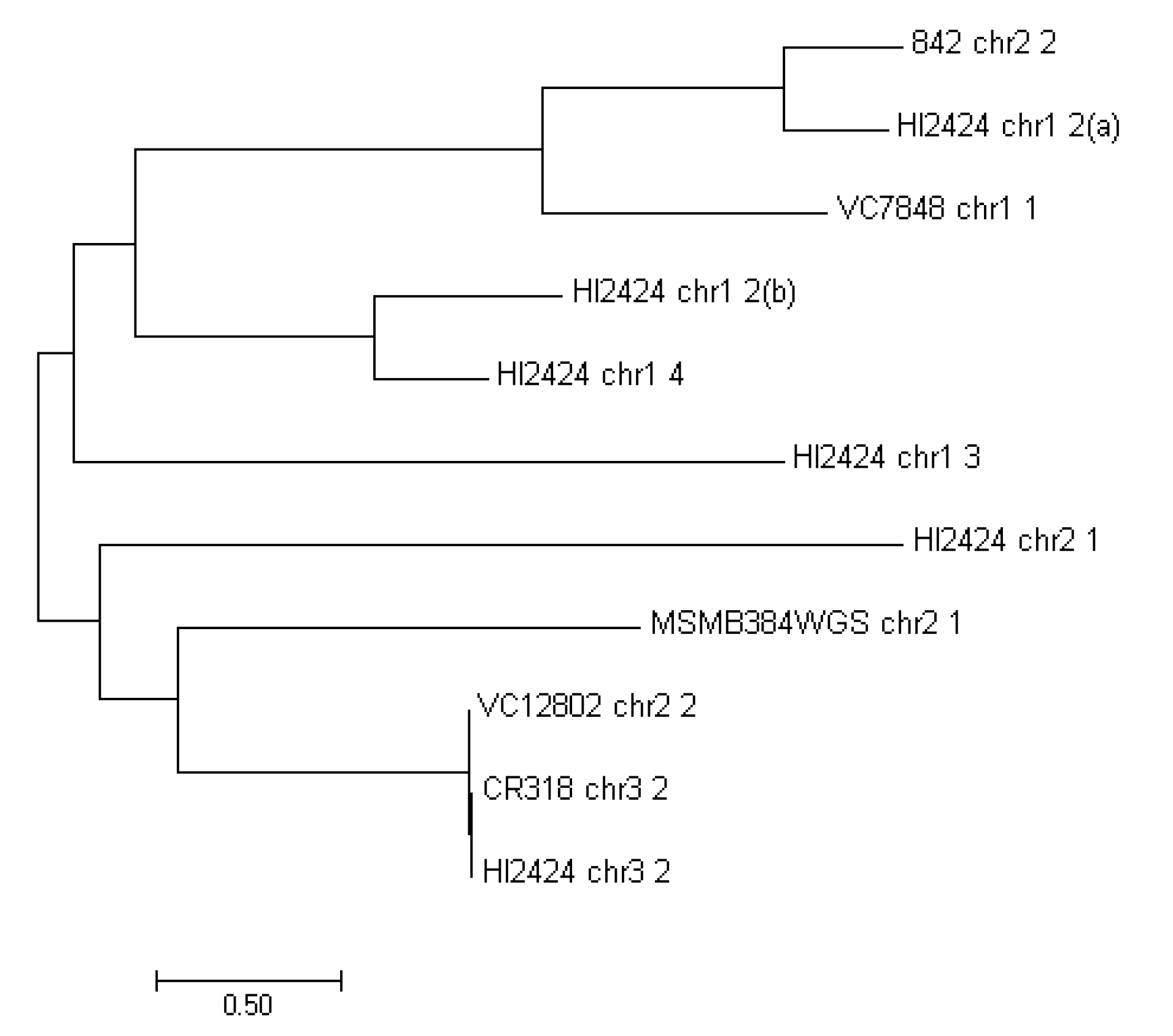
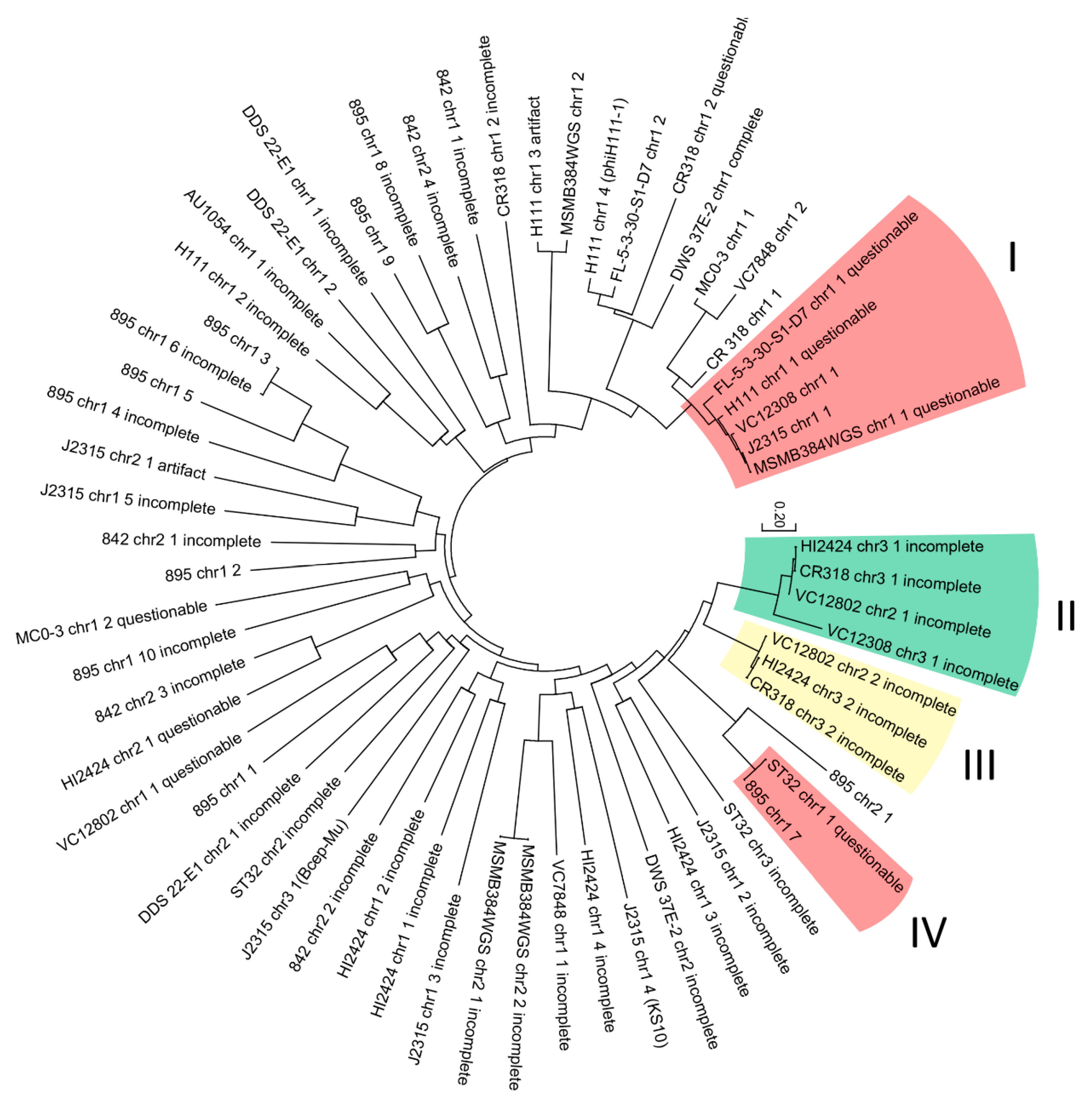
2.3. Phylogeny Analysis
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Burkholder, W.H. Three bacterial plant pathogens: Phytomonas caryophylli, sp. n., Phytomonas alliicola sp. n., and Phytomonas manihotis (Artaud, Berthet and Bondar) Viégas. Phythopathology 1942, 57, 141–149. [Google Scholar]
- Yabuuchi, E.; Kosako, Y.; Oyaizu, H.; Yano, I.; Hotta, H.; Hashimoto, Y.; Ezaki, T.; Arakawa, M. Proposal of Burkholderia gen. nov. and transfer of seven species of the genus Pseudomonas homology group II to the new genus, with the type species Burkholderia cepacia (Palleroni and Holmes 1981) comb. nov. Microbiol. Immunol. 1992, 36, 1251–1275. [Google Scholar] [CrossRef] [PubMed]
- Chiarini, L.; Bevivino, A.; Dalmastri, C.; Tabacchioni, S.; Visca, P. Burkholderia cepacia complex species: Health hazards and biotechnological potential. Trends Microbiol. 2006, 14, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.L.; Fasi, A.C.; Ramette, A.; Smith, J.J.; Hammerschmidt, R.; Sundin, G.W. Identification and onion pathogenicity of Burkholderia cepacia complex isolates from the onion rhizosphere and onion field soil. Appl. Environ. Microbiol. 2008, 74, 3121–3129. [Google Scholar] [CrossRef] [PubMed]
- Coventry, H.S.; Dubery, I.A. Lipopolysaccharides from Burkholderia cepacia contribute to an enhanced defensive capacity and the induction of pathogenesis-related proteins in Nicotianae tabacum. Physiol. Mol. Plant Pathol. 2001, 58, 149–158. [Google Scholar] [CrossRef]
- Vandamme, P.; Holmes, B.; Vancanneyt, M.; Coenye, T.; Hoste, B.; Coopman, R.; Revets, H.; Lauwers, S.; Gillis, M.; Kersters, K.; et al. Occurrence of multiple genomovars of Burkholderia cepacia in cystic fibrosis patients and proposal of Burkholderia multivorans sp. nov. Int. J. Syst. Bacteriol. 1997, 47, 1188–1200. [Google Scholar] [CrossRef] [PubMed]
- Courtney, J.; Dunbar, K.E.; McDowell, A.; Moore, J.; Warke, T.; Stevenson, M.; Elborn, J. Clinical outcome of Burkholderia cepacia complex infection in cystic fibrosis adults. J. Cyst. Fibros. 2004, 3, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Folescu, T.W.; da Costa, C.H.; Cohen, R.W.F.; da Conceição Neto, O.C.; Albano, R.M.; Marques, E.A. Burkholderia cepacia complex: Clinical course in cystic fibrosis patients. BMC Pulm. Med. 2015, 15, 158. [Google Scholar] [CrossRef] [PubMed]
- Chernish, R.N.; Aaron, S.D. Approach to resistant Gram-negative bacterial pulmonary infections in patients with cystic fibrosis. Curr. Opin. Pulm. Med. 2003, 9, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Beckman, W.; Lessie, T.G. Response of Pseudomonas cepacia to β-lactam antibiotics: Utilization of penicillin G as the carbon source. J. Bacteriol. 1979, 140, 1126–1128. [Google Scholar] [PubMed]
- Taylor, K.; McCullough, B.; Clarke, D.J.; Langley, R.J.; Pechenick, T.; Hill, A.; Campopiano, D.J.; Barran, P.E.; Dorin, J.R.; Govan, J.R.W. Covalent dimer species of β-defensin Defr1 display potent antimicrobial activity against multidrug-resistant bacterial pathogens. Antimicrob. Agents Chemother. 2007, 51, 1719–1724. [Google Scholar] [CrossRef] [PubMed]
- Leitão, J.H.; Sousa, S.A.; Ferreira, A.S.; Ramos, C.G.; Silva, I.N.; Moreira, L.M. Pathogenicity, virulence factors, and strategies to fight against Burkholderia cepacia complex pathogens and related species. Appl. Microbiol. Biotechnol. 2010, 87, 31–40. [Google Scholar] [CrossRef] [PubMed]
- McClean, S.; Callaghan, M. Burkholderia cepacia complex: Epithelial cell-pathogen confrontations and potential for therapeutic intervention. J. Med. Microbiol. 2009, 58, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Saldias, M.S.; Valvano, M.A. Interactions of Burkholderia cenocepacia and other Burkholderia cepacia complex bacteria with epithelial and phagocytic cells. Microbiology 2009, 155, 2809–2817. [Google Scholar] [CrossRef] [PubMed]
- Hutchison, M.L.; Bonell, E.C.; Poxton, I.R.; Govan, J.R. Endotoxic activity of lipopolysaccharides isolated from emergent potential cystic fibrosis pathogens. FEMS Immunol. Med. Microbiol. 2000, 27, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Venturi, V.; Friscina, A.; Bertani, I.; Devescovi, G.; Aguilar, C. Quorum sensing in the Burkholderia cepacia complex. Res. Microbiol. 2004, 155, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Whitby, P.W.; VanWagoner, T.M.; Taylor, A.A.; Seale, T.W.; Morton, D.J.; LiPuma, J.J.; Stull, T.L. Identification of an RTX determinant of Burkholderia cenocepacia J2315 by subtractive hybridization. J. Med. Microbiol. 2006, 55, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Engledow, A.S.; Medrano, E.G.; Mahenthiralingam, E.; LiPuma, J.J.; Gonzalez, C.F. Involvement of a plasmid-encoded type IV secretion system in the plant tissue watersoaking phenotype of Burkholderia cenocepacia. J. Bacteriol. 2004, 186, 6015–6024. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sajjan, S.U.; Carmody, L.A.; Gonzalez, C.F.; LiPuma, J.J. A type IV secretion system contributes to intracellular survival and replication of Burkholderia cenocepacia. Infect. Immun. 2008, 76, 5447–5455. [Google Scholar] [CrossRef] [PubMed]
- Caraher, E.; Reynolds, G.; Murphy, P.; McClean, S.; Callaghan, M. Comparison of antibiotic susceptibility of Burkholderia cepacia complex organisms when grown planktonically or as biofilm in vitro. Eur. J. Clin. Microbiol. Infect. Dis. 2007, 26, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Summer, E.J.; Gill, J.J.; Upton, C.; Gonzalez, C.F.; Young, R. Role of phages in the pathogenesis of Burkholderia, or ‘Where are the toxin genes in Burkholderia phages?’. Curr. Opin. Microbiol. 2007, 10, 410–417. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.W.; Suttle, C.A. Viruses and nutrient cycles in the sea. Bioscience 1999, 49, 781–788. [Google Scholar] [CrossRef]
- Clokie, M.R.; Millard, A.D.; Letarov, A.V.; Heaphy, S. Phages in nature. Bacteriophage 2011, 1, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Ubukata, K.; Konno, M.; Fujii, R. Transduction of drug resistance to tetracycline, chloramphenicol, macrolides, lincomycin and clindamycin with phages induced from Streptococcus pyogenes. J. Antibiot. 1975, 28, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Casjens, S. Prophages and bacterial genomics: What have we learned so far? Mol. Microbiol. 2003, 49, 277–300. [Google Scholar] [CrossRef] [PubMed]
- Campbell, A.M. Cryptic prophages. In Escherichia coli and Salmonella: Cellular and Molecular Biology; Neidhardt, F.C., Curtiss, R., III, Ingraham, J.L., Lin, E.C.C., Low, K.B., Magasanik, B., Reznikoff, W.S., Riley, M., Schaechter, M., Umbarger, H.E., Eds.; ASM Press: Washington, DC, USA, 1996; pp. 2041–2046. [Google Scholar]
- Bobay, L.-M.; Touchon, M.; Rocha, E.P.C. Pervasive domestication of defective prophages by bacteria. Proc. Natl. Acad. Sci. USA 2014, 111, 12127–12132. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M.; Prangishvili, D.; Hendrix, R.W.; Bamford, D.H. Genomics of bacterial and archaeal viruses: Dynamics within the prokaryotic virosphere. Microbiol. Mol. Biol. Rev. 2011, 75, 610–635. [Google Scholar] [CrossRef] [PubMed]
- Lynch, K.H.; Liang, Y.; Eberl, L.; Wishart, D.S.; Dennis, J.J. Identification and characterization of ϕH111-1. Bacteriophage 2013, 3, e26649. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Holden, M.T.G.; Seth-Smith, H.M.B.; Crossman, L.C.; Sebaihia, M.; Bentley, S.D.; Cerdeno-Tarraga, A.M.; Thomson, N.R.; Bason, N.; Quail, M.A.; Sharp, S.; et al. The genome of Burkholderia cenocepacia J2315, an epidemic pathogen of cystic fibrosis patients. J. Bacteriol. 2009, 191, 261–277. [Google Scholar] [CrossRef] [PubMed]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed]
- NCBI. NCBI—Complete Virus Genomes Table. Available online: https://www.ncbi.nlm.nih.gov/genomes/GenomesGroup.cgi?taxid=10239&opt=Virus&sort=genome (accessed on 31/05/2017).
- Zhou, Y.; Liang, Y.; Lynch, K.H.; Dennis, J.J.; Wishart, D.S. PHAST: A fast phage search tool. Nucleic Acids Res. 2011, 39, W347–W352. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, K.; Parkhill, J.; Crook, J.; Horsnell, T.; Rice, P.; Rajandream, M.A.; Barrell, B. Artemis: Sequence visualization and annotation. Bioinformatics 2000, 16, 944–945. [Google Scholar] [CrossRef] [PubMed]
- Besemer, J.; Lomsadze, A.; Borodovsky, M. GeneMarkS: A self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001, 29, 2607–2618. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Gish, W.; States, D.J. Identification of protein coding regions by database similarity search. Nat. Genet. 1993, 3, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef] [PubMed]
- Laslett, D.; Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Schattner, P.; Brooks, A.N.; Lowe, T.M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33, W686–W689. [Google Scholar] [CrossRef] [PubMed]
- Gautheret, D.; Lambert, A. Direct RNA motif definition and identification from multiple sequence alignments using secondary structure profiles. J. Mol. Biol. 2001, 313, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Lavigne, R.; Sun, W.D.; Volckaert, G. STORM towards protein function: Systematic tailored ORF-data retrieval and management. Appl. Bioinform. 2003, 2, 177–179. [Google Scholar]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME Suite: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 1992, 8, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Wagner, P.L.; Waldor, M.K. Bacteriophages in bacterial pathogenesis. In The Bacteriophages; Calendar, R., Abedon, S., Eds.; Oxford University Press: New York, NY, USA, 2007; ISBN 0-19-514850-9. [Google Scholar]
- Blaisdell, B.E.; Campbell, A.M.; Karlin, S. Similarities and dissimilarities of phage genomes. Proc. Natl. Acad. Sci. USA 1996, 93, 5854–5859. [Google Scholar] [CrossRef] [PubMed]
- Goudie, A.D.; Lynch, K.H.; Seed, K.D.; Stothard, P.; Shrivastava, S.; Wishart, D.S.; Dennis, J.J. Genomic sequence and activity of KS10, a transposable phage of the Burkholderia cepacia complex. BMC Genom. 2008, 9, 615. [Google Scholar] [CrossRef] [PubMed]
- Summer, E.J.; Gonzalez, C.F.; Carlisle, T.; Mebane, L.M.; Cass, A.M.; Savva, C.G.; LiPuma, J.; Young, R. Burkholderia cenocepacia phage BcepMu and a family of Mu-like phages encoding potential pathogenesis factors. J. Mol. Biol. 2004, 340, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Dortet, L.; Girlich, D.; Virlouvet, A.-L.; Poirel, L.; Nordmann, P.; Iorga, B.I.; Naas, T. Characterization of BRPMBL, the bleomycin resistance protein associated with the carbapenemase NDM. Antimicrob. Agents Chemother. 2017, 61, e02413-16. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.K.; Keithly, M.E.; Sulikowski, G.A.; Armstrong, R.N. Diversity in fosfomycin resistance proteins. Perspect. Sci. 2015, 4, 17–23. [Google Scholar] [CrossRef]
- Reddy, V.S.; Shlykov, M.A.; Castillo, R.; Sun, E.I.; Saier, M.H., Jr. The major facilitator superfamily (MFS) revisited. FEBS J. 2012, 279, 2022–2035. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Mukherjee, M.M.; Varela, M.F. Modulation of bacterial multidrug resistance efflux pumps of the major facilitator superfamily. Int. J. Bacteriol. 2013, 2013, 204141. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Grishin, N.V.; Koonin, E.V. The HicAB cassette, a putative novel, RNA-targeting toxin-antitoxin system in archaea and bacteria. Bioinformatics 2006, 22, 2581–2584. [Google Scholar] [CrossRef] [PubMed]
- Mojica, F.J.M.; Díez-Villaseñor, C.; García-Martínez, J.; Almendros, C. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology 2009, 155, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Zegans, M.E.; Wagner, J.C.; Cady, K.C.; Murphy, D.M.; Hammond, J.H.; O’Toole, G.A. Interaction between bacteriophage DMS3 and host CRISPR region inhibits group behaviors of Pseudomonas aeruginosa. J. Bacteriol. 2009, 191, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Goldfarb, T.; Sberro, H.; Weinstock, E.; Cohen, O.; Doron, S.; Charpak-Amikam, Y.; Afik, S.; Ofir, G.; Sorek, R. BREX is a novel phage resistance system widespread in microbial genomes. EMBO J. 2015, 34, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Grissa, I.; Vergnaud, G.; Pourcel, C. CRISPRFinder: A web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007, 35, W52–W57. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, M.; Kurokawa, K.; Hayashi, T. Diversification of Escherichia coli genomes: Are bacteriophages the major contributors? Trends Microbiol. 2001, 9, 481–485. [Google Scholar] [CrossRef]
- Beres, S.B.; Sylva, G.L.; Barbian, K.D.; Lei, B.; Hoff, J.S.; Mammarella, N.D.; Liu, M.-Y.; Smoot, J.C.; Porcella, S.F.; Parkins, L.D.; et al. Genome sequence of a serotype M3 strain of group A Streptococcus: Phage-encoded toxins, the high-virulence phenotype, and clone emergence. Proc. Natl. Acad. Sci. USA 2002, 99, 10078–10083. [Google Scholar] [CrossRef] [PubMed]
- Bodilis, J.; Denet, E.; Brothier, E.; Graindorge, A.; Favre-Bonté, S.; Nazaret, S. Comparative genomics of environmental and clinical Burkholderia cenocepacia strains closely related to the highly transmissible epidemic ET12 lineage. Front. Microbiol. 2018, 9, 383. [Google Scholar] [CrossRef] [PubMed]
- Ronning, C.M.; Losada, L.; Brinkac, L.; Inman, J.; Ulrich, R.L.; Schell, M.; Nierman, W.C.; Deshazer, D. Genetic and phenotypic diversity in Burkholderia: Contributions by prophage and phage-like elements. BMC Microbiol. 2010, 10, 202. [Google Scholar] [CrossRef] [PubMed]
- DeShazer, D. Genomic diversity of Burkholderia pseudomallei clinical isolates: Subtractive hybridization reveals a Burkholderia mallei-specific prophage in B. pseudomallei 1026b. J. Bacteriol. 2004, 186, 3938–3950. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Xu, B.; Gao, Z.; Zhou, K.; Liu, P.; Dong, Y.; Zhang, J.; Liu, Q. HicAB toxin–antitoxin complex from Escherichia coli: Expression and crystallization. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2017, 73, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Butt, A.; Müller, C.; Harmer, N.; Titball, R.W. Identification of type II toxin-antitoxin modules in Burkholderia pseudomallei. FEMS Microbiol. Lett. 2013, 338, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Butt, A.; Higman, V.A.; Williams, C.; Crump, M.P.; Hemsley, C.M.; Harmer, N.; Titball, R.W. The HicA toxin from Burkholderia pseudomallei has a role in persister cell formation. Biochem. J. 2014, 459, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Cooper, V.S.; Carlson, W.A.; LiPuma, J.J. Susceptibility of Caenorhabditis elegans to Burkholderia infection depends on prior diet and secreted bacterial attractants. PLoS ONE 2009, 4, e7961. [Google Scholar] [CrossRef] [PubMed]
- Roszniowski, B.; Latka, A.; Maciejewska, B.; Vandenheuvel, D.; Olszak, T.; Briers, Y.; Holt, G.S.; Valvano, M.A.; Lavigne, R.; Smith, D.L.; et al. The temperate Burkholderia phage AP3 of the Peduovirinae shows efficient antimicrobial activity against B. cenocepacia of the IIIA lineage. Appl. Microbiol. Biotechnol. 2017, 101, 1203–1216. [Google Scholar] [CrossRef] [PubMed]
- Holden, M.T.G.; Titball, R.W.; Peacock, S.J.; Cerdeño-Tárraga, A.M.; Atkins, T.; Crossman, L.C.; Pitt, T.; Churcher, C.; Mungall, K.; Bentley, S.D.; et al. Genomic plasticity of the causative agent of melioidosis, Burkholderia pseudomallei. Proc. Natl. Acad. Sci. USA 2004, 101, 14240–14245. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Strain | Origin | Chromosome | A/N | Genome Size (bp) |
|---|---|---|---|---|---|
| 1 | B. cenocepacia J2315 | CF | 1 | NC_011000.1 | 3,870,082 |
| 2 | NC_011001.1 | 3,217,062 | |||
| 3 | NC_011002.1 | 875,977 | |||
| 2 | B. cenocepacia H111 | CF | 1 | NZ_HG938370.1 | 3,572,953 |
| 2 | NZ_HG938371.1 | 3,102,677 | |||
| 3 | NZ_HG938372.1 | 1,039,263 | |||
| 3 | B. cenocepacia DWS 37E-2 | Soil | 1 | NZ_CP007781.1 | 3,241,886 |
| 2 | NZ_CP007780.1 | 2,375,865 | |||
| 3 | NZ_CP007779.1 | 994,670 | |||
| 4 | B. cenocepacia FL-5-3-30-S1-D7 | Soil | 1 | CP013397.1 | 3,461,321 |
| 2 | CP013396.1 | 2,869,430 | |||
| 5 | B. cenocepacia 895 | Sepsis (neonatal)/Cord blood | 1 | NZ_CP015036.1 | 7,459,003 |
| 2 | NZ_CP015037.1 | 1,072,666 | |||
| 6 | B. cenocepacia ST32 | CF | 1 | NZ_CP011917.1 | 3,822,749 |
| 2 | NZ_CP011918.1 | 3,086,109 | |||
| 3 | NZ_CP011919.1 | 989,585 | |||
| 7 | B. cenocepacia 842 | Nasal inflammation/non-CF | 1 | NZ_CP015033.1 | 3,526,250 |
| 2 | NZ_CP015034.1 | 3,107,451 | |||
| 3 | NZ_CP015035.1 | 1,271,875 | |||
| 8 | B. cenocepacia MSMB384WGS | Water | 1 | NZ_CP013450.1 | 3,588,848 |
| 2 | NZ_CP013452.1 | 3,069,864 | |||
| 3 | NZ_CP013451.1 | 1,121,886 | |||
| 9 | B. cenocepacia HI2424 | Soil | 1 | NC_008542.1 | 3,483,902 |
| 2 | NC_008543.1 | 2,998,664 | |||
| 3 | NC_008544.1 | 1,055,417 | |||
| 10 | B. cenocepacia CR318 | Plant root | 1 | NZ_CP017238.1 | 3,511,146 |
| 2 | NZ_CP017239.1 | 3,097,552 | |||
| 3 | NZ_CP017240.1 | 1,056,196 | |||
| 11 | B. cenocepacia AU 1054 | CF | 1 | NC_008060.1 | 3,294,563 |
| 2 | NC_008061.1 | 2,788,459 | |||
| 3 | NC_008062.1 | 1,196,094 | |||
| 12 | B. cenocepacia MC0-3 | Soil | 1 | NC_010508.1 | 3,532,883 |
| 2 | NC_010515.1 | 3,213,911 | |||
| 3 | NC_010512.1 | 1,224,595 | |||
| 13 | B. cenocepacia DDS 22E-1 | Aerosol sample | 1 | NZ_CP007783.1 | 3,668,832 |
| 2 | NZ_CP007784.1 | 3,209,624 | |||
| 3 | NZ_CP007782.1 | 1,166,794 | |||
| 14 | B. cenocepacia VC7848 | CF | 1 | NZ_CP019668.1 | 7,499,459 |
| 15 | B. cenocepacia VC12308 | CF | 1 | NZ_CP019674.1 | 3,668,000 |
| 2 | NZ_CP019672.1 | 2,984,720 | |||
| 3 | NZ_CP019673.1 | 964,521 | |||
| 16 | B. cenocepacia VC12802 | CF | 1 | NZ_CP019670.1 | 6,339,862 |
| 2 | NZ_CP019669.1 | 1,055,047 |
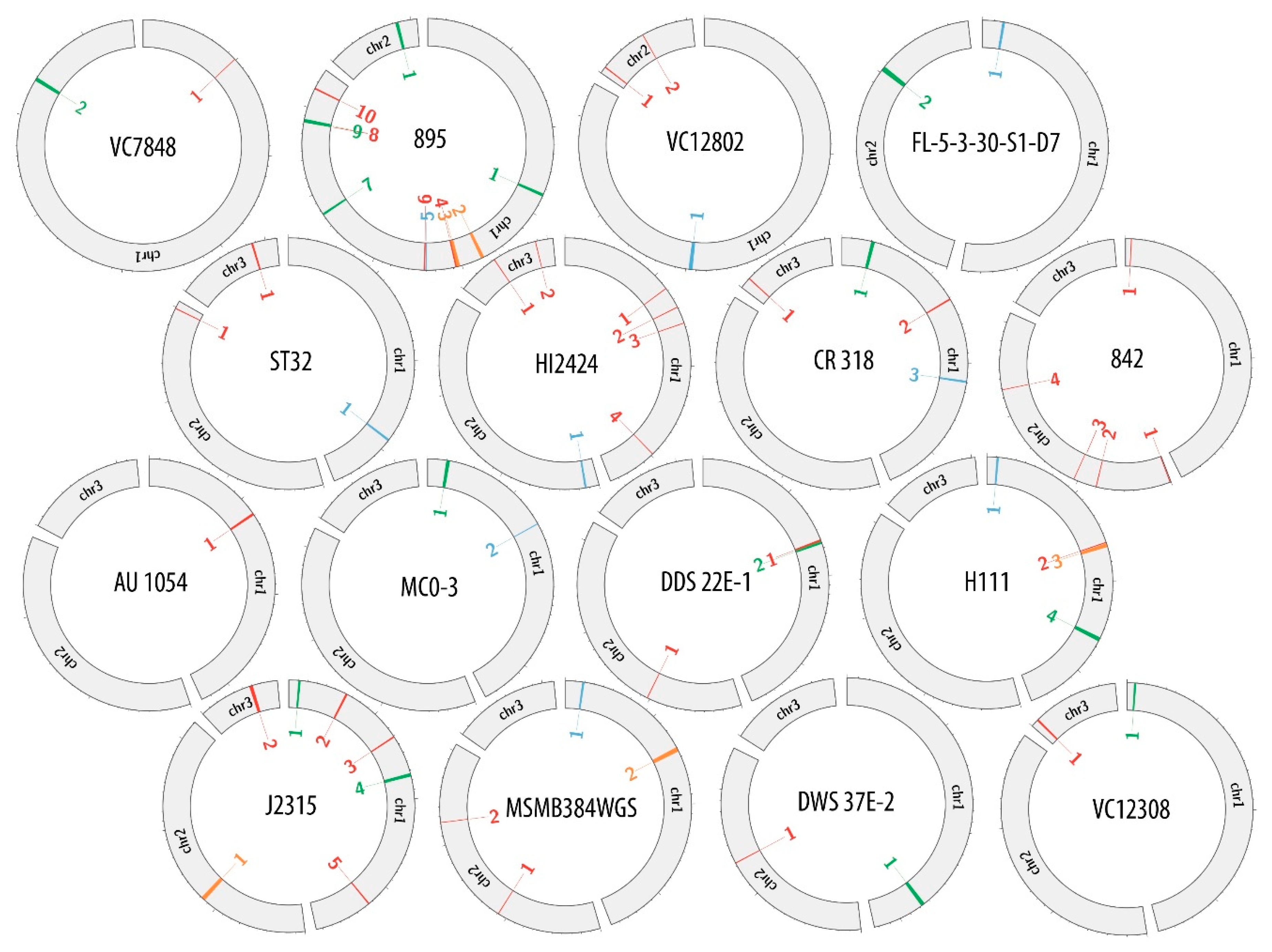
| Host Name | Chromosome | Region Name | Phage Genome Size (bp) | Status | Location in Host Genome | |
|---|---|---|---|---|---|---|
| Start | End | |||||
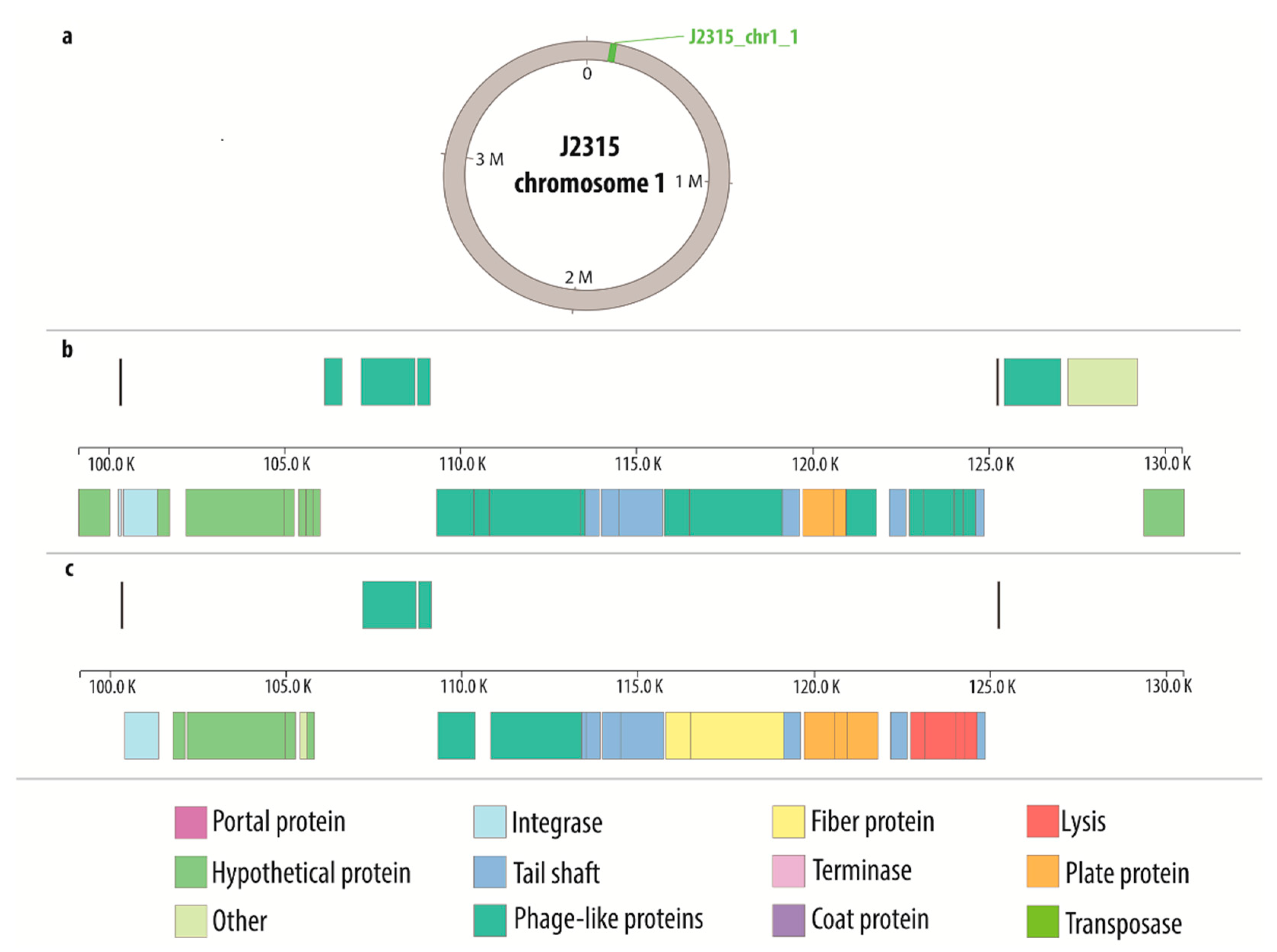
| J2315 | 1 | J2315_chr1_1 | 24,997 | intact | 100,299 | 125,296 |
| J2315_chr1_2 | 22,458 | incomplete | 620,851 | 643,309 | ||
| J2315_chr1_3 | 16,209 | incomplete | 1,301,322 | 1,317,531 | ||
| J2315_chr1_4 (KS10) | 37,369 | intact | 1,729,077 | 1,766,446 | ||
| J2315_chr1_5 | 15,015 | incomplete | 3,241,582 | 3,256,597 | ||
| 2 | J2315_chr2_1 | 46,822 | artifact region | 1,140,168 | 1,186,990 | |
| 3 | J2315_chr3_1 (Bcep-Mu) | 37,581 | intact | 572,009 | 609,590 | |
| H111 | 1 | H111_chr1_1 | 22,907 | questionable | 100,107 | 123,014 |
| H111_chr1_2 | 10,399 | incomplete | 1,593,686 | 1,604,085 | ||
| H111_chr1_3 | 38,387 | artifact region | 1,609,303 | 1,647,690 | ||
| H111_chr1_4 (ϕH111-1) | 43,024 | intact | 2,595,517 | 2,638,541 | ||
| 2 | - | - | - | - | - | |
| 3 | - | - | - | - | - | |
| DWS 37E-2 | 1 | DWS 37E-2_chr1 | 36,859 | intact | 2,747,954 | 2,784,813 |
| 2 | DWS 37E-2_chr2 | 8671 | incomplete | 1,323,368 | 1,332,039 | |
| 3 | - | - | - | - | - | |
| FL-5-3-30-S1-D7 | 1 | FL-5-3-30-S1-D7_chr1_1 | 23,101 | questionable | 167,573 | 190,674 |
| FL-5-3-30-S1-D7_chr1_2 | 44,062 | intact | 2,017,976 | 2,062,038 | ||
| 2 | - | - | - | - | - | |
| 895 | 1 | 895_chr1_1 | 34,705 | intact | 2,755,051 | 2,789,756 |
| 895_chr1_2 | 38,627 | artifact region | 3,737,465 | 3,776,092 | ||
| 895_chr1_3 | 43,300 | artifact region | 4,023,532 | 4,066,832 | ||
| 895_chr1_4 | 14,687 | incomplete | 4,061,645 | 4,076,332 | ||
| 895_chr1_5 | 14,593 | questionable | 4,398,712 | 4,413,305 | ||
| 895_chr1_6 | 14,946 | incomplete | 4,416,973 | 4,431,919 | ||
| 895_chr1_7 | 26,430 | intact | 5,741,502 | 5,767,932 | ||
| 895_chr1_8 | 29,290 | incomplete | 6,823,839 | 6,833,750 | ||
| 895_chr1_9 | 40,988 | intact | 6,823,839 | 6,864,827 | ||
| 895_chr1_10 | 18,325 | incomplete | 7,212,238 | 7,230,563 | ||
| 2 | 895_chr2_1 | 37,652 | intact | 826,395 | 864,047 | |
| ST32 | 1 | ST32_chr1 | 23,895 | questionable | 2,869,763 | 2,893,658 |
| 2 | ST32_chr2 | 10,694 | incomplete | 2,980,754 | 2,991,448 | |
| 3 | ST32_chr3 | 20,608 | incomplete | 850,166 | 870,774 | |
| 842 | 1 | 842_chr1_1 | 10,037 | incomplete | 57,007 | 67,044 |
| 2 | 842_chr2_1 | 8621 | incomplete | 6423 | 15,044 | |
| 842_chr2_2 | 9739 | incomplete | 791,624 | 801,363 | ||
| 842_chr2_3 | 7465 | incomplete | 1,038,705 | 1,046,170 | ||
| 842_chr2_4 | 7448 | incomplete | 2,281,847 | 2,289,295 | ||
| 3 | - | - | - | - | - | |
| MSMB384WGS | 1 | MSMB384WGS_chr1_1 | 24,156 | questionable | 172,483 | 195,500 |
| MSMB384WGS_chr1_2 | 48,964 | artifact region | 1,394,910 | 1,443,874 | ||
| 2 | MSMB384WGS_chr2_1 | 8810 | incomplete | 1,087,426 | 1,096,236 | |
| MSMB384WGS_chr2_2 | 7306 | incomplete | 2,234,278 | 2,241,584 | ||
| 3 | - | - | - | - | - | |
| HI2424 | 1 | HI2424_chr1_1 | 8280 | incomplete | 1,165,247 | 1,173,527 |
| HI2424_chr1_2 | 9114 | incomplete | 1,383,513 | 1,392,627 | ||
| HI2424_chr1_3 | 8107 | incomplete | 1,556,642 | 1,564,749 | ||
| HI2424_chr1_4 | 7746 | incomplete | 2,976,934 | 2,984,680 | ||
| 2 | HI2424_chr2_1 | 20,628 | questionable | 126,942 | 147,570 | |
| 3 | HI2424_chr3_1 | 8809 | incomplete | 421,282 | 430,091 | |
| HI2424_chr3_2 | 8134 | incomplete | 872,989 | 881,123 | ||
| CR 318 | 1 | CR 318_chr1_1 | 38,476 | intact | 300,609 | 339,084 |
| CR 318_chr1_2 | 16,791 | incomplete | 1,309,949 | 1,326,739 | ||
| CR 318_chr1_3 | 22,450 | questionable | 2,180,663 | 2,203,112 | ||
| 2 | - | - | - | - | - | |
| 3 | CR 318_chr3_1 | 11,919 | incomplete | 112734 | 124652 | |
| CR 318_chr3_2 | 7542 | incomplete | 570,308 | 577,849 | ||
| AU 1054 | 1 | AU 1054_chr1_1 | 24,119 | incomplete | 1,172,051 | 1,196,169 |
| 2 | - | - | - | - | - | |
| 3 | - | - | - | - | - | |
| MC0-3 | 1 | MC0-3_chr1_1 | 38,872 | intact | 198,439 | 237,310 |
| MC0-3_chr1_2 | 10,797 | questionable | 1,405,878 | 1,416,674 | ||
| 2 | - | - | - | - | - | |
| 3 | - | - | - | - | - | |
| DDS 22E-1 | 1 | DDS 22E-1_chr1_1 | 24,368 | incomplete | 1,606,100 | 1,630,467 |
| DDS 22E-1_chr1_2 | 31,311 | intact | 1,625,635 | 1,656,945 | ||
| 2 | DDS 22E-1_chr2_1 | 9602 | incomplete | 1,022,571 | 1,032,172 | |
| 3 | - | - | - | - | - | |
| VC7848 | 1 | VC7848_chr1_1 | 7080 | incomplete | 983,606 | 990,685 |
| VC7848_chr1_2 | 38,294 | intact | 6,353,033 | 6,391,326 | ||
| VC12308 | 1 | VC12308_chr1_1 | 22,704 | intact | 70,339 | 93,042 |
| 2 | - | - | - | - | - | |
| 3 | VC12308_chr3_1 | 20,452 | incomplete | 73,079 | 93,531 | |
| VC12802 | 1 | VC12802_chr1_1 | 39,072 | questionable | 3,934,787 | 3,973,858 |
| 2 | VC12802_chr2_1 | 11,921 | incomplete | 63,441 | 75,362 | |
| VC12802_chr2_2 | 7545 | incomplete | 546,849 | 554,394 | ||
| Host | Chromosome | Chromosome Size (bp) | Phage Prevalence in Chromosome (%) | Potential Phage Regions in Chromosome | Total Phage Prevalence in the Host Genome (%) |
|---|---|---|---|---|---|
| J2315 | 1 | 3,870,082 | 2.99 | 5 | 2.51 |
| 2 | 3,217,062 | 1.45 | 1 | ||
| 3 | 875,977 | 4.29 | 1 | ||
| H111 | 1 | 3,572,953 | 3.24 | 4 | 1.50 |
| 2 | 3,102,677 | - | 0 | ||
| 3 | 1,039,263 | - | 0 | ||
| DWS 37E-2 | 1 | 3,241,886 | 0.71 | 1 | 0.81 |
| 2 | 2,375,865 | 0.36 | 1 | ||
| 3 | 994,670 | - | 0 | ||
| FL-5-3-30-S1-D7 | 1 | 3,461,321 | 1.94 | 2 | 1.06 |
| 2 | 2,869,430 | - | 0 | ||
| 895 | 1 | 7,459,003 | 3.70 | 10 | 3.67 |
| 2 | 1,072,666 | 3.51 | 1 | ||
| ST32 | 1 | 3,822,749 | 0.67 | 1 | 0.69 |
| 2 | 3,086,109 | 0.34 | 1 | ||
| 3 | 989,585 | 2.08 | 1 | ||
| 842 | 1 | 3,526,250 | 0.28 | 1 | 0.54 |
| 2 | 3,107,451 | 1.07 | 4 | ||
| 3 | 1,271,875 | - | 0 | ||
| MSMB384WGS | 1 | 3,588,848 | 2.04 | 2 | 1.15 |
| 2 | 3,069,864 | 0.52 | 2 | ||
| 3 | 1,121,886 | - | 0 | ||
| HI2424 | 1 | 3,483,902 | 0.95 | 4 | 0.93 |
| 2 | 2,998,664 | 0.69 | 1 | ||
| 3 | 1,055,417 | 1.60 | 2 | ||
| CR 318 | 1 | 3,511,146 | 2.21 | 3 | 1.27 |
| 2 | 3,097,552 | - | 0 | ||
| 3 | 1,056,196 | 1.84 | 3 | ||
| AU 1054 | 1 | 3,294,563 | 0.73 | 1 | 0.30 |
| 2 | 2,788,459 | - | 0 | ||
| 3 | 1,196,094 | - | 0 | ||
| MC0-3 | 1 | 3,532,883 | 1.40 | 2 | 0.62 |
| 2 | 3,213,911 | - | 0 | ||
| 3 | 1,224,595 | - | 0 | ||
| DDS 22E-1 | 1 | 3,668,832 | 1.52 | 2 | 0.80 |
| 2 | 3,209,624 | 0.30 | 1 | ||
| 3 | 1,166,794 | - | 0 | ||
| VC7848 | 1 | 7,499,459 | 0.60 | 2 | 0.60 |
| VC12308 | 1 | 3,668,000 | 0.62 | 1 | 0.56 |
| 2 | 2,984,720 | - | 0 | ||
| 3 | 964,521 | 2.12 | 1 | ||
| VC12802 | 1 | 6,339,862 | 0.61 | 1 | 0.79 |
| 2 | 1,055,047 | 1.84 | 2 |
| Region | Start | End | Product | Virulence Effect | Accession |
|---|---|---|---|---|---|
| J2315_chr2_1 | 12,317 | 12,700 | Fic | TA system compound | WP_006488862.1 |
| 842_chr2_1 | 6642 | 7541 | class A β-lactamase | drug resistance | WP_034202207.1 |
| 842_chr2_2 | 2065 | 3390 | MFS transporter | drug resistance/virulence | WP_006495119.1 |
| MSMB384WGS_chr2_1 | 1 | 1500 | MFS transporter | drug resistance/virulence | WP_060268128.1 |
| MSMB384WGS_chr2_1 | 2697 | 3080 | VOC family protein | drug resistance/virulence | WP_060268132.1 |
| HI2424_chr1_2 | 1 | 1311 | MFS transporter * (a) | drug resistance/virulence | WP_011545048.1 |
| HI2424_chr1_2 | 3764 | 4969 | MFS transporter * (b) | drug resistance/virulence | WP_011545051.1 |
| HI2424_chr1_3 | 1039 | 2301 | MFS transporter | drug resistance/virulence | WP_011545193.1 |
| HI2424_chr1_4 | 2829 | 4004 | MFS transporter | drug resistance/virulence | WP_011694391.1 |
| HI2424_chr2_1 | 3597 | 4892 | MFS transporter | drug resistance/virulence | WP_011548498.1 |
| HI2424_chr3_2 | 3269 | 4666 | MFS transporter | drug resistance/virulence | WP_011695034.1 |
| CR 318_chr3_2 | 1499 | 2896 | MFS transporter | drug resistance/virulence | WP_011695034.1 |
| DDS 22E-1_chr1_2 | 9157 | 9552 | HicB | TA system compound | AJT61392.1 |
| DDS 22E-1_chr1_2 | 9577 | 9759 | HicA | TA system compound | ADF59182.1 |
| VC7848_chr1_1 | 5764 | 7080 | MFS transporter | drug resistance/virulence | WP_011548265.1 |
| VC12802_chr2_2 | 1499 | 2896 | MFS transporter | drug resistance/virulence | WP_077217595.1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roszniowski, B.; McClean, S.; Drulis-Kawa, Z. Burkholderia cenocepacia Prophages—Prevalence, Chromosome Location and Major Genes Involved. Viruses 2018, 10, 297. https://doi.org/10.3390/v10060297
Roszniowski B, McClean S, Drulis-Kawa Z. Burkholderia cenocepacia Prophages—Prevalence, Chromosome Location and Major Genes Involved. Viruses. 2018; 10(6):297. https://doi.org/10.3390/v10060297
Chicago/Turabian StyleRoszniowski, Bartosz, Siobhán McClean, and Zuzanna Drulis-Kawa. 2018. "Burkholderia cenocepacia Prophages—Prevalence, Chromosome Location and Major Genes Involved" Viruses 10, no. 6: 297. https://doi.org/10.3390/v10060297
APA StyleRoszniowski, B., McClean, S., & Drulis-Kawa, Z. (2018). Burkholderia cenocepacia Prophages—Prevalence, Chromosome Location and Major Genes Involved. Viruses, 10(6), 297. https://doi.org/10.3390/v10060297