Live-Cell Imaging of Early Steps of Single HIV-1 Infection


Abstract
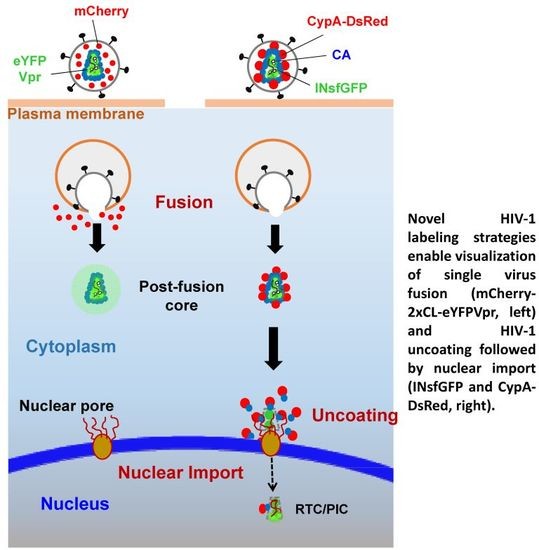
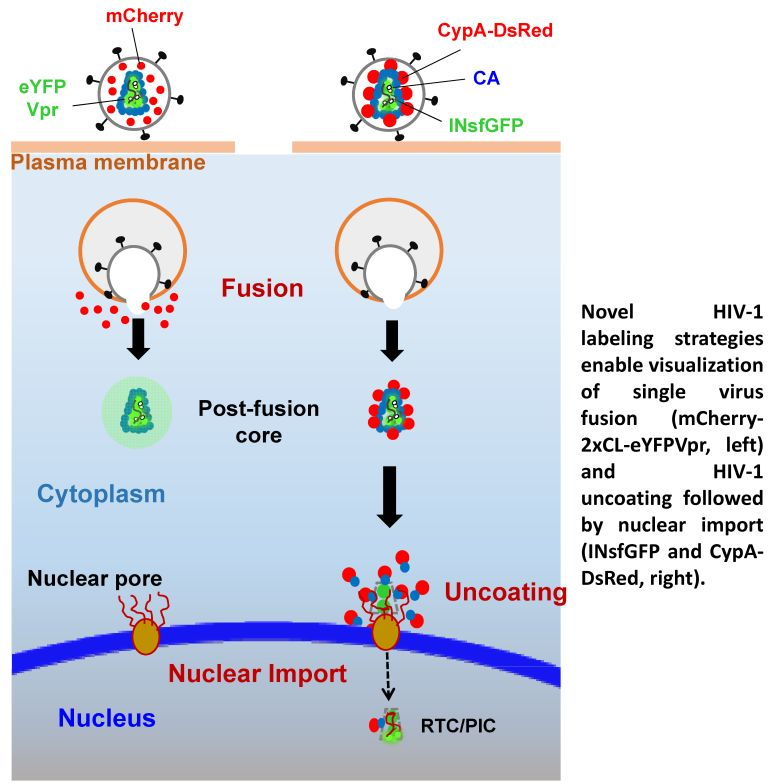{kind=link}
{kind=link}
{kind=link}
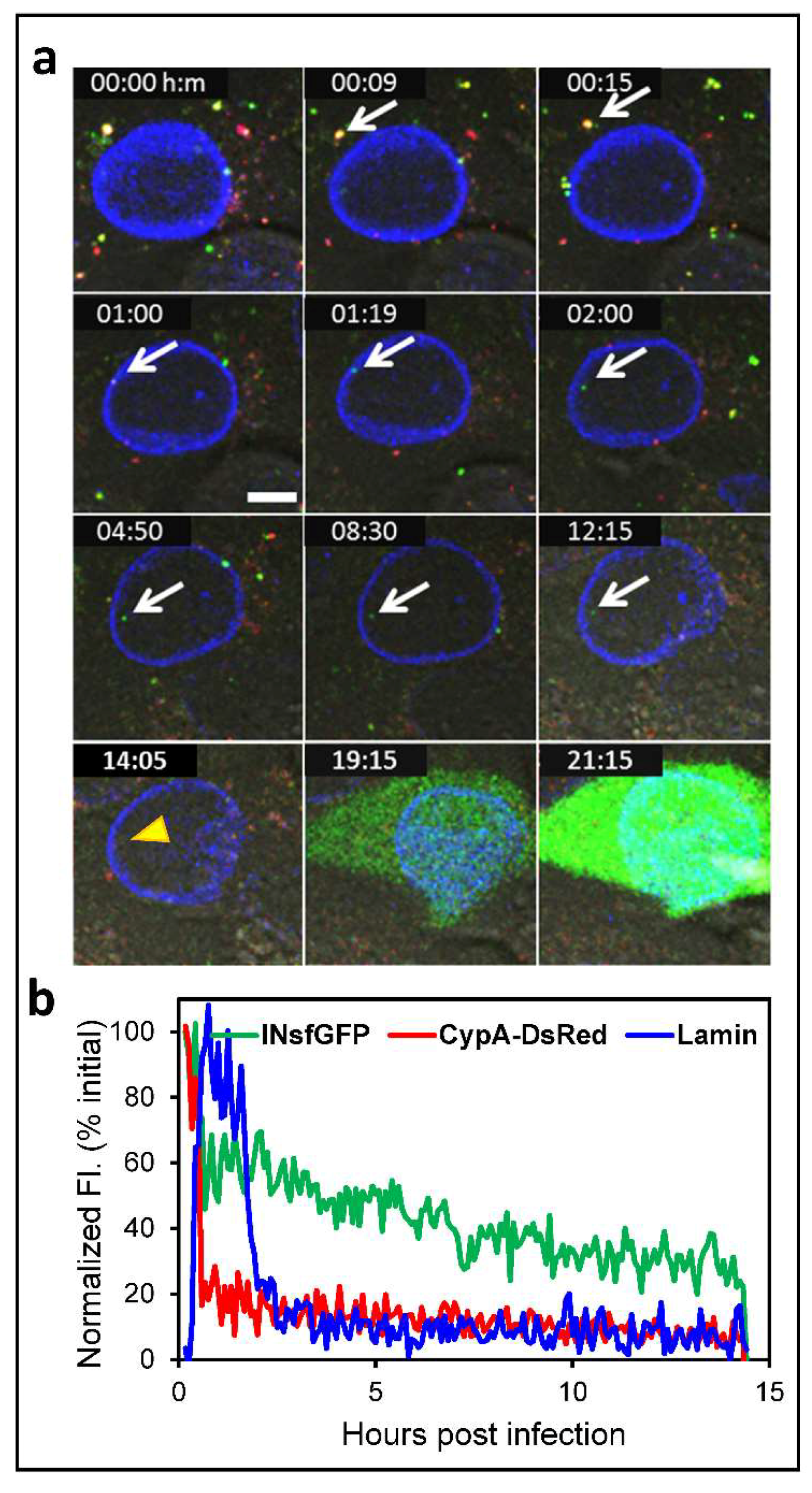{kind=link}
1. Introduction
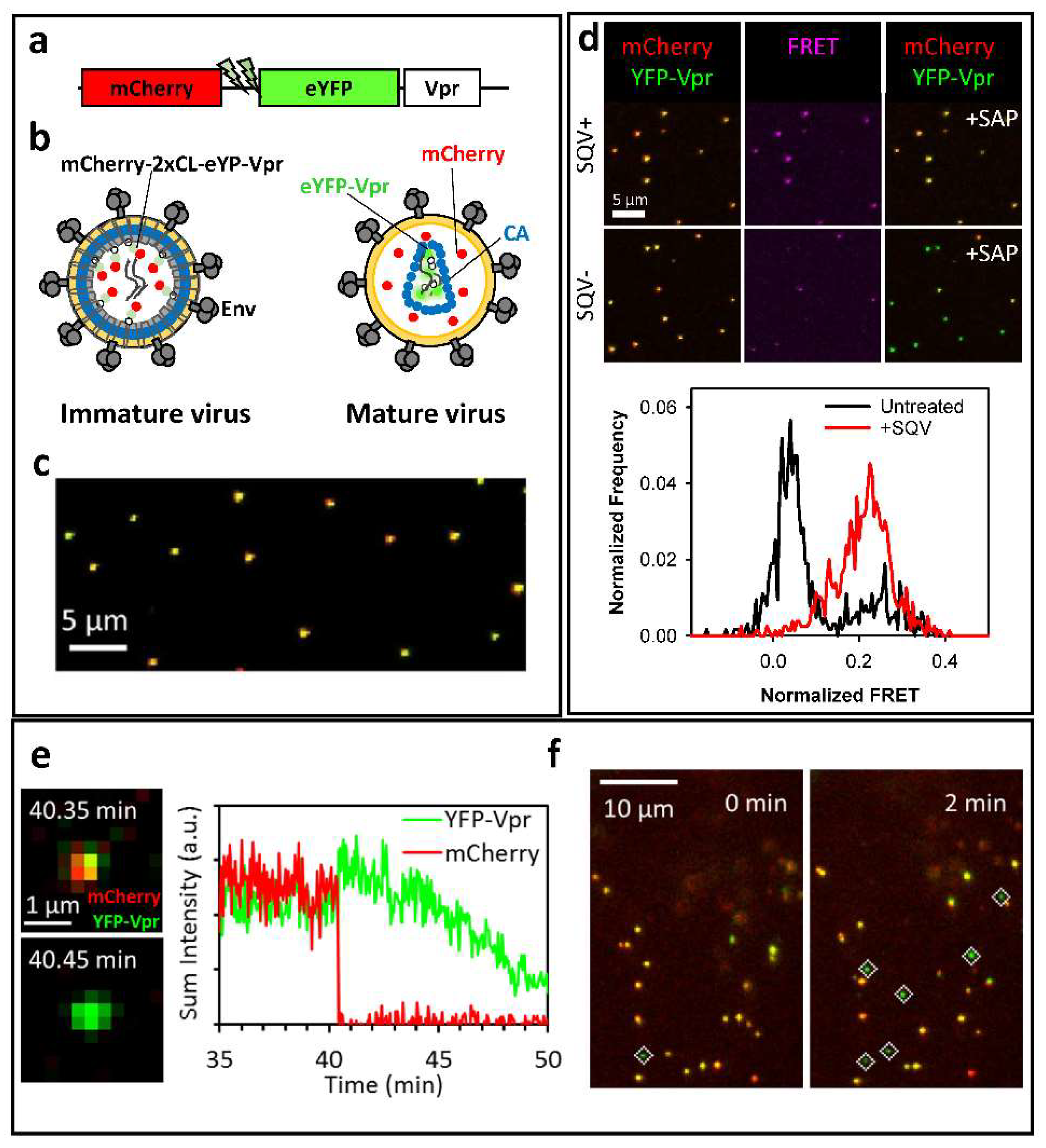
2. A Bi-Functional Reporter of Single Virus Protease Activity and Virus–Cell Fusion
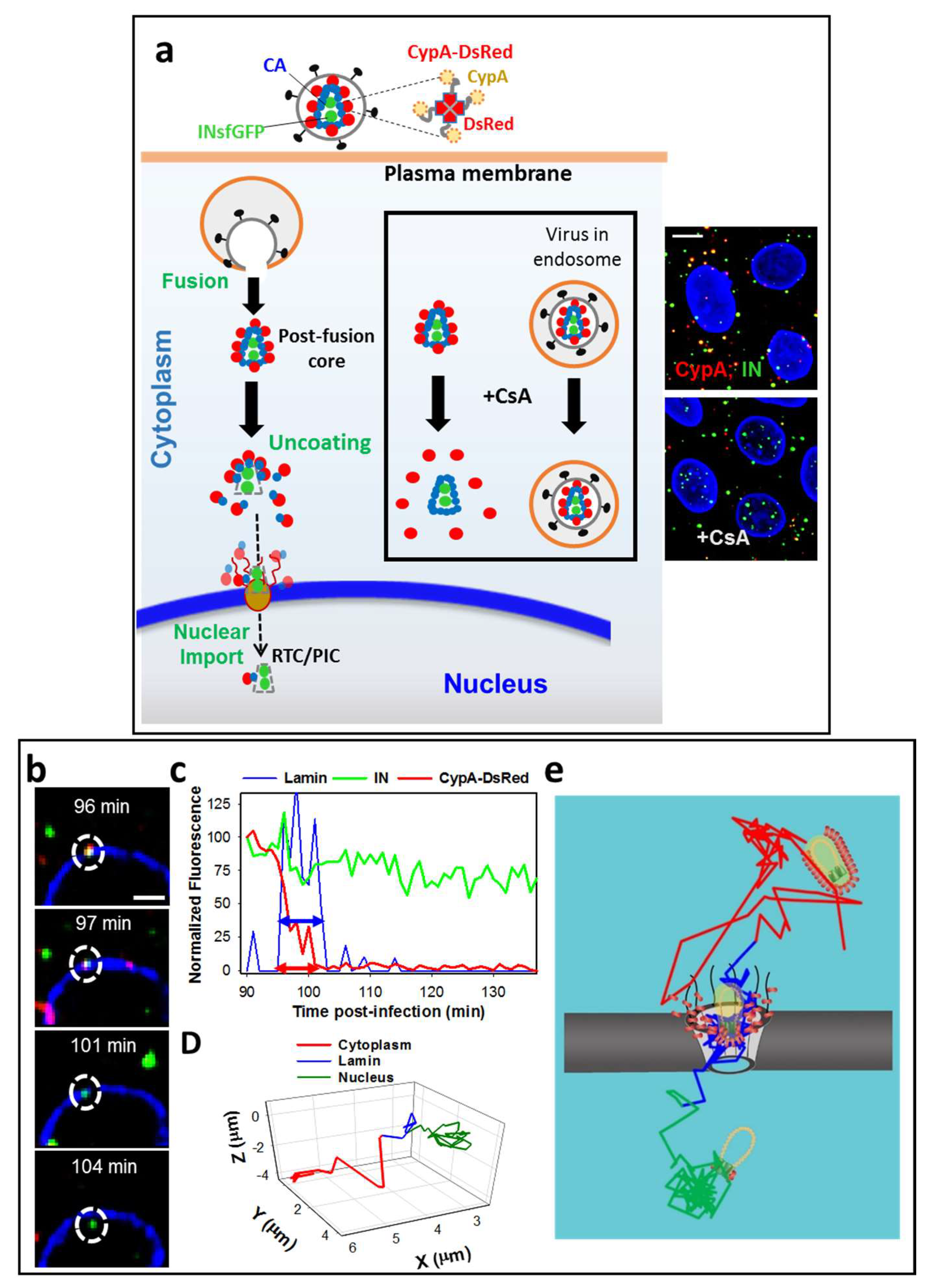
3. Labeling CA to Study Single HIV-1 Uncoating
4. Single HIV-1 Uncoating In Vitro
5. Time-Resolved Imaging of Single HIV-1 Uncoating in the Cytoplasm
6. Docking at the Nuclear Pore
7. Uncoating at the Nuclear Membrane Is a Prerequisite for HIV-1 Nuclear Entry
8. HIV-1 Nuclear Import Occurs after a Pronounced Lag Following the Loss of CypA-DsRed/CA at the NE
9. Visualization of Functionally Relevant Single HIV-1 Entry/Uncoating Events
10. Summary and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Briggs, J.A.; Krausslich, H.G. The molecular architecture of HIV. J. Mol. Biol. 2011, 410, 491–500. [Google Scholar] [CrossRef] [PubMed]
- McDonald, D.; Vodicka, M.A.; Lucero, G.; Svitkina, T.M.; Borisy, G.G.; Emerman, M.; Hope, T.J. Visualization of the intracellular behavior of HIV in living cells. J. Cell Biol. 2002, 159, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Dharan, A.; Talley, S.; Tripathi, A.; Mamede, J.I.; Majetschak, M.; Hope, T.J.; Campbell, E.M. KIF5B and Nup358 Cooperatively Mediate the Nuclear Import of HIV-1 during Infection. PLoS Pathog. 2016, 12. [Google Scholar] [CrossRef] [PubMed]
- Lukic, Z.; Dharan, A.; Fricke, T.; Diaz-Griffero, F.; Campbell, E.M. HIV-1 uncoating is facilitated by dynein and kinesin 1. J. Virol. 2014, 88, 13613–13625. [Google Scholar] [CrossRef] [PubMed]
- Malikov, V.; da Silva, E.S.; Jovasevic, V.; Bennett, G.; de Souza Aranha Vieira, D.A.; Schulte, B.; Diaz-Griffero, F.; Walsh, D.; Naghavi, M.H. HIV-1 capsids bind and exploit the kinesin-1 adaptor FEZ1 for inward movement to the nucleus. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Pawlica, P.; Berthoux, L. Cytoplasmic dynein promotes HIV-1 uncoating. Viruses 2014, 6, 4195–4211. [Google Scholar] [CrossRef] [PubMed]
- Arhel, N. Revisiting HIV-1 uncoating. Retrovirology 2010, 7. [Google Scholar] [CrossRef] [PubMed]
- Ambrose, Z.; Aiken, C. HIV-1 uncoating: Connection to nuclear entry and regulation by host proteins. Virology 2014, 454–455, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Campbell, E.M.; Hope, T.J. HIV-1 capsid: The multifaceted key player in HIV-1 infection. Nat. Rev. Microbiol. 2015, 13, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Schroder, A.R.; Shinn, P.; Chen, H.; Berry, C.; Ecker, J.R.; Bushman, F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell 2002, 110, 521–529. [Google Scholar] [CrossRef]
- Craigie, R.; Bushman, F.D. HIV DNA integration. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef] [PubMed]
- Engelman, A.N.; Singh, P.K. Cellular and molecular mechanisms of HIV-1 integration targeting. Cell. Mol. Life Sci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Campbell, E.M.; Hope, T.J. Live cell imaging of the HIV-1 life cycle. Trends Microbiol. 2008, 16, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Baumgartel, V.; Muller, B.; Lamb, D.C. Quantitative live-cell imaging of human immunodeficiency virus (HIV-1) assembly. Viruses 2012, 4, 777–799. [Google Scholar] [CrossRef] [PubMed]
- Chojnacki, J.; Muller, B. Investigation of HIV-1 assembly and release using modern fluorescence imaging techniques. Traffic 2013, 14, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Hulme, A.E.; Hope, T.J. Live cell imaging of retroviral entry. Annu. Rev. Virol. 2014, 1, 501–515. [Google Scholar] [CrossRef] [PubMed]
- Mamede, J.I.; Hope, T.J. Detection and tracking of dual-labeled HIV particles using wide-field live cell imaging to follow viral core integrity. Methods Mol. Biol. 2016, 1354, 49–59. [Google Scholar] [PubMed]
- De la Vega, M.; Marin, M.; Kondo, N.; Miyauchi, K.; Kim, Y.; Epand, R.F.; Epand, R.M.; Melikyan, G.B. Inhibition of HIV-1 endocytosis allows lipid mixing at the plasma membrane, but not complete fusion. Retrovirology 2011, 8. [Google Scholar] [CrossRef] [PubMed]
- Miyauchi, K.; Kim, Y.; Latinovic, O.; Morozov, V.; Melikyan, G.B. HIV enters cells via endocytosis and dynamin-dependent fusion with endosomes. Cell 2009, 137, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Oum, Y.H.; Desai, T.M.; Marin, M.; Melikyan, G.B. Click labeling of unnatural sugars metabolically incorporated into viral envelope glycoproteins enables visualization of single particle fusion. J. Virol. Methods 2016, 233, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Padilla-Parra, S.; Marin, M.; Gahlaut, N.; Suter, R.; Kondo, N.; Melikyan, G.B. Fusion of mature HIV-1 particles leads to complete release of a gag-gfp-based content marker and raises the intraviral pH. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Sood, C.; Francis, A.C.; Desai, T.M.; Melikyan, G.B. An improved labeling strategy enables automated detection of single-virus fusion and assessment of HIV-1 protease activity in single virions. J. Biol. Chem. 2017, 292, 20196–20207. [Google Scholar] [CrossRef] [PubMed]
- Sood, C.; Marin, M.; Mason, C.S.; Melikyan, G.B. Visualization of content release from cell surface-attached single HIV-1 particles carrying an extra-viral fluorescent ph-sensor. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Desai, T.M.; Marin, M.; Sood, C.; Shi, J.; Nawaz, F.; Aiken, C.; Melikyan, G.B. Fluorescent protein-tagged Vpr dissociates from HIV-1 core after viral fusion and rapidly enters the cell nucleus. Retrovirology 2015, 12. [Google Scholar] [CrossRef] [PubMed]
- Dale, B.M.; McNerney, G.P.; Thompson, D.L.; Hubner, W.; de Los Reyes, K.; Chuang, F.Y.; Huser, T.; Chen, B.K. Cell-to-cell transfer of HIV-1 via virological synapses leads to endosomal virion maturation that activates viral membrane fusion. Cell Host Microbe 2011, 10, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.M.; Alvarez, L.A.; Nolan, R.; Ferriz, M.; Sainz Urruela, R.; Massana-Munoz, X.; Novak-Kotzer, H.; Dustin, M.L.; Padilla-Parra, S. Dynamin-2 stabilizes the HIV-1 fusion pore with a low oligomeric state. Cell Rep. 2017, 18, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Francis, A.C.; Marin, M.; Shi, J.; Aiken, C.; Melikyan, G.B. Time-resolved imaging of single HIV-1 uncoating in vitro and in living cells. PLoS Pathog. 2016, 12. [Google Scholar] [CrossRef] [PubMed]
- Francis, A.C.; Melikyan, G.B. Single HIV-1 imaging reveals progression of infection through CA-dependent steps of docking at the nuclear pore, uncoating, and nuclear transport. Cell Host Microbe 2018, 23, 536–548. [Google Scholar] [CrossRef] [PubMed]
- Mamede, J.I.; Cianci, G.C.; Anderson, M.R.; Hope, T.J. Early cytoplasmic uncoating is associated with infectivity of HIV-1. Proc. Natl. Acad. Sci. USA 2017, 114, E7169–E7178. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; He, Z.; Tan, T.; Li, W.; Zhang, Z.; Song, S.; Zhang, X.; Hu, Q.; Zhou, P.; Wu, Y.; Zhang, X.E.; Cui, Z. Real-time imaging of single HIV-1 disassembly with multicolor viral particles. ACS Nano 2016, 10, 6273–6282. [Google Scholar] [CrossRef] [PubMed]
- Arhel, N.; Genovesio, A.; Kim, K.A.; Miko, S.; Perret, E.; Olivo-Marin, J.C.; Shorte, S.; Charneau, P. Quantitative four-dimensional tracking of cytoplasmic and nuclear HIV-1 complexes. Nat. Methods 2006, 3, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Burdick, R.C.; Delviks-Frankenberry, K.A.; Chen, J.; Janaka, S.K.; Sastri, J.; Hu, W.S.; Pathak, V.K. Dynamics and regulation of nuclear import and nuclear movements of HIV-1 complexes. PLoS Pathog. 2017, 13. [Google Scholar] [CrossRef] [PubMed]
- Burdick, R.C.; Hu, W.S.; Pathak, V.K. Nuclear import of APOBEC3F-labeled HIV-1 preintegration complexes. Proc. Natl. Acad. Sci. USA 2013, 110, E4780–E4789. [Google Scholar] [CrossRef] [PubMed]
- Boireau, S.; Maiuri, P.; Basyuk, E.; de la Mata, M.; Knezevich, A.; Pradet-Balade, B.; Backer, V.; Kornblihtt, A.; Marcello, A.; Bertrand, E. The transcriptional cycle of HIV-1 in real-time and live cells. J. Cell Biol. 2007, 179, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Molle, D.; Maiuri, P.; Boireau, S.; Bertrand, E.; Knezevich, A.; Marcello, A.; Basyuk, E. A real-time view of the TAR:Tat:P-TEFb complex at HIV-1 transcription sites. Retrovirology 2007, 4. [Google Scholar] [CrossRef] [PubMed]
- Dieudonne, M.; Maiuri, P.; Biancotto, C.; Knezevich, A.; Kula, A.; Lusic, M.; Marcello, A. Transcriptional competence of the integrated HIV-1 provirus at the nuclear periphery. EMBO J. 2009, 28, 2231–2243. [Google Scholar] [CrossRef] [PubMed]
- Rudner, L.; Nydegger, S.; Coren, L.V.; Nagashima, K.; Thali, M.; Ott, D.E. Dynamic fluorescent imaging of human immunodeficiency virus type 1 gag in live cells by biarsenical labeling. J Virol. 2005, 79, 4055–4065. [Google Scholar] [CrossRef] [PubMed]
- Perlman, M.; Resh, M.D. Identification of an intracellular trafficking and assembly pathway for HIV-1 gag. Traffic 2006, 7, 731–745. [Google Scholar] [CrossRef] [PubMed]
- Hubner, W.; Chen, P.; Del Portillo, A.; Liu, Y.; Gordon, R.E.; Chen, B.K. Sequence of human immunodeficiency virus type 1 (HIV-1) Gag localization and oligomerization monitored with live confocal imaging of a replication-competent, fluorescently tagged HIV-1. J. Virol. 2007, 81, 12596–12607. [Google Scholar] [CrossRef] [PubMed]
- Gousset, K.; Ablan, S.D.; Coren, L.V.; Ono, A.; Soheilian, F.; Nagashima, K.; Ott, D.E.; Freed, E.O. Real-time visualization of HIV-1 GAG trafficking in infected macrophages. PLoS Pathog. 2008, 4. [Google Scholar] [CrossRef] [PubMed]
- Jouvenet, N.; Bieniasz, P.D.; Simon, S.M. Imaging the biogenesis of individual HIV-1 virions in live cells. Nature 2008, 454, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Jouvenet, N.; Simon, S.M.; Bieniasz, P.D. Imaging the interaction of HIV-1 genomes and Gag during assembly of individual viral particles. Proc. Natl. Acad. Sci. USA 2009, 106, 19114–19119. [Google Scholar] [CrossRef] [PubMed]
- Ku, P.I.; Bendjennat, M.; Ballew, J.; Landesman, M.B.; Saffarian, S. ALIX is recruited temporarily into HIV-1 budding sites at the end of gag assembly. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, J.; Baumgartel, V.; Schrimpf, W.; Ivanchenko, S.; Digman, M.A.; Gratton, E.; Krausslich, H.G.; Muller, B.; Lamb, D.C. Live-cell observation of cytosolic HIV-1 assembly onset reveals RNA-interacting Gag oligomers. J. Cell Biol. 2015, 210, 629–646. [Google Scholar] [CrossRef] [PubMed]
- Ivanchenko, S.; Godinez, W.J.; Lampe, M.; Krausslich, H.G.; Eils, R.; Rohr, K.; Brauchle, C.; Muller, B.; Lamb, D.C. Dynamics of HIV-1 assembly and release. PLoS Pathog. 2009, 5. [Google Scholar] [CrossRef] [PubMed]
- Sardo, L.; Hatch, S.C.; Chen, J.; Nikolaitchik, O.; Burdick, R.C.; Chen, D.; Westlake, C.J.; Lockett, S.; Pathak, V.K.; Hu, W.S. Dynamics of HIV-1 RNA near the plasma membrane during virus assembly. J. Virol. 2015, 89, 10832–10840. [Google Scholar] [CrossRef] [PubMed]
- Hubner, W.; McNerney, G.P.; Chen, P.; Dale, B.M.; Gordon, R.E.; Chuang, F.Y.; Li, X.D.; Asmuth, D.M.; Huser, T.; Chen, B.K. Quantitative 3D video microscopy of HIV transfer across T cell virological synapses. Science 2009, 323, 1743–1747. [Google Scholar] [CrossRef] [PubMed]
- Martin, N.; Welsch, S.; Jolly, C.; Briggs, J.A.; Vaux, D.; Sattentau, Q.J. Virological synapse-mediated spread of human immunodeficiency virus type 1 between T cells is sensitive to entry inhibition. J. Virol. 2010, 84, 3516–3527. [Google Scholar] [CrossRef] [PubMed]
- Symeonides, M.; Murooka, T.T.; Bellfy, L.N.; Roy, N.H.; Mempel, T.R.; Thali, M. HIV-1-Induced small T cell syncytia can transfer virus particles to target cells through transient contacts. Viruses 2015, 7, 6590–6603. [Google Scholar] [CrossRef] [PubMed]
- Sherer, N.M.; Lehmann, M.J.; Jimenez-Soto, L.F.; Horensavitz, C.; Pypaert, M.; Mothes, W. Retroviruses can establish filopodial bridges for efficient cell-to-cell transmission. Nat. Cell Biol. 2007, 9, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Albanese, A.; Arosio, D.; Terreni, M.; Cereseto, A. HIV-1 pre-integration complexes selectively target decondensed chromatin in the nuclear periphery. PLoS ONE 2008, 3. [Google Scholar] [CrossRef] [PubMed]
- Quercioli, V.; Di Primio, C.; Casini, A.; Mulder, L.C.F.; Vranckx, L.S.; Borrenberghs, D.; Gijsbers, R.; Debyser, Z.; Cereseto, A. Comparative analysis of HIV-1 and murine leukemia virus three-dimensional nuclear distributions. J. Virol. 2016, 90, 5205–5209. [Google Scholar] [CrossRef] [PubMed]
- Hulme, A.E.; Kelley, Z.; Foley, D.; Hope, T.J. Complementary assays reveal a low level of CA associated with nuclear HIV-1 viral complexes. J. Virol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Borrenberghs, D.; Dirix, L.; De Wit, F.; Rocha, S.; Blokken, J.; De Houwer, S.; Gijsbers, R.; Christ, F.; Hofkens, J.; Hendrix, J.; et al. Dynamic oligomerization of integrase orchestrates HIV nuclear entry. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Joo, K.I.; Lei, Y.; Lee, C.L.; Lo, J.; Xie, J.; Hamm-Alvarez, S.F.; Wang, P. Site-specific labeling of enveloped viruses with quantum dots for single virus tracking. ACS Nano 2008, 2, 1553–1562. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M.; Engelman, A.N. Capsid-dependent host factors in HIV-1 infection. Trends Microbiol. 2017, 25, 741–755. [Google Scholar] [CrossRef] [PubMed]
- Campbell, E.M.; Perez, O.; Anderson, J.L.; Hope, T.J. Visualization of a proteasome-independent intermediate during restriction of HIV-1 by rhesus TRIM5alpha. J. Cell Biol. 2008, 180, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.F.; Ellenberg, P.C.; Jones, K.L.; Fernandez, T.L.; Smyth, R.P.; Hawkes, D.J.; Hijnen, M.; Vivet-Boudou, V.; Marquet, R.; Johnson, I.; et al. Labeling of multiple HIV-1 proteins with the biarsenical-tetracysteine system. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Luban, J.; Bossolt, K.L.; Franke, E.K.; Kalpana, G.V.; Goff, S.P. Human immunodeficiency virus type 1 Gag protein binds to cyclophilins A and B. Cell 1993, 73, 1067–1078. [Google Scholar] [CrossRef]
- Braaten, D.; Ansari, H.; Luban, J. The hydrophobic pocket of cyclophilin is the binding site for the human immunodeficiency virus type 1 Gag polyprotein. J. Virol. 1997, 71, 2107–2113. [Google Scholar] [PubMed]
- Yoo, S.; Myszka, D.G.; Yeh, C.; McMurray, M.; Hill, C.P.; Sundquist, W.I. Molecular recognition in the HIV-1 capsid/cyclophilin A complex. J. Mol. Biol. 1997, 269, 780–795. [Google Scholar] [CrossRef] [PubMed]
- Javanbakht, H.; Diaz-Griffero, F.; Yuan, W.; Yeung, D.F.; Li, X.; Song, B.; Sodroski, J. The ability of multimerized cyclophilin A to restrict retrovirus infection. Virology 2007, 367, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Neagu, M.R.; Ziegler, P.; Pertel, T.; Strambio-De-Castillia, C.; Grutter, C.; Martinetti, G.; Mazzucchelli, L.; Grutter, M.; Manz, M.G.; Luban, J. Potent inhibition of HIV-1 by TRIM5-cyclophilin fusion proteins engineered from human components. J. Clin. Investig. 2009, 119, 3035–3047. [Google Scholar] [CrossRef] [PubMed]
- Sayah, D.M.; Sokolskaja, E.; Berthoux, L.; Luban, J. Cyclophilin A retrotransposition into TRIM5 explains owl monkey resistance to HIV-1. Nature 2004, 430, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Ambrose, Z.; Martin, T.D.; Oztop, I.; Mulky, A.; Julias, J.G.; Vandegraaff, N.; Baumann, J.G.; Wang, R.; Yuen, W.; et al. Flexible use of nuclear import pathways by HIV-1. Cell Host Microbe 2010, 7, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Braaten, D.; Luban, J. Cyclophilin A regulates HIV-1 infectivity, as demonstrated by gene targeting in human T cells. EMBO J. 2001, 20, 1300–1309. [Google Scholar] [CrossRef] [PubMed]
- Forshey, B.M.; von Schwedler, U.; Sundquist, W.I.; Aiken, C. Formation of a human immunodeficiency virus type 1 core of optimal stability is crucial for viral replication. J Virol. 2002, 76, 5667–5677. [Google Scholar] [CrossRef] [PubMed]
- Campbell, E.M.; Perez, O.; Melar, M.; Hope, T.J. Labeling HIV-1 virions with two fluorescent proteins allows identification of virions that have productively entered the target cell. Virology 2007, 360, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Fricke, T.; Brandariz-Nunez, A.; Wang, X.; Smith, A.B., 3rd; Diaz-Griffero, F. Human cytosolic extracts stabilize the HIV-1 core. J Virol. 2013, 87, 10587–10597. [Google Scholar] [CrossRef] [PubMed]
- Francis, A.C.; Di Primio, C.; Quercioli, V.; Valentini, P.; Boll, A.; Girelli, G.; Demichelis, F.; Arosio, D.; Cereseto, A. Second generation imaging of nuclear/cytoplasmic HIV-1 complexes. AIDS Res. Hum. Retrovir. 2014, 30, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.; Muranyi, W.; Glass, B.; Laketa, V.; Yant, S.R.; Tsai, L.; Cihlar, T.; Muller, B.; Krausslich, H.G. Quantitative microscopy of functional HIV post-entry complexes reveals association of replication with the viral capsid. eLife 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Di Nunzio, F.; Fricke, T.; Miccio, A.; Valle-Casuso, J.C.; Perez, P.; Souque, P.; Rizzi, E.; Severgnini, M.; Mavilio, F.; Charneau, P.; et al. Nup153 and Nup98 bind the HIV-1 core and contribute to the early steps of HIV-1 replication. Virology 2013, 440, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Blair, W.S.; Pickford, C.; Irving, S.L.; Brown, D.G.; Anderson, M.; Bazin, R.; Cao, J.; Ciaramella, G.; Isaacson, J.; Jackson, L.; et al. HIV capsid is a tractable target for small molecule therapeutic intervention. PLoS Pathog. 2010, 6. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Alam, S.L.; Fricke, T.; Zadrozny, K.; Sedzicki, J.; Taylor, A.B.; Demeler, B.; Pornillos, O.; Ganser-Pornillos, B.K.; Diaz-Griffero, F.; et al. Structural basis of HIV-1 capsid recognition by PF74 and CPSF6. Proc. Natl. Acad. Sci. USA 2014, 111, 18625–18630. [Google Scholar] [CrossRef] [PubMed]
- Price, A.J.; Jacques, D.A.; McEwan, W.A.; Fletcher, A.J.; Essig, S.; Chin, J.W.; Halambage, U.D.; Aiken, C.; James, L.C. Host cofactors and pharmacologic ligands share an essential interface in HIV-1 capsid that is lost upon disassembly. PLoS Pathog. 2014, 10. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.R.; Perreira, J.M.; Savidis, G.; Portmann, J.M.; Aker, A.M.; Feeley, E.M.; Smith, M.C.; Brass, A.L. Direct visualization of HIV-1 replication intermediates shows that capsid and cpsf6 modulate hiv-1 intra-nuclear invasion and integration. Cell Rep. 2015, 13, 1717–1731. [Google Scholar] [CrossRef] [PubMed]
- Schaller, T.; Ocwieja, K.E.; Rasaiyaah, J.; Price, A.J.; Brady, T.L.; Roth, S.L.; Hue, S.; Fletcher, A.J.; Lee, K.; KewalRamani, V.N.; et al. HIV-1 capsid-cyclophilin interactions determine nuclear import pathway, integration targeting and replication efficiency. PLoS Pathog. 2011, 7. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Dobro, M.J.; Woodward, C.L.; Levandovsky, A.; Danielson, C.M.; Sandrin, V.; Shi, J.; Aiken, C.; Zandi, R.; Hope, T.J.; et al. Unclosed HIV-1 capsids suggest a curled sheet model of assembly. J. Mol. Biol. 2013, 425, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Di Primio, C.; Quercioli, V.; Allouch, A.; Gijsbers, R.; Christ, F.; Debyser, Z.; Arosio, D.; Cereseto, A. Single-cell imaging of HIV-1 provirus (SCIP). Proc. Natl. Acad. Sci. USA 2013, 110, 5636–5641. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Wang, M.; Li, W.; Zhang, Z.; Zhang, X.; Wu, G.; Tan, T.; Cui, Z.; Zhang, X.E. Live visualization of HIV-1 proviral DNA using a dual-color-labeled crispr system. Anal. Chem. 2017, 89, 12896–12901. [Google Scholar] [CrossRef] [PubMed]
- Huisken, J.; Swoger, J.; Del Bene, F.; Wittbrodt, J.; Stelzer, E.H. Optical sectioning deep inside live embryos by selective plane illumination microscopy. Science 2004, 305, 1007–1009. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Francis, A.C.; Melikyan, G.B. Live-Cell Imaging of Early Steps of Single HIV-1 Infection. Viruses 2018, 10, 275. https://doi.org/10.3390/v10050275
Francis AC, Melikyan GB. Live-Cell Imaging of Early Steps of Single HIV-1 Infection. Viruses. 2018; 10(5):275. https://doi.org/10.3390/v10050275
Chicago/Turabian StyleFrancis, Ashwanth C., and Gregory B. Melikyan. 2018. "Live-Cell Imaging of Early Steps of Single HIV-1 Infection" Viruses 10, no. 5: 275. https://doi.org/10.3390/v10050275
APA StyleFrancis, A. C., & Melikyan, G. B. (2018). Live-Cell Imaging of Early Steps of Single HIV-1 Infection. Viruses, 10(5), 275. https://doi.org/10.3390/v10050275